
ICOTINIB
N-(3-Ethynylphenyl)-7,8,10,11,13,14-hexahydro[1,4,7,10]tetraoxacy
[1,4,7,10]Tetraoxacyclododecino[2,3-g]quinazolin-4-amine, N-(3-ethynylphenyl)-7,8,10,11,13,14-hexahydro-
BPI 2009H 610798-31-7 CAS BASE

Icotinib Hydrochloride, 1204313-51-8, CS-0918, HY-15164, Conmana Zhejiang Beta Pharma Ltd.
Icotinib is a potent and specific EGFR inhibitor with IC50 of 5 nM, including the EGFR, EGFR(L858R), EGFR(L861Q), EGFR(T790M) and EGFR(T790M, L858R). Phase 4.Icotinib hydrochloride is the epidermal growth factor receptor kinase targeting a new generation of targeted anti-cancer drugs, completely independent from the original tumor clinical practitioners and experts of science, through eight years of the development, its first adaptation disease is advanced non-small cell lung cancer. Icotinib is an orally available quinazoline-based inhibitor of epidermal growth factor receptor (EGFR), with potential antineoplastic activity. Icotinib selectively inhibits the wild-type and several mutated forms of EGFR tyrosine kinase. This may lead to an inhibition of EGFR-mediated signal transduction and may inhibit cancer cell proliferation. EGFR, a receptor tyrosine kinase, is upregulated in a variety of cancer cell types. Icotinib was approved in China in 2011
Icotinib has been found to be noninferior to gefitinib in patients with non-small-cell lung cancer (NSCLC), according to reports from the phase III Chinese double-blind ICOGEN study.
“[I]cotinib is a valid therapeutic option for patients with non-small-cell lung cancer as a second-line or third-line treatment, although patients might find taking icotinib three times a day an inconvenience,” write Yan Sun (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China) and colleagues.
Icotinib is an oral epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) that has exhibited good antitumor activity in phase II studies. However, it has a shorter half-life than gefitinib, another TKI, which means that it needs to be taken more often.
Label-free cell phenotypic assessment of the molecular mechanism of action of epidermal growth factor receptor inhibitors. Huayun Deng, Chaoming Wang, Ye Fang, RSC Adv., 2013, 3, 10370
Design and discovery of 4-anilinoquinazoline ureas as multikinase inhibitors targeting BRAF, VEGFR-2 and EGFR. Qingwen Zhang, Yuanyuan Diao, Fei Wang, Ying Fu, Fei Tang, Qidong You, Houyuan Zhou, Med. Chem. Commun., 2013, 4, 979
...
- Tyrosine kinase receptors are trans-membrane proteins that, in response to an extracellular stimulus, propagate a signaling cascade to control cell proliferation, angiogenesis, apoptosis and other important features of cell growth. One class of such receptors, epidermal growth factor receptor (EGFR) tyrosine kinases, are over-expressed in many human cancers, including brain, lung, liver, bladder, breast, head and neck, esophagus, gastrointestinal, breast, ovary, cervix or thyroid cancer.
- EGFR is expressed in many types of tumor cells. Binding of cognate ligands (including EGF, TGFα (i.e., Transforming Growth Factor-α) and neuregulins) to the extracellular domain causes homo- or heterodimerization between family members; the juxtaposition of cytoplasmic tyrosine kinase domains results in transphosphorylation of specific tyrosine, serine and threonine residues within each cytoplasmic domain. The formed phosphotyrosines act as docking sites for various adaptor molecules and subsequent activation of signal transduction cascades (Ras/mitogen-activated, PI3K/Akt and Jak/STAT) that trigger proliferative cellular responses.
- Various molecular and cellular biology and clinical studies have demonstrated that EGFR tyrosine kinase inhibitors can block cancer cell proliferation, metastasis and other EGFR-related signal transduction responses to achieve clinical anti-tumor therapeutic effects. Two oral EGFR kinase inhibitors with similar chemical structures are Gefitinib (Iressa; AstraZeneca), approved by the U.S. FDA for advanced non-small cell lung cancer in 2003 (and later withdrawn), and Erlotinib Hydrochloride (Tarceva; Roche and OSI), approved by the U.S. FDA for advanced non-small cell lung cancer and pancreatic cancer treatment in 2004.
- Chinese Patent Publication No. CN1305860C discloses the structure of 4-[(3-ethynyl-phenyl)amino]-6,7-benzo-12-crown-quinoline (free base) on page 29, Example 15, Compound 23.
Icotinib was launched in China in August 2011, after approval by the State Food and Drug Administration. It is a targeted EGFR tyrosine kinase inhibitor that, like erlotinib (Tarceva) and gefitinib (Iressa), shows benefit in patients with EGFR m+ NSCLC.
............................................ http://www.google.com/patents/EP2392576A1
- Formula I (Icotinib hydrochloride):

Method 1:

Method 2:

Method 3:
- BPI-02 is obtained by recrystallization.

http://www.google.com/patents/EP2392576A1 Example 1Step 1
- Preparation: 16 kg (400 mol) of sodium hydroxide was dissolved in 80 L of water in a 400 L reactor, and then 18.8 L (140 mol) of triethylene glycol, 32 L of THF were added into the reactor. After cooling below 5 °C, a solution of 47.84 kg (260 mol) of tosyl chloride and 50 L of THF was added dropwise. Following the addition, the reaction mixture was kept at this temperature for 2 hours, and it was then poured into 240 L of ice water. The precipitate was formed and filtered, washed with a small amount of water, and dried. 58.64 kg of BPI-01 as a white crystalline powder was yielded at 91.4%. mp: 77-80 °C, HPLC: 97%. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.87.
- NMR data: 1H-NMR (CDCl3): δ ppm: 7.78 (d, 4H, J = 10.4 Hz, benzene protons by sulfonyl group); 7.34 (d, 4H, J = 11.6 Hz, benzene protons by methyl group); 4.129 (dd, 4H, J = 5.6 Hz, ethylene protons by the sulfonyl group); 3.64 (dd, 4H, J = 5.6 Hz, ethylene protons away from the sulfonyl group); 3.517 (s, 4H, ethylene protons in the middle); 2.438 (s, 6H, methyl protons on the benzene).

Step 2
- Preparation: A solution containing 3.64 kg (20 mol) of ethyl 3,4-dihydroxybenzoate and 12.4 kg (89.6 mol) of potassium carbonate in 300 L of N,N-dimethylformamide was stirred and heated to 85-90 °C for about 30 minutes. A solution of 9.17 kg (20 mol) of BPI-01 in 40 L of N,N-dimethylformamide was added dropwise over 1.5-2 hours. After the addition, the reaction was kept for 30 minutes; the reaction completion was confirmed by TLC (developing solvent: petroleum ether:ethyl acetate = 1:1, Rf = 0.58). The reaction mixture was removed from the reactor and filtered. Then, the filtrate was evaporated to remove N,N-dimethylformamide; 240 L of ethyl acetate was added to dissolve the residue. After filtration and vacuum evaporation, the residual solution was extracted with 300 L of petroleum ether. After evaporation of the petroleum ether, the residual solids were re-crystallized with isopropanol in a ratio of 1:2.5 (W/V); 1.68 kg of BPI-02 as a white powder was obtained in a yield of 28%. mp: 73-76 °C, HPLC: 96.4%. NMR data: 1H-NMR (CDCl3): δ ppm: 7.701 (d, 1H, J = 2.4 Hz, benzene proton at position 6); 7.68 (s, 1 H, benzene proton at position 2); 6.966 (d, 1H, J = 10.8 Hz, benzene proton at position 5); 4.374-3.81 (q, 2H, J = 9.6 Hz, methylene protons of the ethyl); 3.78-4.23 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.394 (t, 3H, J = 9.6 Hz, methyl protons of the ethyl). MS: m/z 296.

Step 3
- Preparation: A solution of 592 g (2 mol) of BPI-02 and 600 mL of acetic acid in a 5 L reaction flask was cooled to 0°C; 1640 mL (25.4 mol) of concentrated nitric acid was slowly added. The internal temperature should not exceed 10 °C. While cooled below 0°C, 1 L of concentrated sulfuric acid was added dropwise. The internal temperature should not be higher than 5°C. After the addition, the reaction was kept at 0-5 °C for 1-2 hours. After completion of the reaction, the reaction solution was poured into 15 L of ice water in a plastic bucket. After mixing, filtration, and re-crystallization in ethanol, 449 g of BPI-03 as a light yellow to yellow crystalline powder was obtained in 65.7% yield. mp: 92-95 °C, HPLC: 98.2%. TLC (petroleum ether: ethyl acetate =1:1) Rf = 0.52. NMR data: 1H-NMR (CDCl3): δ ppm: 7.56 (s, 1H, benzene proton at position 5); 7.20 (s, 1H, benzene proton at position 2); 4.402 (q, 2H, J = 9.2 Hz, methylene protons of the ethyl); 4.294 (dd, 12H, J = 4.8 Hz, crown ether protons); 1.368 (t, 3H, J = 9.2 Hz, methyl protons of the ethyl).

Step 4
- Preparation: In a 3 L hydrogenation reactor, 2 L of methanol and 195 g (0.57 mol) of BPI-03 were added, and then 63 mL of acetyl chloride was slowly added. After a short stir, 33 g of Pd/C containing 40% water was added. The reaction was conducted under 4 ATM hydrogen until hydrogen absorption stopped, and then the reaction was kept for 1-2 hours. After completion of the reaction, the reaction mixture was transferred into a 5 L reactor. After filtration, crystallization, and filtration, the product was obtained. The mother liquor was concentrated under vacuum, and more product was obtained. The combined crops were 168 g of BPI-04 as a white to pink crystalline powder in a yield of 85%. mp: 198-201 °C, HPLC: 99.1 %. TLC (petroleum ether: ethyl acetate = 1:1) Rf = 0.33. NMR data: 1H-NMR (DMSO-d6): δ ppm: 8-9 (br., 3H, 2 protons of the amino group and a proton of the hydrochloric acid); 7.37 (s, 1H, benzene proton at position 5); 6.55 (s, 1H , benzene proton at position 2); 4.25 (q, 2H, J = 7.06 Hz, methylene protons of the ethyl); 4.05 (dd, 12H, J = 4.04 Hz, crown ether protons); 1.31 (t, 3H, J = 7.06 Hz, methyl protons of the ethyl).

Step 5
- Preparation: 1105 g (3.175 mol)of BPI-04, 4810 g (106.9 mol) of formamide, and 540 g (8.55 mol) of ammonium formate were added to a 10 L 3-neck bottle. The reaction mixture was heated to 165 °C under reflux for 4 hours. After cooling to room temperature, 3 L of water was added, and then the mixture was stirred for 10 minutes. After filtration, washing, and drying, 742 g of BPI-05 as a white crystalline powder was obtained in a yield of 80%. mp: 248-251 °C, HPLC: 99.78%. TLC (chloroform: methanol = 8:1) Rf = 0.55. NMR data: 1H-NMR (DMSO-d6): δ ppm: 12.06 (s, 1H, NH of the quinazoline); 8.0 (d, 1H, J = 3.28 Hz, proton of the quinazoline position 3); 7.62 (s, 1H, proton of the quinazoline position 6); 7.22 (s, 1H, proton of the quinazoline position 9); 4.25 (dd, 12H, J = 4.08 Hz, crown ether protons).

Step 6
- Preparation: 337 g (1.13 mol) of BPI-05, 7.1 L of chloroform, 1.83 L (19.58mol) of POCI3 and 132 ml of N,N-dimethylformamide were added to a 10 L 3-neck bottle. The reaction mixture was stirred at reflux temperature. After dissolution, reaction completion was checked by TLC (developing solvent: chloroform: methanol = 15:1, Rf = 0.56); the reaction took approximately 8 hours to complete. Then, the reaction solution was cooled and evaporated under vacuum to dryness. The residue was dissolved in 4 L of chloroform; 4 kg of crushed ice was poured into the solution and the mixture was stirred for 0.5 hours. After separation, the aqueous phase was extracted twice with 2 L of chloroform. The organic phases were combined, 4 L of ice water was added and the pH was adjusted with 6 N NaOH to pH 8-9 while the temperature was maintained below 30 °C. After separation, the organic phase was washed with saturated NaCl, dried over anhydrous sodium sulfate and the solvents removed by vacuum evaporation. The residual solids were washed with acetone and filtered; 268 g of BPI-06 as a white crystalline powder was obtained in a yield of 77% with mp: 164-167°C and HPLC purity of 99%. NMR data: 1H-NMR (CDCl3): δ ppm: 8.89 (s, 1H, proton of the quinazoline position 2); 7.68 (s, 1H, proton of the quinazoline position 9); 7.42 (s, 1H, proton of the quinazoline position 6); 4.38-3.81 (dd, 12H, J = 3.88 Hz, crown ether protons).

Step 7
- Preparation of the compound of the present invention: To a suspension of 20.8 g of BPI-06 in 500 mL of ethanol was added 25 mL of N,N-dimethylformamide and a solution of 8.98 g m-acetylene aniline in 200 mL of isopropanol. The reaction mixture was stirred at room temperature for 5 minutes until dissolved completely, and then the reaction solution was heated at reflux for 3 hours. After concentration and drying, the residual solids were dissolved in ethyl acetate, washed with water, and dried over anhydrous sodium sulfate. Thus, 27.1 g of the compound of Formula I was obtained as a white crystalline powder. NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.

.........................................

Synthesis of compound 1 A
1 Synthesis of Compound 2

2
79.5g 3,4 - dihydroxybenzene nitrile, 272g of potassium carbonate, acetonitrile (6L) was added to a 10L three-necked reaction flask, and dissolved with stirring, heated to reflux and reflux was added dropwise an acetonitrile solution of the compound 1 (compound 1, 200 g; acetonitrile , 2L), and completion of the dropping, the HPLC monitoring of the completion of the reaction, the mixture was cooled to room temperature, filtered, and the solvent was removed, and the resulting solid was washed with ethyl acetate was dissolved, filtered, and the filtrate was concentrated, the resulting residue was dissolved in petroleum ether by rotary evaporation, the resulting solid was purified to give 18.9g of the compound 2.
1 LAI MR (CDC1 3-Sppm): 7.30 ~ 7.33 (m, 1H); 7.25 (s, 1H); 6.97-6.99 (d, 1H); 4.19 - 4.23 (m, 4H); 3.83 ~ 3.91 (m, 4H); 3.77 (s, 4H). MS: (M + H) +250 2 Synthesis of compound A

2 A
41.6g of compound 2 was dissolved in 580ml of acetic acid, dropwise addition of 83ml of fuming nitric acid at 30 ° C under completion of the dropping, the dropwise addition of 42ml of concentrated sulfuric acid at 30 ° C under the reaction at room temperature overnight, TLC monitoring completion of the reaction, the reaction solution was poured into ice water 4L , the precipitated solid was filtered, washed with cold water (500 mL X 2), vacuum 35 ° C and dried crude A compound 46g, isopropanol recrystallization was purified to give 33g of compound A.
1 LAI MR (CDC1 3-Sppm): 7.90 (s, 1H); 7.36 (s, 1H); 4.33 ~ 4.36 (m, 4H); 3.87 ~ 3.89 (m, 4H); 3.737 (s, 4H). Embodiment of Example 2 Synthesis of Compound B

AB
32g of compound A, 30.5g of iron powder, 5% acetic acid solution in methanol 1070ml 2L reaction flask was heated to reflux
TLC monitoring of the end of the reaction cooled and concentrated, dissolved in ethyl acetate, filtered, dried over anhydrous NaS0 4 23g of compound B. The solvent was removed.
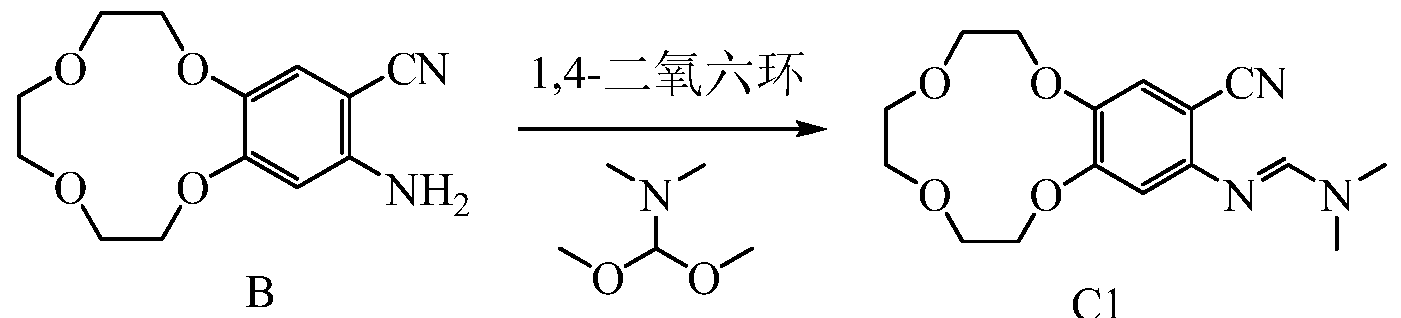
1HNMR (d 6-DMSO-Sppm): 7.07 (s, 1H); 6.36 (s, 1H); 5.73 (s, 2H); 3.95 ~ 4.22 (m, 4H); 3.77-3.78 (m, 2H); 3.34 3.62 (m, 6H).Embodiment of Example 3 Synthesis of Compound CI

B CI
500mL three-necked flask, the Add 5g compound B, 5g v, v-dimethyl formamide dimethyl acetal and 160ml of dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time is about 12 hours, after the end of the reaction The reaction solution was cooled to room temperature, spin-dry to give 5.8g of compound Cl.
1 LAI MR (CDCl 3-Sppm): 7.56 (s, 1H); 7.15 (s, 1H); 6.51 (s, 1H); 4.12-4.18 (m, 4H); 3.89-3.91 (m, 2H); 3.78 -3.80 (m, 6H); 3.07 (s, 6H); Example 4 Icotinib Synthesis

5 g of the compound Cl, 2.2 g inter-aminophenyl acetylene, 230ml of acetic acid was added to a 500 ml reaction flask was heated to 100 ° c,
TLC monitoring of the reaction. The end of the reaction, the reaction system spin dry methanol was added, and shock dispersion, filtration, wash with methanol, 5g Icotinib.
^ M (d 6-DMSO-5ppm): 11.98 (s, IH); 9.50 (s, IH); 8.53 (s 1H); 8.14 (s, IH); 8.04-8.05 (m, IH); 7.90-7.92 (m, IH); 7.38-7.42 (m, IH); 7.31 (s IH); 7.20-7.22 (m, IH); 4.29-4.30 (m, 4H); 4.21 (s, IH); 3.74-3.81 ( m, 4H); 3.64 (s, 4H); 1.91 (s, 3H); Synthesis Example 5 Exe hydrochloride erlotinib

Exeter for Nick for; s
700mg Icotinib Add to a 100 ml reaction flask, add 40 ml of methanol, stirred pass into the hydrogen chloride gas or concentrated hydrochloric acid, and filtered to give crude hydrochloric acid Icotinib after, and purified by recrystallization from isopropanol to give 760mg hydrochloride Icotinib.
1HNMR (d 6-DMSO-Sppm): 11.37 (s, IH); 8.87 (s, IH); 8.63 (s, IH); 7.90 (s, IH); 7.78-7.80 (d, IH); 7.48-7.52 (m, IH); 7.40-7.41 (m, 2H); 4.36-4.38 (d, 4H); 4.30 (s, IH); 3.75-3.81 (d, 4H); 3.61 (s, 4H); Example 6 Synthesis of Compound B

AB
25g of compound A, 25 g of iron powder, 3% acetic acid in methanol solution 900ml with Example 2 are the same, to give 16.6g of compound B.
Embodiment of Example 7 Synthesis of Compound B

AB
40 g of compound A, 40 g of iron powder and 7% acetic acid in methanol solution was 1200ml, in Example 2, to give 28.4g of compound B.
Example 8 Compound B Synthesis

AB
25 g of compound A, 5 g of Pd / C in 3% acetic acid in methanol solution 900ml Add 2L reaction flask, of the hydrogen, TLC monitoring of the end of the reaction, filtered, and the solvent was removed to give 17g of compound B.
Example 9 Compound B Synthesis

AB
40g of compound A, 17 g of magnesium and 5% acetic acid in methanol solution 1200ml, in Example 2, to give 25.2g of compound B. Example 10 Compound B Synthesis

AB
25 g of compound A, 32.5g of zinc powder and 5% acetic acid in methanol solution 900ml with Example 2 are the same, to give 17.1g of compound B.
Example Synthesis of compound 11 B

AB
25g of compound A, 28 g of iron powder, 5% trifluoroacetic acid in methanol solution 700ml, in Example 2, 16g of compound B.
Embodiment Example 12 Synthesis of Compound C1

3g compound B, 3G v, v-dimethyl formamide dimethyl acetal and 140ml of dioxane, reflux the reaction time is 10-11 hours, the other in the same manner as in Example 3 to give 3.2g of the compound Cl.
Example 13 Synthesis of Compound C1
8g compound B, 8G N, v-dimethyl formamide dimethyl acetal and 180ml of dioxane under reflux for a reaction time of approximately 12-13 hours, with the same manner as in Example 3 to give 8.7g of compound C. Embodiment Example 14 Synthesis of Compound CI

3g compound B, 3 g of N, N-dimethyl formamide dimethyl acetal and 140ml of toluene, the reaction time is 13-15 hours under reflux, with the same manner as in Example 3 to give 2.9g of the compound Cl.
Example 15 Synthesis of Compound C1

The same as in Example 14, except that reaction time is 10 hours, to obtain 2.6g compound Cl t
Embodiment Example 16 Synthesis of Compound C1

500mL three-necked flask, add 3 g of compound B, 3.7 g v, v-dimethylformamide, diethyl acetal and 140ml of dioxane was heated to reflux, TLC monitoring the progress of the reaction, the reaction time of approximately 11-12 hours, After completion of the reaction, the mixture was cooled to room temperature, spin-dry the reaction solution to give 2.5g of the compound Cl.
Example 17 Synthesis of Compound C1

G of compound B, 5.1 g of the N, N-dimethyl formamide di-t-butyl acetal was dissolved in 140ml dioxane was heated to reflux the TLC monitoring progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.6g of the compound Cl.
Embodiment Example 18 Synthesis of Compound CI

3g compound B, 4.4g N, N-dimethyl formamide diisopropyl acetal was dissolved in 140ml dioxane was heated to reflux, tlc monitoring the progress of the reaction, the reaction time of approximately 11-12 hours after the completion of the reaction, was cooled to room temperature, the reaction solution was spin-dry to give 2.4g of the compound Cl.
The implementation of the synthesis of Example 19 Icotinib

3g compound Cl, 1.3 g inter-aminophenyl acetylene, 130 ml of acetic acid was added 250 ml reaction flask and heated to 70-80
V, TLC monitoring of the reaction. Spin dry the reaction system, methanol was added, and shock dispersion, filtered, and the methanol wash was 2.8g Icotinib. Implementation of Example 20 Icotinib synthesis

C1 Icotinib
. Example 25 Icotinib Hydrochloride synthesis

Icotinib Hydrochloride
The 500mg Icotinib Add to a 100 ml reaction flask, add 30ml of ethanol was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 515mg hydrochlorideIcotinib. Example 26 Icotinib Hydrochloride Synthesis
500mg Icotinib Add 100 ml reaction flask, add 40 ml of tetrahydrofuran was stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochlorideIcotinib. EXAMPLE 27 Icotinib Hydrochloride Synthesis

500mg Icotinib Add 100 ml reaction flask, add 50 ml of isopropanol and stirred under hydrogen chloride gas was passed into the after, filtered crude hydrochloride Icotinib recrystallized from isopropanol to give 500mg hydrochloride Icotinib.
............................................................................
http://www.google.com/patents/EP2392576A1 NMR data: 1H-NMR (Bruker APX-400, solvent: DMSO-d6, TMS as internal standard): δ ppm: 3.58 (dd, 2H, two protons of the crown position 12); 3.60 (dd, 2H, two protons of the crown position 13); 3.73 (dd, 2H, two protons of the crown position 10); 3.80 (dd, 2H, two protons of the crown position 15); 4.30 (s, 1H, proton of the alkynyl); 4.34 (dd, 2H, two protons of the crown position 16); 4.40 (dd, 2H, two protons of the crown position 9); 7.39 (d, 1H, benzene proton at position 25); 7.46 (dd, 1H, benzene proton at position 26); 7.49 (s, 1H, proton of the quinazoline position 6); 7.82 (d, 1H, benzene proton at position 27); 7.94 (t due dd, 1H, proton of the quinazoline position 19); 8.85 (s, 1H, benzene proton at the position 23); 8.87 (s, 1H, proton of the quinazoline position 2); 11.70 (s, 1H, proton of the aromatic amine as salt); 14-16 (bs, 1H, hydrochloride), see Figure 5. NMR data: 13C-NMR (DMSO-d6), see Figure 6. Mass spectrometry (MS): Instrument: ZAB-HS, testing conditions: EI, 200°C, 700ev, MS measured molecular weight: m/z 427.
No comments:
Post a Comment