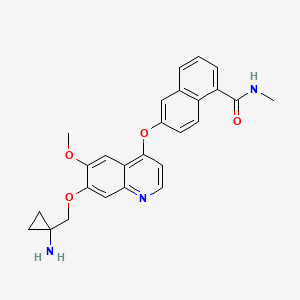
LUCITANIB
6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide
6-(7-((l-aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)- N-methyl- 1 -naphthamide
1058137-23-7 (E-3810 free base); 1058137-84-0 (E-3810 HCl salt)
E-3810, E-3810 amine, UNII-PP449XA4BH, E3810, Lucitanib [INN], AL3810
Molecular Weight:443.4944 g/mol
| PATENT | SUBMITTED | GRANTED |
|---|
| Spiro Substituted Compounds As Angiogenesis Inhibitors [US8163923] | 2008-09-18 | 2012-04-24 |
A 4-(3-methoxypropoxy)-3-methylpyridinyl derivative of timoprazole that is used in the therapy of STOMACH ULCERS and ZOLLINGER-ELLISON SYNDROME. The drug inhibits H(+)-K(+)-EXCHANGING ATPASE which is found in GASTRIC PARIETAL CELLS.
For in advanced solid tumors.
Lucitanib (E-3810): Lucitanib, also known as E-3810, is a novel dual inhibitor targeting human vascular endothelial growth factor receptors (VEGFRs) and fibroblast growth factor receptors (FGFRs) with antiangiogenic activity. VEGFR/FGFR dual kinase inhibitor E-3810 inhibits VEGFR-1, -2, -3 and FGFR-1, -2 kinases in the nM range, which may result in the inhibition of tumor angiogenesis and tumor cell proliferation, and the induction of tumor cell death. Both VEGFRs and FGFRs belong to the family of receptor tyrosine kinases that may be upregulated in various tumor cell type
Overview
Lucitanib is an oral, potent inhibitor of the tyrosine kinase activity of fibroblast growth factor receptors 1 through 3 (FGFR1-3), vascular endothelial growth factor receptors 1 through 3 (VEGFR1-3) and platelet-derived growth factor receptors alpha and beta (PDGFR α-ß). We own exclusive development and commercial rights to lucitanib on a global basis, excluding China. Lucitanib rights to markets outside of the U.S. and Japan have been sublicensed to Les Laboratoires Servier (Servier). We are collaborating with Servier on the global clinical development of lucitanib.

A Phase I/IIa clinical trial of lucitanib was initiated in 2010 and has demonstrated multiple objective responses in FGFR1 gene-amplified breast cancer patients, and objective responses were also observed in patients with tumors often sensitive to VEGFR inhibitors, such as renal cell and thyroid cancer. FGFR amplification is common in a number of tumor types, including breast cancer and squamous non-small cell lung cancer, and we intend to study lucitanib in these cancers as well as other solid tumors exhibiting FGFR pathway activation. A broad Phase II development program has been initiated by us and Servier in multiple indications, including advanced breast cancer and squamous NSCLC. For more information or to participate in the trials, contact the Clovis Oncology Clinical Trial Navigation Service at 1-855-262-3040, or 303-625-5010, or
clovistrials@emergingmed.com.

WO 2008/112408 Al and US 2008/0227812 Al disclose angiogenesis inhibitors with quinoline structure, useful for the treatment of neoplasias. One of the disclosed products is 6-(7-((l-aminocyclopropyl)methoxy)-6- methoxyquinolin-4-yloxy)-N-methyl-l-naphthamide of formula (I), described in example 3 of the above mentioned patent applications.
According to said documents, compound (I) is prepared by removing the benzyloxycarbonyl protective group from the compound benzyl l-((6- methoxy-4-(5-(methylcarbamoyl)-naphthalen-2-yloxy)quinolin-7- yloxy)methyl)cyclopropyl carbamate (II):
in acid medium or by hydrogenolysis, to give compound (I).
Compound (II) is obtained in a number of steps with different processes in which the benzyloxycarbonyl protected 1 -amino- 1-cyclopropylmethyl moiety is introduced by subjecting the acyl azide obtained from l-((6- methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7- yloxy)methyl)cyclopropanecarboxylic acid of formula (III):
to Curtius rearrangement, in the presence of benzyl alcohol, or by alkylation of 6-(7-hydroxy-6-methoxyquinolin-4-yloxy)-N- methyl-1-naphthamide of formula (IV):
with 1 -benzyloxy carbony lamino- 1 -methylsolfonyloxymethyl- cyclopropane of formula (V):
The above mentioned applications do not provide yields concerning both the preparation of compound (II) by the two above mentioned reactions, and the conversion of compound (II) to (I).
Compound (III) is prepared by a process in which the 1-carboxy-l- cyclopropylmethyl moiety is introduced in 4-hydroxy-3-methoxyacetophenone as in the form of the ethyl ester, followed by formation of the 4- hydroxyquinoline ring and, finally, by the introduction of the 1- naphthylcarboxyamido fragment.
It is well known that the reactions requiring the use of azides, such as the formation of acyl azides, or Curtius rearrangement of the latter, are potentially hazardous as they involve risk of explosions, therefore they are not suitable for use in preparations on large scale. The synthetic methods reported in WO 2008/1 12408 and US
2008/0227812 include, inter alia, a general synthetic scheme in which the cycloalkyl-alkyl portion of the products is introduced by reaction between a cycloalkyl-alkyl mesylate and an hydroxy or amino acetophenone, followed by nitration to give a nitroacetofenone, reduction of the nitro group to amino group, formation of the 4-hydroxyquinoline ring and further work up of the latter to the final products. The above mentioned applications do not provide examples of the use of this process for compound (I) or the other described products.
………………………..
Patent
…………………………….
sequence of intermediates……………

LUCITANIB
Example 1: Preparation of l-[(4-acetyl-2-methoxyphenoxy)methyl]- N-benzyloxycarbonyl-1-aminocyclopropane

A 10 L reactor equipped with mechanical stirrer was loaded with triphenylphosphine (340.0 g, 1.296 mol) and THF (2 L) and the suspension was cooled with an ice bath. The stirred suspension was then slowly added with DIAD (264 g, 1.296 mol) over 30 minutes. After stirring for 30 min at 00C, the stirred suspension was added dropwise with a solution of 4-hydroxy- 3-methoxyacetofenone (180 g, 1.08 mol) and DIPEA (210 g, 1.62 mol) in THF (1500 mL). The suspension was left under stirring for 45 min at 00C, then added dropwise with a solution of 1-benzyloxycarbonylamino-l- hydroxymethylcyclopropane (China Gateway) (240 g, 1.08 mol) in THF (1500 mL). After Ih, LC-MS analysis of a sample from the reaction mixture showed the complete disappearance of 1-benzyloxycarbonylamino-l- hydroxymethylcyclopropane. The reaction mixture was evaporated and the crude product was recrystallized with EtOH 95% (4000 mL) to give l-[(4- acetyl-2-methoxyphenoxy)methyl]-N-benzyloxycarbonyl- 1 – aminocyclopropane (214 g, yield: 53.5%) as a white solid.
1H-NMR (300 MHz, CDCl3): δ: 7.41-7.45 (m, 2 H), 7.26 (s, 5 H), 6.77 (d, 1 H), 5.43 (s, 1 H), 5.00 (s, 2 H), 4.04 (s, 2 H), 3.82 (s, 3 H), 2.49 (s, 3H), 0.92 (m, 4 H).
LC-MS: M+H+: 370.4
Example 2: Preparation of l-[(4-acetyl-2-methoxy-5- nitrophenoxy)methyl]-N-benzyloxycarbonyl-l-aminocycIopropane
A solution of HNO3 (65%, 3 mL) in Ac2O (2 mL) at 0°C was slowly added with a suspension of the compound of Example 1 (1.1 g, 2.9 mmol) in
Ac2O (3 mL). After stirring at 00C for 2 h, the reaction mixture was poured into 50 mL of ice/water and the precipitate was recovered by filtration. The resulting yellow solid was recrystallized with 95% EtOH (5 mL) to give l-[(4- acetyl-2-methoxy-5-nitrophenoxy)methyl]-N-benzyloxycarbonyl-l- aminocyclopropane (0.69 g, yield: 56%) as a yellow solid.
1H-NMR (300 MHz, CDCl3): δ: 7.52 (s, 1 H), 7.26 (s, 5 H), 6.67 (s, 1 H), 5.36 (s, IH), 5.02 (s, 2 H), 4.05 (s, 2 H), 3.86 (s, 3 H), 2.42 (s, 3 H), 0.94 (m, 4 H).
LC-MS: M+H+: 414.41
Example 3: Preparation of l-[(4-(3-dimethylaminopropenoyl)-2- methoxy-5-nitrophenoxy)methyl]-N-benzyloxycarbonyl-l- aminocyclopropane
A mixture of the compound of Example 2 (1.7 g, 4.1 mmol) and N5N- dimethylformamide dimethylacetal (0.9 g, 8.2 mmol) in DMF (6 mL) was stirred at 1000C for 2 h. After cooling at room temperature, the reaction mixture was diluted with water (30 mL) and extracted with AcOEt (3 x 50 mL). The combined organic phases were washed with brine (2 x 50 mL), dried and evaporated to give l-[(4-(3-dimethylaminopropenoyl)-2-methoxy-5- nitrophenoxy)methyl]-N-benzyloxycarbonyl-l -aminocyclopropane (1.9 g, yield: 95%) as a yellow solid. 1H-NMR (300 MHz, CDCl3): δ: 7.50 (s, 1 H), 7.27 (s, 5 H), 6.75 (s, 1
H), 5.44 (s, 1 H), 5.23 (s, 1 H), 5.1 1 (br, 1 H), 5.01 (s, 2 H), 4.04 (s, 2 H), 3.83 (s, 3 H), 2.78-3.00 (m, 6 H), 0.94 (m, 4 H) LC-MS: M+H+: 470.49
Example 4: Preparation of l-[(4-hydroxy-6-methoxyquinolin-7- yloxy)methyl]-N-benzyloxycarbonyl-l-aminocyclopropane
A mixture of the compound of Example 3 (1.5 g, 3.2 mmol) and powder iron (1.8 g, 32 mmol) in AcOH (15 mL) was stirred a 800C for 2 h. The reaction mixture was cooled at room temperature, diluted with AcOEt (150 mL), filtered and washed with 50 ml of AcOEt. The filtration liquors were combined, washed with water (2 x 100 mL) and an NaHCO3 saturated solution (2 x 100 mL), dried and evaporated to give l-[(4-hydroxy-6-methoxyquinolin-7-yloxy)methyl]-N- benzyloxycarbonyl-1 -aminocyclopropane (1.2 g, yield: 95%) as a yellow solid.
1H-NMR (300 MHz, MeOD): δ: 7.75 (d, 1 H), 7.51 (s, 1 H), 7.15 (m, 5 H), 6.80 (br, 1 H), 6.20 (d, 1 H), 4.97 (s,2 H), 4.05 (s, 2 H), 3.84 (s, 3 H), 0.87 (m, 4 H).
LC-MS: M+H+: 395.2
Example 5: Preparation of l-[(4-chloro-6-methoxyquinolin-7- yloxy)methyl]-N-benzyIoxycarbonyl-l-aminocyclopropane
a) By chlorination of the compound of Example 4
A 50 ml round-bottom flask fitted with magnetic stirrer, thermometer, condenser and kept under nitrogen atmosphere, was loaded at 20°/25°C with 3.90 g (9.89 mmol) of the compound of Example 4 and 25 ml of POCl3. The resulting suspension became a solution after stirring for a few minutes. The solution was heated at 85°C inner T and after 30 minutes the reaction was monitored by TLC, showing the disappearance of the starting product. The solution was cooled and dropwise added, over about 30 minutes and keeping the temperature below 100C, to a mixture of 250 ml of DCM and 250 ml of water, cooled at 00C. After completion of the addition, stirring was maintained for 30 minutes at 0°-10°C. The phases were separated and the aqueous phase was washed with 150 ml of DCM; the phases were separated and the organic phases combined. The combined organic phase was added with 150 ml of water, stirred at 20°/25°C for 15 minutes and pH was adjusted to 7-8 with a sodium bicarbonate saturated solution. The phases were separated and the organic phase was washed with 150 ml of water; the phases were separated, the organic phase was dried with sodium sulfate, filtered and the solvent evaporated off by distillation under vacuum. Stripping with ethyl ether afforded 3.8 g of a brownish solid. The solid residue was dissolved in 20 ml of tert-butyl methyl ether, stirring at 20°/25°C for an hour; filtered and washed with ter /-butyl methyl ether, then dried to obtain l-[(4-chloro-6- methoxyquinolin-7-yloxy)methyl]-N-benzyloxycarbonyl- l- aminocyclopropane (3.4 g; yield: 87%) having (1H-NMR) titre of 95%.
1H-NMR (500 MHz, DMSO-d6) δ ppm: 8.61 (d, 1 H), 7.91 (s, 1 H), 7.56 (s, 1 H), 7.44 (s, 1 H), 7.38 (s, 1 H), 7.29 (m, 5 H), 4.99 (s, 2 H), 4.23 (s, 2 H), 3.97 (s, 3 H), 0.87 (m, 4 H). b) by Mitsunobu reaction between 4-chloro-7-hydroxy-6- methoxyquinoline and 1 -benzyloxycarbonylamino- 1 – hydroxymethylcyclopropane 20 ml of DCM were added with 4-chloro-7-hydroxy-6- methoxyquinoline (300 mg, 1.43 mmol; from China Gateway),
1 -benzyloxycarbonylamino- 1 -hydroxymethylcyclopropane (412 mg,
1.87 mmol, 1.3 eq; from China Gateway) and triphenylphosphine (490 mg,
1.87 mmol, 1.3 eq). The resulting solution was dropwise added with a solution of DEAD (378 mg, 1.87 mmol, 1.3 eq) in 3 ml of DCM, keeping the temperature at 00C for 2 hours. The mixture was then left at 100C for 20 hours, then filtered to recover the unreacted 4-chloro-7-hydroxy-6- methoxyquinoline. The filtrate was evaporated under vacuum and the resulting residue was added with 20 ml of 95% EtOH and left under stirring for 30 min. The solid was collected by filtration, washed with 5 ml of 95% EtOH and dried under vacuum to give l-[(4-chloro-6-methoxyquinolin-7-yloxy)methyl]-
N-benzyloxycarbonyl-1-aminocyclopropane (273 mg; yield: 46%).
LC-MS: M+H+: 413.1
Example 6: Preparation of benzyl l-[(6-methoxy-4-(5- (methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)]cyclopropyl carbamate (II)
A solution of 0.51 g (2.53 mmol) of 6-hydroxy-N-methyl- 1 – naphthamide prepared according to WO2008/112408, 2, 7 ml of 2,6-lutidine and 0.3 g (2.42 mmol) of DMAP, kept at 20°/25°C and under nitrogen atmosphere, was added with the compound of Example 5 (1.0 g, NMR titre 95%, 2.30 mmol). The suspension was heated to 1400C inner temperature for
6 hours; then cooled to 20°/25°C and added with 80 ml of water and kept under stirring a 20°/25°C for 1 hour; the suspension was filtered and washed with water, to afford 0.88 g (yield: 66%) of benzyl l-[(6-methoxy-4-(5-
(methylcarbamoyl)naphthalen-2-yloxy)quinolin-7-yloxy)methyl)]cyclopropyl carbamate (II).
1H-NMR (500 MHz, DMSO-d6) δ ppm: δ: 8.56 (d, 1 H), 8.50 (d, 1 H), 8.39 (d, 1 H), 8.04 (d, 1 H), 7.94 (s, 1 H), 7.87 (s, 1 H), 7.59 (m, 4 H), 7.41 (s, 1 H), 7.44 (s, 1 H), 7.30 (m, 5 H), 6.56 (d, 1 H), 5.01 (s, 2 H), 4.48 (s, 2 H), 4.23 (s, 2 H), 3.95 (s, 3 H), 0.87 (m, 4 H). LC-MS: M+H+: 578.3
Example 7: Preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6- methoxyquinolin-4-yloxy)-N-methyl-l-naphthamide (I)
A mixture of the compound of Example 6 (0.24 g, 0.42 mmol) in 2 ml of a solution of 40% HBr in acetic acid was stirred at 300C for 3h, then added with 10 ml of water and the reaction mixture was extracted with AcOEt (2 x 10 mL). The organic phases were removed. The aqueous solution was dropwise added with a solution of 50% NaOH to reach pH 10. The mixture was extracted with DCM (3 x 20 mL) and the combined organic phases were dried and evaporated to give a crude containing 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxyquinolin-4- yloxy)-N-methyl-l-naphthamide (I) with purity higher than >94% by LC-MS analysis. This crude was further purified by chromatography on a silica gel column eluting with DCM/MeOH 10: 1), to afford 6-(7-((l- aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-N-methyl-l- naphthamide (I) having purity higher than 98% by LC-MS analysis (140 mg, yield: 76%).
1H-NMR (500 MHz, DMSO-d6) δ ppm: 8.47 (d, 2 H), 7.87 (d, 1 H), 7.53 (m, 3 H), 7.51 (m, 1 H), 7.44 (d, 1 H), 7.38 (s, 1 H), 6.50 (d, 1 H), 6.16 (d, 1 H), 5.01 (s, 2 H), 4.05 (s, 2 H), 4.03 (s, 3 H), 3.12 (d, 3 H), 2.09 (m, 2 H), 0.80 (m, 4 H).
LC-MS: M+H+: 444.0
…………………………………………
6-(7-((l -Aminocyclopropyl)-methoxy)-6-methoxyquinolin-4-yloxy)-N-methyl- 1 -naphthamide (AL3810), or a pharmaceutically acceptable salt (such as hydrochloride salt) thereof, has been developed as an anti-tumor agent also named as E3810 and lucitanib, see “Journal of Cellular and Molecular Medicine vol. 16 issue 10 October 2012. p. 2321-2330 “, “Cancer Res February 15, 2011 vol. 71 no A 1396-1405 “.
This compound has been structurally disclosed in WO20081 12408 as an agiogenesis inhibitor with few preparation methods. A new process has been disclosed in WO2010105761 with the removal of use of sodium azide. Both above disclosed processes have involved a deprotection of benzyl carbmate protected precursor by HBr/Acetic acid solution that is a strong, fuming and high corrosive acidic condition. No crystalline form has been disclosed.
A
B
Process C
Formula III Scheme IV
Example 1
Representation of Process A and Process B
Process for preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy-quinolin-4- yloxy)-N-methyl- 1 -naphthamide (AL3810)
To a stirred mixture of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II (150 g) in DCM (1.5 L) was added TFA (150 ml) through an additional funnel for about 30 min at RT. The reaction was stirred at 30°C for 4 hours and added into water (3 L). The aqueous layer was extracted with DCM twice (1.5L X 2) and basified with 3N NaOH (620 ml) to adjust pH 1 1-12 with a fine white solid precipitation. The solid was filtered and washed with water, further suction dry. The solid was dissolved into a mixture of chloroform/methanol (5 L, 3.5L/1.5L) and further washed with brine (2 L). It was dried with MgS04 and filtered. The solution was evaporated with EtOAc (2 L) three times to a slurry solution and cooled to RT. It was filtered and the filter cake was washed with ether, further air dried to give the crude titled compound 105g, yield: 95.9%. MS: (M+l) 444.
Example 2
Representation of Process A and Process B
Process for preparation of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy-quinolin-4- yloxy)-N-methyl- 1 -naphthamide (AL3810)
To a stirred mixture of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)naph- thalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II (1 g) in ACN (15 ml) was added TSA.H20 (3 eq). The reaction was stirred at RT for 24 hours and it was basified with 3N NaOH. The solution was extracted with DCM three times, washed with brine and dried with MgS04. The solution then was filtered and evaporated, further recrystalized from IPA to give pure titled compound 550 mg, yield: 75%. MS: (M+l) 444. Example 3
Representation of Process C
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II
To a stirred mixture of 6-hydroxy- 1 -naphthoic acid (19 g, formula 10) in DMF (150 ml) was added CDI (22 g). The reaction was heated at 80°C for 30 min and CH3NH2.HC1 (40 g) was added into the reaction. The reaction was heated for 3 hours at 80°C and cooled to RT and further diluted with water (300 ml). It was acidified with IN HC1 to pH 2-3 and extracted three times with EtOAc (150 ml). The combined organic layer was washed with saturated NaHC03solution followed by water and brine. The solution was dried with Na2S04 and evaporated to give the 4- rmula 1 1 compound 12 g.

(i) To a mixture of formula 1 1 (6.5 g), formula 12 (6.5 g) and DMAP (5.5 g) was added 1,6- lutidine (20 ml). The reaction was stirred and heated at 135°C for 5 hours from heterogeneous to homogeneous. The reaction was cooled and IPA (35 ml) was added into the reaction under slow stirring for 2 hours at RT. The solid was filtered and further washed with IPA, dried to give the formula 13 compound 5.8 g as a gray solid, yield 57%, or
(ii) To a mixture of formula 1 1 (500 mg), formula 12 (500 mg), Cul (80 mg), Cs2C03 (1 g) and 1-picolinic acid (150 mg) was added DMF (0.5 ml). The reaction was stirred and heated at 120 °C for 24 hours. It was directed loaded on silica gel column to purify to give the formula 13 compound 370 mg, yield 48%, or (iii) To a mixture of formula 1 1 (500 mg), formula 12 (500 mg), Cul (80 mg), CS2CO3 (1 g) and 2,4-pentanedione (10 mg) was added DMF (0.5 ml). The reaction was stirred and heated at 120 °C for 24 hours. It was directed loaded on silica gel column to purify to give the formula 13
A mixture of formula 13 (5.8 g) and TFA (12 ml) was heated at 90°C for one hour. The reaction was evaporated under reduced pressure and triturated with EtOAc. The solid was filtered e formula 14 as a TFA salt 5.5 g, yield 95%.

To a mixture of acid-ester (8.2 g, formula 15) and 4-methoxybenzyl alcohol (9.5 g) in toluene (50 ml) was added DPPA (15 g), the reaction was stirred and TEA was added into the reaction through an additional funnel at RT. The reaction then was refluxed for 20 hours and cooled to RT. To the reaction was added 2N NaOH (30 ml) and followed by extraction with EtOAc three times. The combined organic layer was washed with water to neutral and dried with Na2SOzt. The solution was filtered and evaporated followed by addition of EtOAc/PE (petroleum ether) and stored in a refrigerator overnight. The crystals were filtered and washed with cold EtOAc/PE to give an off white powder. The product formula 15b was vacuum oven dried at 30°C to give 8.0 g as ethyl l-((4-methoxybenzyloxy)carbonylamino)cyclopropanecarboxylate (formula
294.
To a mixture of formula 15b (8.0 g) and THF (50 ml) was added NaBH4 (8 g). The reaction was refluxed for 12 hours. Methanol (15 ml) was slowly added to the reaction and refluxed for 4 hour. The solvent was evaporated and cooled. NH4C1 (6.3 g) and water (60 ml) were added and stirred. The mixture was extracted with DCM three times and dried with Na2S04. The solution was filtered and evaporated followed by addition of ethanol to recrystalize overnight. The crystal was filtered to give an off white powder and further dried in oven to give the product 4.0 g as 4-methoxybenzyl l-(hydroxymethyl)cyclopropylcarbamate (formula 15c),

To a stirred mixture of formula 15c (100 g) and DCM (400 ml) was added DIPEA (78g). The result solution was cooled to 0-5°C with ice/water and further stirred under this temperature for 15 min. MsCl (60g) was added via an addition funnel dropwise keeping temperature below 5°C for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5°C for 30 min and quenched with saturated NaHC03 (300 ml). The solution was extracted with 200 ml DCM twice. The combined DCM layer was washed with 0.1 N HC1 (400 ml) followed by brine. It was dried over Na2S04 and concentrated to obtain an off- white solid 123 g
330.
To a stirred mixture of formula 15d (3.3 g) and KI (3.3 g) was added acetone (30 ml), the reaction was refluxed for 2 hours and cooled. The reaction was evaporated and extracted with EtOAc (30 ml) twice and washed with brine, further evaporated under reduced pressure to give the crude product 2.3 g of formula 15e, MS: (M+l) 362.
Formula II Method A:
To a stirred mixture of formula 14 (500 mg), formula 15d (450 mg), K2C03 (400 mg) and Nal (180 mg) was added acetone (10 ml), the reaction suspension was heated to reflux for 20 hours as one pot reaction. The reaction was evaporated and purified on silica gel column to give the product 510 mg of Formula II. MS: (M+1) 608. lH NMR (DMSO-d6): δ: 8.53-8.54 (m, 2H), 8.37-8.39 (d, 1H), 8.00-8.02 (d, 1H), 7.83-7.88 (m, 2H), 7.53-7.61 (m, 4H), 7.42 (s, 1H), 7.22- 7.24 (d, 2H), 6.83-6.85 (d, 2H), 6.61-6.62 (d, 1H), 4.91 (s, 2H), 4.23 (s, 2H), 3.95 (s, 3H), 3.70 (s, 3H), 2.86-2.87 (d, 3H), 0.83-0.93 (d, 4H).
Method B:
To a stirred mixture of formula 14 (500 mg), formula 15e (500 mg) and K2C03 (400 mg) was added acetone (10 ml), the reaction suspension was heated to reflux for 20 hours. The reaction was evaporated and purified on silica gel column to give the product 560 mg of Formula II. MS: (M+1) 608. ¾ NMR conforms to Formula II from above Method A.
Method C:
To a stirred mixture of formula 14 (33 g), formula 15d (43 g), K2C03 (41 g) and KI (16.6 g) was added acetone (400 ml). The reaction suspension was heated to reflux for about 30 hr. The reaction was concentrated and to the residue was added water (700 ml). The result suspension was stirred for 1 hour slowly to get a brown solid. The solid was filtered and rinsed with water twice further rinsed with ethanol. The crude product was dried in oven at 40°C for 2-3 hours. The product was purified with IPA by recrystalization to give 29 g of Formula II. MS: (M+1) 608. lH NMR conforms to Formula II from above Method A.
Example 4
Representation of Process D
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarba- moyl)naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropyl-carbamate Formula II
A mixture of 2-(l-((6-methoxy-4-(5-(methylcarbamoyl)naphthalen-2-yloxy)quino-lin-7- yloxy)methyl)cyclopropyl)acetyl azide formula 17 (WO2008112408, 150 mg) and 4-methoxybenzyl alcohol (0.15 ml) in toluene (10 ml) was refluxed for 1.5 hour. The reaction was evaporated and purified with silica gel column to give the titled product. Mass: (M + 1), 608
Example 5
Representation of Process E Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarba- moyl)naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II
A mixture of 6-Hydroxy- 1 -naphthoic acid (1 g) and H2SO4 (0.2ml) in EtOH (25 ml) was refluxed overnight and evaporated, followed by dissolving into EtOAc. The solution was washed with water, IN NaHC03 solution and brine, further dried by Na2S04. The solution was evaporated to give crude ethyl 6-hydroxy- 1 -naphthoate 0.9 g which was reacted with formula 12 at similar preparation conditions to formula 13 of Example 3 to give the above product of formula 18. Formula 19 was similarly prepared to formula 14 of Example 3.
A reaction between formula 19 and formula 15d similarly to the preparation of Formula II of Method A gave ethyl 6-(6-methoxy-7-((l-((4-methoxybenzyloxy)carbonylamino)cyclopro- pyl)methoxy)quinolin-4-yloxy)- 1 -naphthoate which was hydro lyzed with 10% NaOH in EtOH at RT to give 6-(6-methoxy-7-((l-((4-methoxybenzyloxy)carbonylamino)cyclopropyl)methoxy)- quinolin-4-yloxy)-l -naphthoic acid. The resulting acid was acylated similarly to the preparation of formula 1 1 of Example 3 with CH3NH2.HC1 under the heat pre- activation at the presence of CDI to give the titled product.
Example 6
Representation of Process F
Process for preparation of 4-methoxybenzyl l-((6-methoxy-4-(5-(methylcarbamoyl)- naphthalen-2-yloxy)-quinolin-7-yloxy)methyl)cyclopropylcarbamate Formula II
To a mixture of 4-chloro-6-methoxyquilolin-7-ol (formula 21, 5.2g), l-((4-methoxyben- zyloxy)carbonylamino)cyclopropanecarboxylate (formula 15b, 8.3g) and triphenylphosphine (9.8 g) in THF (250 ml) was added DEAD (6.5 g) dropwise at RT in 1.5 hours, the reaction was further stirred for 20 hours at RT and evaporated. The residue was purified with silica gel column to give the 4-methoxybenzyl 1 -((4-chloro-6-methoxy-quinolin-7-yloxy)methyl)cyclopropylcarba- mate formula 21b product 6.5 g.
The titled compound of Formula II was then similarly prepared by using formula 21b to react with 4-hydroxy-N-methyl-naphamide formula 1 1 according to formula 13 of Example 3.
Example 7
Preparation of the crystalline form of 6-(7-((l-aminocyclopropyl)methoxy)-6-methoxy- quinolin-4-yloxy)-N-methyl- 1 -naphthamide (AL3810)
The crude product from Example 1 (105 g) was mixed with isopropanol (2.5 L) and active carbon (5 g), the mixture was heated to reflux for 0.5 hour to dissolve all crude product followed by filtration while it was hot, then the filtrate was refluxed again for 10 minutes and it was cooled to room temperature overnight under a slow stirring condition. The precipitate was filtered and washed with ethyl ether (500 ml x 2), further dried under high vacuum at 80°C to give the pure product (85 g) with melting point at 192°C- 196°C.
HI NMR shown in Fig 1.
DSC shown in Fig 2 having observable endotherm from about 193°C-202°C
TGA shown in Fig 3 demonstrating as an unsolvated material with weight loss at about 230°C
………………………………………….
synthesis…….will be updated
1: Colzani M, Noberini R, Romanenghi M, Colella G, Pasi M, Fancelli D, Varasi M, Minucci S, Bonaldi T. Quantitative chemical proteomics identifies novel targets of the anti-cancer multi-kinase inhibitor E-3810. Mol Cell Proteomics. 2014 Jun;13(6):1495-509. doi: 10.1074/mcp.M113.034173. Epub 2014 Apr 2. PubMed PMID: 24696502; PubMed Central PMCID: PMC4047469.
2: Zangarini M, Ceriani L, Bello E, Damia G, Cereda R, Camboni MG, Zucchetti M. HPLC-MS/MS method for quantitative determination of the novel dual inhibitor of FGF and VEGF receptors E-3810 in tumor tissues from xenograft mice and human biopsies. J Mass Spectrom. 2014 Jan;49(1):19-26. doi: 10.1002/jms.3305. PubMed PMID: 24446259.
3: Bello E, Taraboletti G, Colella G, Zucchetti M, Forestieri D, Licandro SA, Berndt A, Richter P, D’Incalci M, Cavalletti E, Giavazzi R, Camboni G, Damia G. The tyrosine kinase inhibitor E-3810 combined with paclitaxel inhibits the growth of advanced-stage triple-negative breast cancer xenografts. Mol Cancer Ther. 2013 Feb;12(2):131-40. doi: 10.1158/1535-7163.MCT-12-0275-T. Epub 2012 Dec 27. PubMed PMID: 23270924.
4: Damia G, Colella G, Camboni G, D’Incalci M. Is PDGFR an important target for E-3810? J Cell Mol Med. 2012 Nov;16(11):2838-9. doi: 10.1111/j.1582-4934.2012.01601.x. PubMed PMID: 22805298.
5: Sala F, Bagnati R, Livi V, Cereda R, D’Incalci M, Zucchetti M. Development and validation of a high-performance liquid chromatography-tandem mass spectrometry method for the determination of the novel inhibitor of angiogenesis E-3810 in human plasma and its application in a clinical pharmacokinetic study. J Mass Spectrom. 2011 Oct;46(10):1039-45. doi: 10.1002/jms.1985. PubMed PMID: 22012670.
6: Bello E, Colella G, Scarlato V, Oliva P, Berndt A, Valbusa G, Serra SC, D’Incalci M, Cavalletti E, Giavazzi R, Damia G, Camboni G. E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Res. 2011 Feb 15;71(4):1396-405. doi: 10.1158/0008-5472.CAN-10-2700. Epub 2011 Jan 6. PubMed PMID: 21212416.
7: Kawai T, Ikeda H, Harada Y, Saitou T. [Changes in the rat stomach after long-term administration of proton pump inhibitors (AG-1749 and E-3810)]. Nihon Rinsho. 1992 Jan;50(1):188-93. Japanese. PubMed PMID: 1311785.
| CITED PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2008112408A1* | Feb 24, 2008 | Sep 18, 2008 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| WO2010105761A1* | Mar 11, 2010 | Sep 23, 2010 | Eos Ethical Oncology Science S.P.A. In Abbreviated Form Eos S.P.A. | A process for the preparation of 6-(7-((1-aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-n-methyl-1-naphthamide and synthetic intermediates thereof |
| REFERENCE |
|---|
| 1 | * | BELLO, E. ET AL.: ‘E-3810 Is a Potent Dual Inhibitor of VEGFR and FGFR that Exerts Antitumor Activity in Multiple Preclinical Models‘ CANCER RES. vol. 71, no. 4, 2011, pages 1396 – 1405 |
| 2 | * | SALA, F. ET AL.: ‘Development and validation of a high-performance liquid chromatography-tandem mass spectrometry method for the determination of the novel inhibitor of angiogenesis E-3810 in human plasma and its application in clinical pharmacokinetic study‘ J. MASS. SPECTROM. vol. 46, 2011, pages 1039 – 1045 |
| 3 | * | ZHOU, Y. ET AL.: ‘AL3810, a multi-tyrosine kinase inhibitor, exhibits potent anti-angiogenic and anti-tumour activity via targeting VEGFR, FGFR and PDGFR‘ J. CELL . MOL. MED. vol. 16, no. 10, 2012, pages 2321 – 2330 |
| CITED PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2008112408A1 | Feb 24, 2008 | Sep 18, 2008 | Advenchen Lab Llc | Spiro substituted compounds as angiogenesis inhibitors |
| US20080227812 | Feb 23, 2008 | Sep 18, 2008 | Advenchen Laboratories, Llc | Spiro Substituted Compounds As Angiogenesis Inhibitors |
| REFERENCE |
|---|
| 1 | | J. MED. CHEM. vol. 51, 2008, pages 5766 – 5779 |
| 2 | | ORG. REACT. vol. 42, 1992, pages 335 – 656 |
| 3 | | ORGANIC SYNTHESES vol. 63, 1985, page 314 |
| 4 | | SYNTHESIS 1981, pages 1 – 28 |
| 5 | | TETRAHEDRON LETT. vol. 38, 1997, page 191 |
| 6 | | TETRAHEDRON LETTERS vol. 46, 2005, pages 735 – 737 |
| 7 | * | TOIS J ET AL: “Novel and convenient synthesis of 4(1H)quinolones” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, vol. 46, no. 5, 31 January 2005 (2005-01-31), pages 735-737, XP004705840 ISSN: 0040-4039 |
| 8 | * | WEILIN SUN ET AL: “Biososteric Replacement in the Design and Synthesis of Ligands for Nicotinic Acetylcholine Receptors” MEDICINAL CHEMISTRY RESEARCH, BIRKHÄUSER-VERLAG, BO, vol. 14, no. 5, 1 July 2005 (2005-07-01), pages 241-259, XP019428169 ISSN: 1554-8120 |
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2014113616A1* | Jan 17, 2014 | Jul 24, 2014 | Advenchen Pharmaceuticals, LLC | Process for preparing the anti-tumor agent 6-(7-((1-aminocyclopropyl) methoxy)-6-methoxyquinolin-4-yloxy)-n-methyl-1-naphthamide and its crystalline |
Patent Reference:
EOS ETHICAL ONCOLOGY SCIENCE S.p.A. in abbreviated form EOS S.p.A.; SPINELLI, Silvano; LIVI, Valeria Patent: WO2010/105761 A1, 2010 ; Location in patent: Page/Page column 21 ;
H-NMR spectral analysis
![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide NMR spectra analysis, Chemical CAS NO. 1058137-23-7 NMR spectral analysis, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/567/717/1058137-23-7-1h.png)
CAS NO. 1058137-23-7, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide H-NMR spectral analysis |
C-NMR spectral analysis
![6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide NMR spectra analysis, Chemical CAS NO. 1058137-23-7 NMR spectral analysis, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/567/717/1058137-23-7-13c.png)
CAS NO. 1058137-23-7, 6-[7-[(1-aminocyclopropyl)methoxy]-6-methoxyquinolin-4-yl]oxy-N-methylnaphthalene-1-carboxamide C-NMR spectral analysis |
Advenchen Laboratories is a small pharmaceutical company focusing on pharmaceutical research and development involving small molecule cancer drug discovery …


%20Structure.gif)