
TAMOXIFEN
10540-29-1 CAS
READ ABOUT TITLE AT……….http://www.rsc.org/chemistryworld/sites/default/files/CIIE_Tamoxifen.mp3

Molecular Formula: C26H29NO•C6H8O7
CAS Number: 54965-24-1
Brands: Nolvadex, TAMOXIFEN CITRATE
CAS Number: 54965-24-1
Brands: Nolvadex, TAMOXIFEN CITRATE
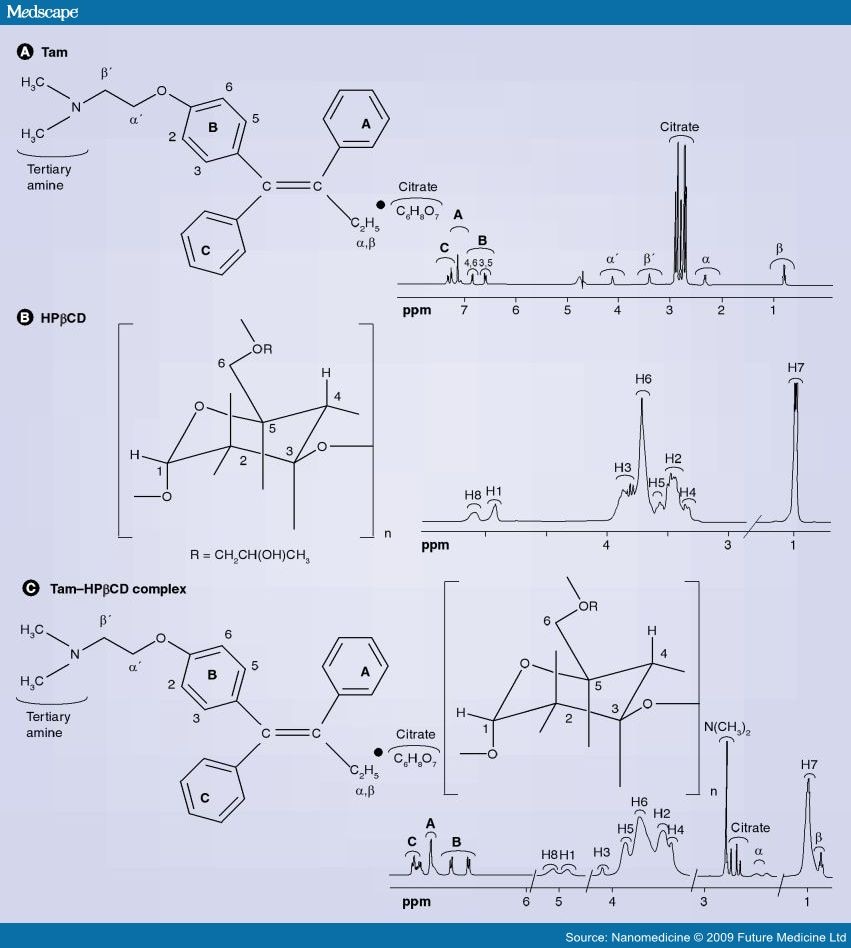
Chemically, NOLVADEX (tamoxifen citrate) is the trans-isomer of a triphenylethylene derivative. The chemical name is (Z)2-[4-(1,2-diphenyl-1-butenyl) phenoxy]-N, N-dimethylethanamine 2 hydroxy-1,2,3- propanetricarboxylate (1:1). The structural and empirical formulas are:
 |
Tamoxifen citrate has a molecular weight of 563.62, the pKa’ is 8.85, the equilibrium solubility in water at 37°C is 0.5 mg/mL and in 0.02 N HCl at 37°C, it is 0.2 mg/mL.
NDA021807 APPR2005-10-29 DARA BIOSCIENCES,
| SOLTAMOX |
US PATENT 6,127,425
| US 6127425 | APPROVED 1998-06-26 | EXPIRY 2018-06-26 |
Tamoxifen is an antagonist of the estrogen receptor in breast tissue via its active metabolite, hydroxytamoxifen. In other tissues such as the endometrium, it behaves as an agonist, and thus may be characterized as a mixed agonist/antagonist. Tamoxifen is the usual endocrine (anti-estrogen) therapy forhormone receptor-positive breast cancer in pre-menopausal women, and is also a standard in post-menopausal women although aromatase inhibitors are also frequently used in that setting.
Some breast cancer cells require estrogen to grow. Estrogen binds to and activates the estrogen receptor in these cells. Tamoxifen is metabolized into compounds that also bind to the estrogen receptor but do not activate it. Because of this competitive antagonism, tamoxifen acts like a key broken off in the lock that prevents any other key from being inserted, preventing estrogen from binding to its receptor. Hence breast cancer cell growth is blocked.
Tamoxifen was discovered by pharmaceutical company Imperial Chemical Industries (now AstraZeneca) and is sold under the trade names Nolvadex, Istubal, and Valodex. However, the drug, even before its patent expiration, was and still is widely referred to by its generic name “tamoxifen.
Breast cancer
Tamoxifen is currently used for the treatment of both early and advanced ER+ (estrogen receptor positive) breast cancer in pre- and post-menopausal women.Additionally, it is the most common hormone treatment for male breast cancer. It is also approved by the FDA for the prevention of breast cancer in women at high risk of developing the disease. It has been further approved for the reduction of contralateral (in the opposite breast) cancer.
In 2006, the large STAR clinical study concluded that raloxifene is equally effective in reducing the incidence of breast cancer, but after an average 4-year follow-up there were 36% fewer uterine cancers and 29% fewer blood clots in women taking raloxifene than in women taking tamoxifen, although the difference is not statistically significant.

Nolvadex (tamoxifen) 20 mg tablets
In 2005, the ATAC trial showed that after average 68 months following a 5 year adjuvant treatment, the group that received anastrozole (Arimidex) had significantly better results than the tamoxifen group in measures like disease free survival, but no overall mortality benefit. Data from the trial suggest that anastrozole should be the preferred medication for postmenopausal women with localized breast cancer that is estrogen receptor(ER) positive.Another study found that the risk of recurrence was reduced 40% (with some risk of bone fracture) and that ER negative patients also benefited from switching to anastrozole.


Crystallographic structure of 4-hydroxy-tamoxifen (carbon = white, oxygen = red, nitrogen = blue) complexed with ligand binding domain of estrogen receptor alpha (cyan ribbon)
Tamoxifen
lTamoxifen was first developed in 1962 as a morning-after birth control pill that was successful in experiments with laboratory rats.
lTamoxifen (brand name Nolvadex) is the best-known hormonal treatment and the most prescribed anti-cancer drug in the world.
lUsed for over 20 years to treat women with advanced breast cancer, tamoxifen also is commonly prescribed to prevent recurrences among women with early breast cancer.
lIs a SERMs.
Anti-estrogens work by binding to estrogen receptors, blocking estrogen from binding to these receptors, stopping cell proliferation
lBreast-cancer prevention occurred in 1998 when the National Cancer Institute (NCI) announced results of a six-year study showing that tamoxifen reduced the incidence of breast cancer by 45 percent among healthy but high-risk women.
l13,388 healthy women considered at high risk for breast cancer were recruited
l85 developed breast cancer compared to 154 of those on the placebo or dummy pill.
lpotentially life-threatening side effects. There were 33 cases of endometrial cancer in the tamoxifen group
lThere were 30 cases of blood clots in major veins (deep-vein thrombosis)
lBecause these problems developed exclusively among postmenopausal women
–60-year-old, an age at which 17 out of every 1,000 women can be expected to develop breast cancer within five years
–ages of 35 and 59 were eligible to participate if their risks matched or exceeded those of a 60-year-old
lAlthough tamoxifen has been useful both in treating breast cancer patients and in decreasing the risk of getting breast cancer.
lSide effects arise from the fact that while tamoxifen acts as an antiestrogen that blocks the effects of estrogen on breast cells, it mimics the actions of estrogen in other tissues such as the uterus. Its estrogen-like effects on the uterus stimulate proliferation of the uterine endometrium and increase the risk of uterine cancer.
Adequate patent protection is required to develop an innovation in a timely manner. In 1962, ICI Pharmaceuticals Division filed a broad patent in the United Kingdom (UK) (Application number GB19620034989 19620913). The application stated, “The alkene derivatives of the invention are useful for the modification of the endocrine status in man and animals and they may be useful for the control of hormone-dependent tumours or for the management of the sexual cycle and aberrations thereof. They also have useful hypocholesterolaemic activity”.
This was published in 1965 as UK Patent GB1013907, which described the innovation that different geometric isomers of substituted triphenylethylenes had either oestrogenic or anti-oestrogenic properties. Indeed, this observation was significant, because when scientists at Merrell subsequently described the biological activity of the separated isomers of their drug clomiphene, they inadvertently reversed the naming. This was subsequently rectified.
Although tamoxifen was approved for the treatment of advanced breast cancer in post-menopausal women in 1977 in the United States (the year before ICI Pharmaceuticals Division received the Queen’s Award for Technological Achievement in the UK), the patent situation was unclear. ICI Pharmaceuticals Division was repeatedly denied patent protection in the US until the 1980s because of the perceived primacy of the earlier Merrell patents and because no advance (that is, a safer, more specific drug) was recognized by the patent office in the United States. In other words, the clinical development advanced steadily for more than a decade in the United States without the assurance of exclusivity. This situation also illustrates how unlikely the usefulness of tamoxifen was considered to be by the medical advisors to the pharmaceutical industry in general. Remarkably, when tamoxifen was hailed as the adjuvant endocrine treatment of choice for breast cancer by the National Cancer Institute in 1984, the patent application, initially denied in 1984, was awarded through the court of appeals in 1985. This was granted with precedence to the patent dating back to 1965! So, at a time when world-wide patent protection was being lost, the patent protecting tamoxifen started a 17 year life in the United States. The unique and unusual legal situation did not go uncontested by generic companies, but AstraZeneca (as the ICI Pharmaceuticals Division is now called) rightly retained patent protection for their pioneering product, most notably, from the Smalkin Decision in Baltimore, 1996. (Zeneca, Ltd. vs. Novopharm, Ltd. Civil Action No S95-163 United States District Court, D. Maryland, Northern Division, March 14, 1996.)
Title: Tamoxifen
CAS Registry Number: 10540-29-1
CAS Name: (Z)-2-[4-(1,2-Diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine
Additional Names: 1-p-b-dimethylaminoethoxyphenyl-trans-1,2-diphenylbut-1-ene
Molecular Formula: C26H29NO
Molecular Weight: 371.51
Percent Composition: C 84.06%, H 7.87%, N 3.77%, O 4.31%
Literature References: Nonsteroidal estrogen antagonist.
Prepn: BE 637389 (1964 to ICI). Identification and separation of isomers: G. R. Bedford, D. N. Richardson, Nature 212, 733 (1966); BE 678807; M. J. K. Harper et al., US 4536516 (1966, 1985 both to ICI). Stereospecific synthesis: R. B. Miller, M. I. Al-Hassan, J. Org. Chem. 50, 2121 (1985). Review of chemistry and pharmacology: B. J. A. Furr, V. C. Jordan, Pharmacol. Ther. 25, 127-205 (1984). Reviews of clinical experience in treatment and prevention of breast cancer: I. A. Jaiyesimi et al., J. Clin. Oncol. 13, 513-529 (1995); C. K. Osborne, N. Engl. J. Med. 339, 1609-1618 (1998).
Properties: Crystals from petr ether, mp 96-98°.
Melting point: mp 96-98°
Derivative Type: Citrate
CAS Registry Number: 54965-24-1
Manufacturers’ Codes: ICI-46474
Trademarks: Kessar (Pharmacia); Nolvadex (AstraZeneca); Tamofène (Aventis); Zemide (Alpharma); Zitazonium (Servier)
Molecular Formula: C26H29NO.C6H8O7
Molecular Weight: 563.64
Percent Composition: C 68.19%, H 6.62%, N 2.49%, O 22.71%
Properties: Fine, white, odorless crystalline powder, mp 140-142°. Slightly sol in water; sol in ethanol, methanol, acetone. Hygroscopic at high relative humidities. Sensitive to uv light. LD50 in mice, rats (mg/kg): 200, 600 i.p.; 62.5, 62.5 i.v.; 3000-6000, 1200-2500 orally (Furr, Jordan).
Melting point: mp 140-142°
Toxicity data: LD50 in mice, rats (mg/kg): 200, 600 i.p.; 62.5, 62.5 i.v.; 3000-6000, 1200-2500 orally (Furr, Jordan)
Derivative Type: (E)-Form
CAS Registry Number: 13002-65-8
Properties: mp 72-74° from methanol.
Melting point: mp 72-74° from methanol
Derivative Type: (E)-Form citrate
Manufacturers’ Codes: ICI-47699
Properties: mp 126-128°.
Melting point: mp 126-128°
CAUTION: Tamoxifen is listed as a known human carcinogen: Report on Carcinogens, Eleventh Edition (PB2005-104914, 2004) p III-239.
Therap-Cat: Antineoplastic (hormonal).
Keywords: Antineoplastic (Hormonal); Antiestrogens; Selective Estrogen Receptor Modulator (SERM).
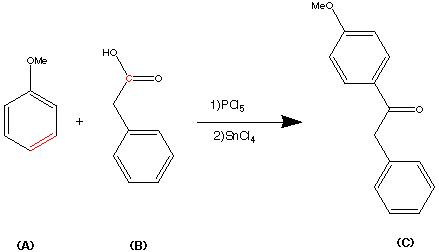
Step 2.
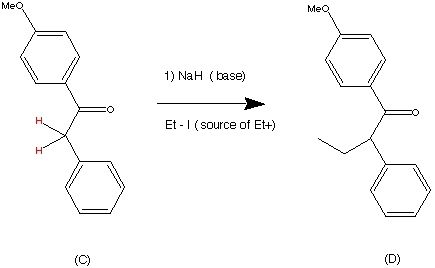
Step 3.
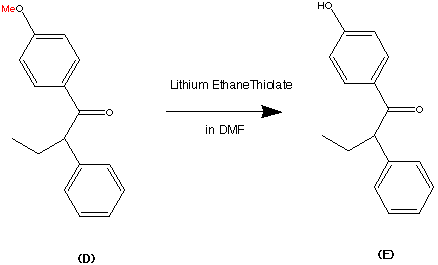
Step 4.
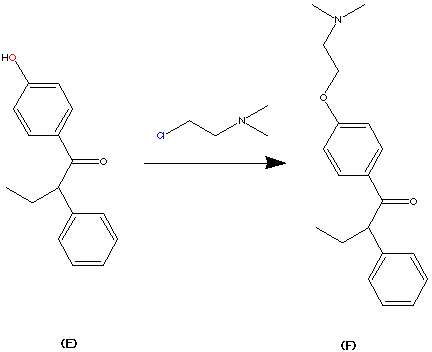
Step 5.
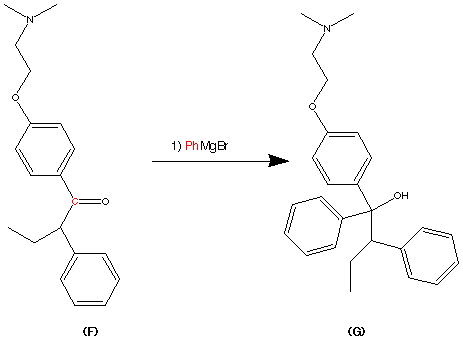
step 6.
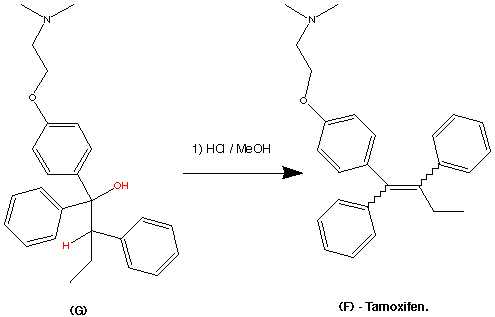
Synthetic Route 2: A Stereospecific Approach.
Stereospecific Synthesis of (Z) – Tamoxifen via carbometalation of Alkynylsilanes.
Studied for historical reasons rather than synthetic brilliance. This synthesis was the first stereo specific synthesis of (Z) Trans Tamoxifen. Comparison between this synthesis and the previous route I believe can illustrate the development of synthetic approaches to large molecules. In particular the quest for stereo specific reactions. So starting from an alkynylsilane (A) and through a series of reactions we can generate only the (Z) – Trans isomer of Tamoxifen.
Again for ease of understanding the complete synthesis has been broken down into a number of steps.
Step1.
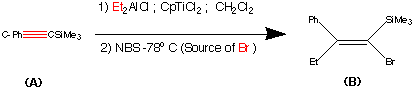
Step1.
This step contains the vital stereo specific step. Namely the carbometalation of the alkynylsilane.It is this step which establishes the stereochemistry about the double bond. The phenyl (trimethyl silyl) – acetylene was carbometalated with diethylaluminium chloride – titanocene dichloride reactant to produce an organometallic intermediate. This organometallic intermediate was then cleaved with N bromosucciniamide to produce the alkene (B) in 85% yield.
The stereochemistry was assigned as E (Cis) mechanistic evidence suggests that this is linked to some steric reasons.
(Earlier work dedicated to this reaction see : Miller, R.B. Al-Hassan.M.I J.Org.Chem. 1984, 49, 725)
Step2.
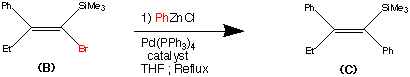
Step 2.
The second step shows the stereo specific replacement of the Br group by a phenyl group. This was achieved by use of Palladium – catalysed coupling of compound (B) with phenyl zinc chloride to form (C) the vinylsilane in a 95% yield.
Step3.
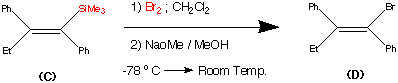
Step3.
This step during the synthesis was reported to be tricky and several approaches were attempted before a successful technique was discovered.
The objective of this step was to replace the trimethyl Silyl group by a suitable halogen atom (e.g. Bromine or Iodine)
However a facile reaction was reported when (C) was treated with bromine – sodium methoxide at -78�C to produce the vinyl bromide (D) in a yield of 85%
Step 4.
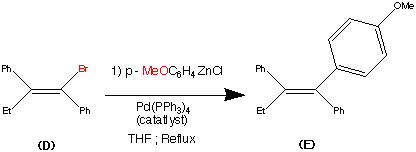
Step 4.
The vinyl bromide (D) coupled well with a Zinc organometallic species to produce (E) the ethyl triaryl olefin in a yield of 84%.
Step 5.
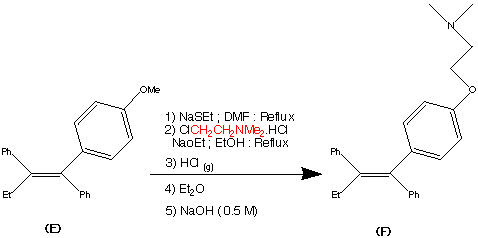
Step 5.
The formation of (F) Tamoxifen was achieved by demethylation with sodium ethylthoilate in refluxing dimethyl formamide. then reaction of the phenoxide ion with 2-( dimethylamino)ethyl chloride via a SN2 substitution.
Purification of the crude product was achieved via it’s hydrochloride salt ( via a reaction with HCl (g)) then F was regenerated by treatment with dilute base this produced the stereospecific (Z)- Trans isomer in an overall yield of 60%.
a synthesis
Palladium-Catalyzed Fluoride-Free Cross-Coupling of Intramolecularly ActivatedAlkenylsilanes and Alkenylgermanes: Synthesis of Tamoxifen as a Synthetic Application (pages 642–650)Kenji Matsumoto and Mitsuru ShindoArticle first published online: 23 FEB 2012 | DOI: 10.1002/adsc.201100627









EP 0883587 A1 WO1997026234A1)
Preparation of Z isomer of Tamoxifen
A solution of bromobenzene (3.92g, 25mmol) in ether (5ml) containing a crystal of iodine was added dropwise to a suspension of magnesium turnings (0.63g, 26mmol) in ether (5ml) at reflux, under nitrogen. After the addition was complete, the reaction mixture was cooled to room temperature and a solution of l- [ 4- ( 2- chloroethoxy)phenyl]-2-phenyl-l-butanone (3.75g, 12.4mmol) in ether (15ml) was added over 1 hour. The resulting mixture was refluxed for 16 hours, then poured into dilute hydrochloric acid (50ml) and extracted with ether (3x40ml) . The combined ether layers were concentrated, the residual oil was dissolved in ethanol (10ml) and refluxed with concentrated hydrochloric acid (5ml) for 4 hours. The organic phase was separated, dried (Na2S04) and evaporated to dryness to give a yellow oil. Η NMR (see Figures 1 to 4 and discussion below) showed this to be a 2:1 mixture of the Z and E isomers. The oil was then dissolved in warm methanol (about 40°C) and allowed to cool to room temperature. The colourless crystals formed proved to be pure Z isomer of 2-chloroethoxy tamoxifen (4.12g, 11.4mmol, 92% yield) . M.p. 107-109°C, m/z 362/364 (chlorine atom present), <SH 0.92 (3H, t, J = 7.33 Hz, CH3) , 2.46 (2H, q, J = 7.33 Hz, CH2CH3) , 3.72 (2H, t, J = 5.86 Hz, 0CH2CH2C1) , 4.09 (2H, t, J = 5.86 Hz, 0CH2CH2C1) , 6.55 (2H, d, J = 8.79 Hz, aromatic protons ortho to 0CH2CH2C1) , 6.79 (2H, d, J = 8.79 Hz, aromatic protons meta to 0CH2CH2C1) , 7.10-7.38 (10H, m, the two remaining C6H5 ,s) (see Figure 5) . The 2-chloroethoxy tamoxifen was reacted with dimethylamine in ethanol, under reflux, to produce the desired Z isomer of tamoxifen.
Analysis of Η NMR data
Figures 1 to 4 represent a mixture of the E- and Z- forms of compound XI described above.
The expansion of the region ό* 0.80 to 1.05 shows two overlapping triplets corresponding to the CH3 groups in the
Z- and E- derivatives respectively. The critical point is the ratio of the heights of the peaks at 0.92 (for the Z) and 0.94 (for the E) , which is approximately 2:1. The expansion of the 4.00 to 4.35 region reveals similar information where ratios are 10:6.4 and 5.56:3.43.
Similarly expansion of the region 3.6 to 3.9 shows the ratio to be 2.46:1. All of these measurements suggest an approximate 2:1 ratio.
Referring to Figure 5, this shows almost pure Z- isomer. It should be noted that there is 660 mg of this from an original mixture of a 2:1 ratio mixture of 780 mg which would contain only 520 mg of the Z-isomer.
Z isomer of tamoxifen and 4-hydroxytamoxi en include stereoselective syntheses (involving expensive catalysts) as described in J. Chem. Soc, Perkin Trans I 1987, 1101 and J. Org. Chem. 1990, 55, 6184 or chromatographic separation of an E/Z mixture of isomers as described in J. Chem. Res., 1985 (S) 116, (M) 1342, 1986 (S) 58, (M) 771.
(Z)-tamoxifen (1) as a white solid, mp: 95.8-96.3 ºC. 1H-NMR (500 MHz, CDCl3) d 0.92 (3H, t, J 7.3 Hz), 2.29 (6H, s), 2.45 (2H, q, J 7.3 Hz), 2.65 (2H, t, J 5.8 Hz), 3.93 (2H, t, J 5.8 Hz), 6.68 (2H, d, J 9.5 Hz), 6.78 (2H, d, J 9.5 Hz), 7.08-7.28 (10H, m).13C-NMR (125 MHz, CDCl3) d 13.6 (CH3), 29.0 (CH2), 45.8 (CH3), 58.2 (CH2), 65.5 (CH2), 113.4 (C), 126.0 (C), 126.5 (CH), 127.8 (CH), 128.1 (C), 129.7 (C), 131.8 (CH), 135.6 (CH), 138.2 (CH), 141.3 (CH), 142.4 (CH), 143.8 (C), 156.7 (C). IR (KBr film) nmax/cm-1: 3055, 2979, 2925, 2813, 2769, 1606, 1509, 1240, 1035, 707. GC-MS (EI) m/z 371(5%), 58(100%).
(Z)-tamoxifen (1) and (E)-tamoxifen (2) in 52% yield. 1H-NMR (300 MHz, CDCl3) d 0.91 (Z isomer. 3H, t, J 7.3 Hz), 0.94 (E isomer. 3H, t, J 7.3 Hz), 2.28 (Z isomer. 6H, s), 2.34 (E isomer. 6H, s), 2.42-2.52 (Z and Eisomers. 4H, m), 2.63 (Z isomer. 2H, t, J 5.9 Hz), 2.74 (E isomer. 2H, t, J 5.9 Hz), 3.94 (Z isomer. 2H, t, J 5.9 Hz), 4.07 (E isomer. 2H, t, J 5.9 Hz), 6.68 (Z isomer. 2H, d, J 9.7 Hz), 6.76 (E isomer. 2H, d, J 9.3 Hz), 6.86-7.36 (Z and E isomers. 10H, m). IR (KBr film) nmax/cm-1: 3081, 3056, 2974, 2826, 2770, 1611, 1509, 1238, 1044. GC-MS (EI) m/z: Z isomer, 371(4%), 72 (24%), 58(100%); E isomer, 371(3%), 72 (24%), 58(100%). (the diastereoisomeric ratio was determined by capillary GC analysis and the configuration of the major diastereoisomer established by comparison of the NMR data of the synthetic mixture with an authentic sample of (Z)-tamoxifen (1).
nmr
ir
FTIR
shows the typical spectra’s of pure tamoxifen citrate, PCL, a physical mixture of tamoxifen citrate and PCL and drug-loaded implants. The spectrum of tamoxifen citrate shows characteristic absorption bands at 3027 cm−1 (=C-H stretching), 1507 and 1477 (C=C ring stretching) and 3180 cm -1 (-NH2). PCL displays a characteristic absorption band at strong bands such as the carbonyl stretching mode around 1727 cm−1 (C=O), asymmetric stretching 2949 cm−1 (CH 2 ) symmetric stretching 2865 cm−1 (CH 2 ). No changes in the spectrum of the physical mixture and drug-loaded microspheres were evident by FTIR spectroscopy. The strong bands such as the carbonyl peak were clear at all points.
| Figure 2: Transmission FTIR spectra of (a) tamoxifen-loaded implant, (b) physical mixture of drug+PCL, (c) pure PCL, (d) pure tamoxifen citrate |
enlarged view
FTIR spectra of A) tamoxifen citrate; B) PLGA; C) mixture of drug and excipients; D) freshly prepared nanoparticles in the formulation (BS-3HS).
Mentions: The pure drug tamoxifen citrate, PLGA-85:15, PVA, a mixture of PLGA and PVA, and a mixture of tamoxifen citrate, PLGA, and PVA; and a freshly prepared formulation were mixed separately with IR grade KBr in the ratio of 1:100 and corresponding pellets were prepared by applying 5.5 metric ton pressure with a hydraulic press. The pellets were scanned in an inert atmosphere over a wave number range of 4000–400 cm−1 in Magna IR 750 series II, FTIR instrument (Nicolet, Madison, WI, USA).
dsc
DSC thermograms of pure tamoxifen (a), pure PCL (b), physical mixture of drug+PCL (c) and (d) drug-loaded implant. The experiment was carried with crimped aluminum pans and a heating rate of 10ºC/min
xrd
X-ray diffraction studies of pure drug (a), pure PCL (b), physical mixture of drug+PCL (c) and (d) drug-loaded implant
synthesis
J.Chem. Research,1985(S) 116, (M) 1342 and 1986 (S) 58, (M) 0771.
No comments:
Post a Comment