PROST SERIES
1 ILOPROST(ciloprost)
2 CICAPROST
3 BIMATOPROST
4 TRAVOPROST
5 LATANOPROST
6 TRAVOPROST
7 TAFLUPROST
8 BERAPROST
9
WILL BE UPDATED




1 Iloprost (ciloprost)


 Iloprost Molecule
Iloprost Molecule
 iloprost
iloprost
 ILOPROST
ILOPROST
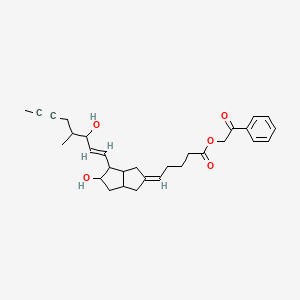 iloprost phenacyl ester
iloprost phenacyl ester


 cicaprost
cicaprost
 cicaprost
cicaprost


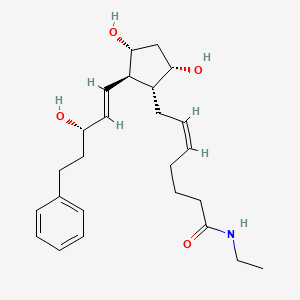
3 BIMATOPROST







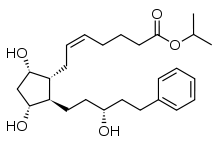
5 LATANOPROST

XALATAN Sterile Ophthalmic Solution (latanoprost ophthalmic solution) is supplied as a sterile, isotonic, buffered aqueous solution of latanoprost with a pH of approximately 6.7 and an osmolality of approximately 267 mOsmol/kg. Each mL of XALATAN contains 50 micrograms of latanoprost. Benzalkonium chloride, 0.02% is added as a preservative. The inactive ingredients are: sodium chloride, sodium dihydrogen phosphate monohydrate, disodium hydrogen phosphate anhydrous, and water for injection. One drop contains approximately 1.5 μg of latanoprostLatanoprost is a colorless to slightly yellow oil that is very soluble in acetonitrile and freely soluble in acetone, ethanol, ethyl acetate, isopropanol, methanol, and octanol. It is practically insoluble in water.








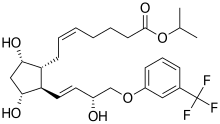
6 TRAVOPROST

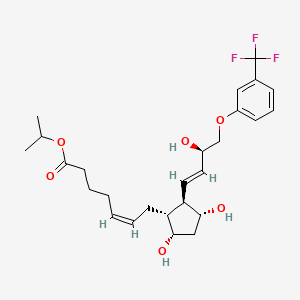







7 TAFLUPROST

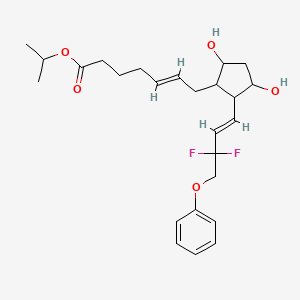











............................................
8 BERAPROST

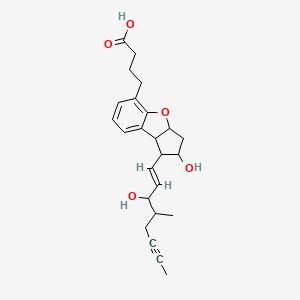





1 ILOPROST(ciloprost)
2 CICAPROST
3 BIMATOPROST
4 TRAVOPROST
5 LATANOPROST
6 TRAVOPROST
7 TAFLUPROST
8 BERAPROST
9
WILL BE UPDATED
1 Iloprost (ciloprost)
Iloprost (ciloprost)
| MF | C22H32O4 |
|---|---|
| Formula Wgt | 360.5 |
6,9ALPHA-METHYLENE-11ALPHA,15S-DIHYDROXY-16-METHYL-PROSTA-5E,13E-DIEN-18-YN-1-OIC ACID
6,9α-

ILOPROST (Ventavis®) is used to treat a serious heart and lung disorder called pulmonary arterial hypertension. While iloprost inhalation solution will not cure this disorder, it is designed to improve symptoms and the quality of life. Generic iloprost inhalation solution is not yet available.
Iloprost is a second generation structural analog of prostacyclin (PGI) with about ten-
73873-87-7 CAS NO
78919-13-8 PHENACYL ESTER
Launched - 1992 bayer
Ilomedin®, Ventavis™
iloprost
An eicosanoid, derived from the cyclooxygenase pathway of arachidonic acid metabolism. It is a stable and synthetic analog of EPOPROSTENOL, but with a longer half-life than the parent compound. Its actions are similar to prostacyclin. Iloprost produces vasodilation and inhibits platelet aggregation.
BAY-q-6256 E-1030 SH-401 ZK-36374
- BAY Q6256
- Ciloprost
- Iloprost
- Iloprostum
- Iloprostum [Latin]
- UNII-AHG2128QW6
- UNII-JED5K35YGL
- Ventavis
- ZK 00036374
- ZK 36374
Endoprost Ilomedin Ilomédine Ventavis Iloprost is a synthetic prostacyclin analog discovered and developed by Schering AG and Berlex which has been available for more than ten years as therapy for peripheral arterial occlusive disease (PAOD), including Raynaud's phenomenon and Buerger's disease.
Iloprost improves blood flow, relieves the pain associated with circulatory disturbances and improves the healing of ulcers, which can develop as a result of poor arterial blood flow. Iloprost also produces vasodilatation of the pulmonary arterial bed, with subsequent significant improvement in pulmonary artery pressure, pulmonary vascular resistance and cardiac output, as well as mixed venous oxygen saturation. In 2003, Schering AG received approval in the E.U. for an inhaled formulation of iloprost (Ventavis[R]) for the treatment of primary pulmonary hypertension and the following year, the product was launched in Germany and the U.K.
Introduction on the U.S. market took place in March 2005 by CoTherix for the same indication in patients with NYHA Class III or IV symptoms. Iloprost is also available for the treatment of pulmonary hypertension and peripheral vascular disease. CoTherix had been developing a dry powder for potential use in the treatment of pulmonary hypertension; however, no recent development has been reported for this research. In Japan, phase III clinical trials are ongoing for the treatment of pulmonary arterial hypertension.  In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
In 2003, CoTherix licensed exclusive rights from Schering AG to market iloprost in the U.S. for primary pulmonary hypertension while Schering AG retained rights to the product outside the U.S. In April 2005, CoTherix established a collaborative research and development agreement with Quadrant to develop an extended-release formulation of iloprost inhalation solution. Iloprost was designated as an orphan medicinal product for the treatment of pulmonary hypertension in December 2000 by the EMEA and will fall under orphan drug protection until 2013.
The FDA has assigned to iloprost several orphan drug designations. In 1989, iloprost solution for infusion was granted orphan drug designation for the treatment of Raynaud's phenomenon secondary to systemic sclerosis followed by another orphan drug designation in 1990 for iloprost solution for injection for the treatment of heparin-associated thrombocytopenia. In 2004, an additional orphan drug designation for iloprost inhalation solution for the treatment of pulmonary arterial hypertension was assigned.
The status has also been assigned in the E.U. for this indication. In 2012, orphan drug designation was assigned in the U.S. for the treatment of purpura fulminans in combination with eptifibatide and for the treatment of pulmonary arterial hypertension. In 2007, Cotherix was acquired by Actelion.
ILOPROSTiloprost phenacyl ester
Ventavis (TN), Iloprost phenacyl ester, Iloprost-PE, Iloprost (INN), CHEMBL138694, CHEMBL236025, AC1O6009, DAP000273, CID5311181
Molecular Formula: C30H38O5 Molecular Weight: 478.61972
2-oxo-2-phenylethyl 5-[(2Z)-5-hydroxy-4-[(1E)-3-hydroxy-4-methyloct-1-en-6-yn-1-yl]-octahydropentalen-2-ylidene]pentanoate
IMPORTANT PUBLICATIONS
Ciloprost Drugs Fut 1981, 6(11): 676
A carbohydrate approach for the formal total synthesis of the prostacyclin analogue (16S)-iloprost Tetrahedron Asymmetry 2012, 23(5): 388
Angewandte Chemie, 1981 , vol. 93, 12 pg. 1080 - 1081
Tetrahedron Letters, 1992 , vol. 33, 52 pg. 8055 - 8056
Helvetica Chimica Acta, 1986 , vol. 69, 7 pg. 1718 - 1727
Journal of Medicinal Chemistry, 1986 , vol. 29, 3 pg. 313 - 315
US5286494 A1
| US 2013253049 |
uS 2013184295
WO 1992014438
WO 1993007876
WO 1993015739
WO 1994008584
WO 2013040068
WO 2012174407
WO 2011047048
| EP0011591A1 * | Oct 18, 1979 | May 28, 1980 | Schering Aktiengesellschaft | Prostane derivatives, their production and pharmaceutical compositions containing them |
| EP0084856A1 * | Jan 19, 1983 | Aug 3, 1983 | Toray Industries, Inc. | 5,6,7-Trinor-4, 8-inter-m-phenylene prostaglandin I2 derivatives |
| EP0099538A1 * | Jul 11, 1983 | Feb 1, 1984 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
..........................................
- 5,6,7-trinor-4,8-inter-m-phenylene prostaglandin 12derivatives.
- Prostaglandin I2, hereinafter referred to as PGI2, of
was first found by J.R. Vane et.al. in 1976 and is biosynthe- sized from arachidonic acid via endoperoxide(PGH2 or PGG2) in the vascular wall. PGI2 is well known to show potent activity to inhibit platelet aggregation and to dilate peripheral blood vessels(C & EN, Dec. 20, 1976, page 17 and S. Moncade et al., Nature, 263,633(1976)).
- [0003]Because of the unstable exo-enolether structure thereof, PGI2 is extremely unstable even in a neutral aqueous solution and is readily converted to 6-oxo-PGF1α which is almost physiologically inactive. Such instability of PGI2 is a big obstacle to its use as a drug. Furthermore, PGI2 is unstable in vivo as well and shows only short duration of action.
- The compounds of the present invention are novel PGI2 derivatives in which the exo-enolether moiety characteristic of PGI2 is transformed into "inter-m-phenylene" moiety. In this sense the compounds may be regarded as analogs of PGI2.
- The compounds of the present invention feature much improved stability in vitro as well as in vivo in comparison with PGI2. The compounds are highly stable even in an aqueous solution and show long duration of action in vivo. Further, the compounds have advantages over PGI2 for pharmaceutical application because they exhibit more selective physiological actions than PGI2, which has multifarious, inseperable biological activities.
- Some prostaglandin I2 derivatives which have 5,6,7-tri- nor-4,8-inter-m-phenylene structure have already been described in publication by some of the present authors. (Kiyotaka Ohno, Hisao Nishiyama and Shintaro Nishio, U.S.P. 4,301,164 (1981)). But, the compounds of the present invention, which feature the presence of alkynyl side chain, have more potent physiological activities as well as decreased side effects than the already disclosed compounds analogous to those of the present invention.
- It is an object of this invention to provide novel prostaglandin I2derivatives which are stable and possess platelet aggregation-inhibiting, hypotensive, anti-ulcer and other activities.
- is named as 16-methyl-18,19-tetradehydro-5,6,7-trinor-4,8-inter-m-phenylene PGI2.

- Alternatively, the compound of the formula (II) may be named as a derivative of butyric acid by the more formal nomenclature. In such a case, the condensed ring moiety is named after the basical structure of 1H-cyclopenta[b]benzofuran of the following formula:
 The term "synthetic prostacyclins" as used herein can refer to any prostacyclin that can be prepared via synthetic organic chemistry, including those prostacyclins that are also naturally occurring, such as Prostacyclin (PGI2):
The term "synthetic prostacyclins" as used herein can refer to any prostacyclin that can be prepared via synthetic organic chemistry, including those prostacyclins that are also naturally occurring, such as Prostacyclin (PGI2):
which is also known as Epopreostenol.
Thus, examples of synthetic prostacyclins include, but are not limited to: Prosta
lloprost, which has the structure:
Trepro inil (also known as Rumodolin), which has the structure:
Beraprost, which has the structure:
as well as the esters, stereoisomers, and salts thereof, or other analogues or derivatives of the recited synthetic prostacyclins, such as compounds comprising other aliphatic linker groups linking the carboxylic acid group to the cyclic components of the synthetic prostacyclins, compounds containing additional alkene and/or alkyne bonds, and/or compounds containing additional substituents on the cyclic components of the synthetic prostacyclins.
 iloprost, in contrast to PGI.sub.2 a stable prostacyclin derivative, has been known since 1980 by European patent application EP 11591, no other prostacyclin derivative has previously been tested in this indication. It is therefore reasonable to assume that a spontaneous healing is involved in the published case.
iloprost, in contrast to PGI.sub.2 a stable prostacyclin derivative, has been known since 1980 by European patent application EP 11591, no other prostacyclin derivative has previously been tested in this indication. It is therefore reasonable to assume that a spontaneous healing is involved in the published case.
It has now been found, surprisingly, that iloprost is effective in the case of cerebral malaria.
For salt formation of iloprost, inorganic and organic bases are suitable, as they are known to one skilled in the art for the formation of physiologically compatible salts. For example, there can be mentioned: alkali hydroxides, such as sodium and potassium hydroxide, alkaline-earth hydroxides, such as calcium hydroxide, ammonia, amines, such as ethanolamine, diethanolamine, triethanolamine, N-methylglucamine, morpholine, tris-(hydroxymethyl)-methylamine, etc.
The β-cyclodextrin clathrate formation takes place according to EP 259468.
The production of iloprost is described in detail in EP 11591.- Nileprost iloprost, and a process for preparing these compositions.
- From EP 11 591 already carbacyclin derivatives of the cytoprotective effect on the gastric and intestinal mucosa, and the myocardial cytoprotection using carbacyclin derivatives is known.
- It has now been found that iloprost (I) and Nileprost (II)
and their salts with physiologically acceptable bases and cytoprotective effect in the kidney.
- Forming salts of iloprost and Nileprost inorganic and organic bases are suitable, as are known to those skilled in the formation of physiologically compatible salts known. Examples which may be mentioned are: alkali metal hydroxides, such as sodium and potassium hydroxide, alkaline earth metal hydroxides such as calcium hydroxide, ammonia, amines, such as ethanolamine, diethanolamine, triethanolamine, N-methylglucamine, morpholine, tris (hydroxymethyl) methylamine, etc.
- The production of iloprost and is described in detail in EP Nileprost 2234 and EP 11591.
....................J. Med. Chem., 1986, 29 (3), pp 313–315DOI: 10.1021/jm00153a001
see paper
......................................
The formal total synthesis of the synthetic and stable analogue of prostacyclin, (16S)- iloprost is described via a convergent synthesis starting from readily available d-glucose. Julia olefination and the aldol reaction are the key steps involved in the synthesis.
..........................................
- Used as the starting material for the method described above ketone of general formula II can be prepared by reacting an alcohol of the formula IV
(EJCorey et al., J. Amer. Chem. 93, 1490 (1971)) transformed with dihydropyran in the presence of catalytic amounts of p-toluenesulfonic acid in the tetrahydropyranyl ether V.

- [0026]Lactone V with Diisobatylauminiumnydrid reduced at -70 ° C to the lactol VI, which is converted by Wittiereaktion Triphenylphosphoniummethylen with the olefin VII. After conversion to the tosylate with p-toluenesulfonyl chloride in the presence of pyridine is obtained by reaction with potassium nitrite in the dimethylsulfoxide 9SS-configured alcohol IX, which is converted with p-toluenesulfonyl chloride in the presence of pyridine in the tosylate X. Reaction with Malonsäurediäthylester in presence of potassium tert-butoxide gives the diester XI, which is converted by decarbalkoxylation with sodium cyanide in dimethyl sulfoxide in the ester XII.

- [0027]Oxidative cleavage of the double bond in the compound XII with Hatrium p j o dat it out in the presence of catalytic amounts of osmium tetroxide to give the aldehyde XIII, which is oxidized with Jones reagent to the acid XIV which is then esterified with diazomethane to the compound XV. By Dieckmann condensation of XV with potassium tert-butoxide in tetrahydrofuran is obtained a mixture of isomers of the ketocarboxylic acid ester XVI and XVII, which by means of a decarbalkoxylation with 1,4-diazabicyclo [2,2,2] octane in xylene converted into ketone XVIII as the only reaction product is.

- [0028]The removal of the Tetrahydropyranylätherschutzgruppe delivers the alcohol XIX, which is esterified with benzoyl chloride in the presence of pyridine to give the ester XX.

- [0029]Benzyläthers hydrogenolytic cleavage of a catalytic amount of acid gives the alcohol XXI, which is according to ketalization compound XXII oxidized with Collins reagent to aldehyde XXIII.
- [0030]This aldehyde XXIV with a phosphonate of the general formula
wherein D, E and R 2 have the meanings given above is reacted in a Olefinicrungsreaktion to a ketone of the formula XXV.

- [0031]After reduction of the 15-keto group with zinc borohydride or sodium borohydride or reaction with alkylmagnesium bromide or alkyllithium and. Epimerentrennung obtain the 15α-alcohols XXVI (PG numbering).

- [0032]After hydrolysis of the ester group, for example with potassium carbonate in methanol and ketal cleavage with aqueous acetic acid yields the ketone of the formula XXVII,

..........................................
2 CICAPROST
Cicaprost
94079-80-8 , as in entry 4 , J. Org. Chem. 1988,53,1227-1231
ZK-96480
phase 2
Bayer Schering Pharma (Originator)
2-
13,14-Didehydro-16,20-dimethyl-3-oxa-18,18,19,19-tetradehydro-6-carbaprostaglandin I2;
5-(7-Hydroxy-6-(3-hydroxy-4-methylnona-1,6-diynyl)-bicyclo(3.3.0)octan-3-yliden)-3-oxapentanoic acid;
2-[(2E,3aβ,6aβ)-4β-[(3S,4S)-3-Hydroxy-4-methyl-1,6-nonadiynyl]-5α-hydroxyoctahydropentalene-2-ylidene]ethoxyacetic acid;
[2-[(2E,3aβ,4S,6aβ)-4β-[(3S,4S)-3-Hydroxy-4-methyl-1,6-nonadiynyl]-5α-hydroxyoctahydropentalene-2-ylidene]ethoxy]acetic acid;
[2-[[(2E,3aS,3aβ,6aβ)-5α-Hydroxy-4β-[(3S,4S)-3-hydroxy-4-methyl-1,6-nonanediynyl]octahydropentalen]-2-ylidene]ethoxy]acetic acid;
Acetic acid, ((2E)-2-((3as,4S,5R,6as)-hexahydro-5-hydroxy-4-((3S,4S)-3-hydroxy-4-methyl-1,6-nonadiynyl)-2(1H)-pentalenylidene)ethoxy)-;
2-[2-[(2E,3aS,4S,5R,6aS)-Hexahydro-5-hydroxy-4-[(3S,4S)-3-hydroxy-4-Methyl-1,6-nonadiyn-1-yl]-2(1H)-pentalenylidene]ethoxy]acetic Acid;
Acetic acid, (2-(hexahydro-5-hydroxy-4-(3-hydroxy-4-methyl-1,6-nonadiynyl)-2(1H)-pentalenylidene)ethoxy)-, (3as-(2E,3aalpha,4alpha(3R*,4R*),5beta,6aalpha))-
| Molecular Formula | C22H30O5 |
|---|---|
| Formula Weight | 374.5 |
Prostaglandin I2 (PGI2, prostacyclin) is the most potent endogenous vasodilator that affects both the systemic and pulmonary circulation.Cicaprost is a PGI2 analog that is orally active with prolonged availabilityin vivo, having a terminal half life in plasma of one hour. In addition to their effects on smooth muscle, PGI2 analogs, including cicaprost, have been shown to inhibit the pro-
cicaprost
references
1. Drugs Fut 1986, 11(11): 913
2. Synthesis of a new chemically and metabolically stable prostacyclin analogue with high and long-lasting oral activity
J Med Chem 1986, 29(3): 313
J Med Chem 1986, 29(3): 313
3. Journal of Organic Chemistry, 1988 , vol. 53, 6 p. 1227 - 1231 entry4
4. Journal of the American Chemical Society, 2003 , vol. 125, 32 p. 9653 - 9667, nmr
5. WO 2009068190
6. US 5013758
7. WO 2005009446
8. WO 1992014438
9. US2007/196510 A1
10. US2007/293552 A1
11. US2009/221549 A1
12. US2009/54473 A1
| EP0041661A2 * | May 29, 1981 | Dec 16, 1981 | Schering Aktiengesellschaft | Preparation of intermediates of carbaprostacyclines |
| EP0057660A2 * | Feb 1, 1982 | Aug 11, 1982 | Schering Aktiengesellschaft | Prostacycline derivatives, their preparation and applications as medicines |
| EP0086404A1 * | Feb 3, 1983 | Aug 24, 1983 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
| EP0086612A1 * | Feb 7, 1983 | Aug 24, 1983 | The Upjohn Company | 9-Substituted carbacyclin analogues |
| EP0087237A1 * | Feb 7, 1983 | Aug 31, 1983 | The Upjohn Company | Carbacyclin analogues |
| EP0098793A1 * | Jul 1, 1983 | Jan 18, 1984 | Schering Aktiengesellschaft | Carbacycline amides, process for their preparation and their use as medicines |
| EP0155901A1 * | Mar 6, 1985 | Sep 25, 1985 | Schering Aktiengesellschaft | Carbacyclines, process for their preparation and their use as medicines |
| EP0195379A2 * | Mar 14, 1986 | Sep 24, 1986 | G.D. Searle & Co. | Allenic prostacyclins |
| EP0195668A2 * | Mar 19, 1986 | Sep 24, 1986 | Sankyo Company Limited | Carbacyclin derivatives |
| EP0721783A1 * | Jun 6, 1995 | Jul 17, 1996 | Toray Industries, Inc. | Preventive and remedy for diseases caused by fibrinoid or thrombus formation in the lung and model animal for said diseases |
| EP2065054A1 | Nov 29, 2007 | Jun 3, 2009 | Bayer Schering Pharma Aktiengesellschaft | Combinations comprising a prostaglandin and uses thereof |
| DE3427797A1 * | Jul 25, 1984 | Feb 6, 1986 | Schering Ag | Zytoprotektive wirkung von prostacyclin-derivaten an leber, bauchspeicheldruese und niere |
| DE3448256C2 * | Jul 25, 1984 | Aug 18, 1988 | Schering Ag, 1000 Berlin Und 4709 Bergkamen, De | Cytoprotective action of prostacyclin derivatives on the pancreas |
| DE3448257C2 * | Jul 25, 1984 | Aug 18, 1988 | Schering Ag, 1000 Berlin Und 4709 Bergkamen, De | Cytoprotective action of prostacyclin derivatives on the kidney |
| DE4135193C1 * | Oct 22, 1991 | Mar 11, 1993 | Schering Ag Berlin Und Bergkamen, 1000 Berlin, De | Title not available |
| US5405870 * | Nov 4, 1993 | Apr 11, 1995 | Sankyo Company, Limited | Carbacyclin compounds; pharmaceutical compositions and method of use |
| US5489613 * | Jan 21, 1992 | Feb 6, 1996 | Sankyo Company, Limited | Carbacyclin derivatives, process for their preparation and compositions containing them |
| US5716989 * | Nov 27, 1991 | Feb 10, 1998 | Schering Aktiengesellschaft | Bicyclo 3.3.0!octane derivatives, process for their production and their pharmaceutical use |
| US5891910 * | Jun 6, 1995 | Apr 6, 1999 | Schering Aktiengesellschaft | 9-halogen-(Z) prostaglandin derivatives, process for their production and their use as pharmaceutical agents |
| US6040336 * | Aug 6, 1996 | Mar 21, 2000 | Schering Aktiengesellschaft | Prostane derivatives and the combination thereof with antibiotics in the treatment of bacterial infections |
| US6225347 | Sep 27, 1994 | May 1, 2001 | Schering Aktiengesellschaft | 9-halogen-(Z)-prostaglandin derivatives, process for their production and their use as pharmaceutical agents |
| WO1986000808A1 * | Jul 18, 1985 | Feb 13, 1986 | Schering Ag | Prostacycline derivatives with a cytoprotective action on the liver, the pancreas and the kidney |
| WO1987005294A1 * | Mar 9, 1987 | Sep 11, 1987 | Schering Ag | Cyclodextrinclathrates of carbacycline derivatives and their use as medicinal drugs |
| WO1988001867A1 * | Sep 1, 1987 | Mar 24, 1988 | Schering Ag | Topical agent containing prostacycline derivatives |
| WO1991014675A1 * | Mar 27, 1991 | Sep 29, 1991 | Schering Ag | Bicyclo[3.3.)]octane derivatives, process for producing them and their pharmaceutical use |
| WO1992014438A2 * | Feb 11, 1992 | Aug 13, 1992 | Schering Ag | Prostacycline and carbacycline derivatives as agents for treating feverish complaints |
| WO1994003175A1 * | Aug 9, 1993 | Feb 17, 1994 | Schering Ag | Use of prostane derivatives of formulae i and ii for the production of a medicament for the treatment of chronic polyarthritis |
| WO1997006806A1 * | Aug 6, 1996 | Feb 27, 1997 | Schering Ag | Use of prostane derivatives and the combination thereof with antibiotics in the treatment of bacterial infections |
cicaprost
..............................
Journal of Organic Chemistry, 1988 , vol. 53, 6 p. 1227 - 1231 entry4
ZK 96 480 (4). A solution of 19 (68 mg, 0.13 mmol) in eth-
er-toluene (3 mL, 2:l) was added to tetrabutylammonium hydrogen sulfate containing HzO (2 drops). After adding 50% aqueous NaOH (0.8 mL), the whole reaction mixture was stirred at 55 "C for 48 h. The reaction was quenched with HzO, acidified with 5% aqueous HC1, extracted with ethyl acetate, washed withH20 and brine, and concentrated to give ZK 96 480 (4) (42 mg, 86%) as a colorless viscous oil:
er-toluene (3 mL, 2:l) was added to tetrabutylammonium hydrogen sulfate containing HzO (2 drops). After adding 50% aqueous NaOH (0.8 mL), the whole reaction mixture was stirred at 55 "C for 48 h. The reaction was quenched with HzO, acidified with 5% aqueous HC1, extracted with ethyl acetate, washed withH20 and brine, and concentrated to give ZK 96 480 (4) (42 mg, 86%) as a colorless viscous oil:
[alZzD +138.25O (c 1.025, CHCI,). see pdf file for correct cut paste
Other spectral data were identical with those of an authentic
sample.'
sample.'
(1) Skuballa, W.; Schillinger, E.; Stiinebecher, C.-St.; Vorbriiggen, H.
J. Med. Chem. 1986,29, 313.
J. Med. Chem. 1986,29, 313.
.....................................
Skuballa, W.; Schillinger, E.; Stiinebecher, C.-St.; Vorbriiggen, H.
J. Med. Chem. 1986,29, 313.
J. Med. Chem. 1986,29, 313.
see original pdf file for structures
we replaced the methylene group in the
3-position of 1, iloprost by an oxygen atom to prevent the 6-oxi-
dation of the upper side chain. The resulting decrease in
intrinsic activity was compensated for by modification of
the lower side chain. We converted the 13,14-double bond
into a triple bond, introduced a further methyl group at
(2-20, and synthesized selectively the pure 16(S)-methyl
diastereomer. These modifications resulted in the struc-
ture of 2 cicaprost (ZK 96 480), a carbacyclin analogue with a bio-
logical activity at least as high as that of prostacyclin and
iloprost.
The synthesis of 2 started with the preparation of the
lower side chain by resolving racemic 2-methyl-4-heptynoic
acid (3).7 By application of the method of Helmchen et
al.," 3 was converted with phosphorus trichloride into the
acid chloride 4, which gave with D-(-)-a-phenylglycinol a
pair of diastereomeric amides. After chromatographic
separation on SOz, the more polar amide 5 (mp 124 'C)
was hydrolyzed with 3 N H2S04 in dioxane to furnish the
optically pure 2s-configurated acid 6 ([a]D -1.2' (c 1,
EtOH), bp 128 'C (12 mm)). The 2s configuration of 6
was determined by hydrogenation of 6 to 2(S)-methyl-
heptanoic acid (["ID +17.7' (c 1, EtOH)), which was com-
pared with 2-methyl-alkanoic acids of known absolute
config~ration.~ Esterification of 6 with diazomethane
followed by reaction of the methyl ester 7 ([a]D +12.2' (c
1, EtOH), bp 70 'C (12 mm)) with the lithium salt of ethyl
methylphosphonate afforded the optically pure phospho-
nate 8 (['Y]D +35.3' (c 1, EtOH), bp 123 "C (0.3 mm)).
3-position of 1, iloprost by an oxygen atom to prevent the 6-oxi-
dation of the upper side chain. The resulting decrease in
intrinsic activity was compensated for by modification of
the lower side chain. We converted the 13,14-double bond
into a triple bond, introduced a further methyl group at
(2-20, and synthesized selectively the pure 16(S)-methyl
diastereomer. These modifications resulted in the struc-
ture of 2 cicaprost (ZK 96 480), a carbacyclin analogue with a bio-
logical activity at least as high as that of prostacyclin and
iloprost.
The synthesis of 2 started with the preparation of the
lower side chain by resolving racemic 2-methyl-4-heptynoic
acid (3).7 By application of the method of Helmchen et
al.," 3 was converted with phosphorus trichloride into the
acid chloride 4, which gave with D-(-)-a-phenylglycinol a
pair of diastereomeric amides. After chromatographic
separation on SOz, the more polar amide 5 (mp 124 'C)
was hydrolyzed with 3 N H2S04 in dioxane to furnish the
optically pure 2s-configurated acid 6 ([a]D -1.2' (c 1,
EtOH), bp 128 'C (12 mm)). The 2s configuration of 6
was determined by hydrogenation of 6 to 2(S)-methyl-
heptanoic acid (["ID +17.7' (c 1, EtOH)), which was com-
pared with 2-methyl-alkanoic acids of known absolute
config~ration.~ Esterification of 6 with diazomethane
followed by reaction of the methyl ester 7 ([a]D +12.2' (c
1, EtOH), bp 70 'C (12 mm)) with the lithium salt of ethyl
methylphosphonate afforded the optically pure phospho-
nate 8 (['Y]D +35.3' (c 1, EtOH), bp 123 "C (0.3 mm)).
Condensation of the phosphonate 8 with the readily
available optically pure bicyclic aldehyde 93,4 (NaH, DME,
available optically pure bicyclic aldehyde 93,4 (NaH, DME,
-20 "C) in the presence of N-bromosuccinimide furnished
the a,P-unsaturated bromo ketone 10 in 60% yield: oil;
(3 H, d, J = 7 Hz, CHCH,), 3.91 (4 H, m, OCH2CH20), 5.21
(1 H, m, H-llp), 7.09 (1 H, d, J = 10 Hz, H-13), 7.42-7.92
(5 H, m, COPh); IR (neat) 1720 (COPh), 1690 (COC=C)
cm-'. Reduction of 10 (NaBH,, CH,OH, -40 "C) gave a
ca. 1:l mixture of the allylic alcohols lla and llb, which
was separated chromatographically.'O Dehydrobromina-
tion (50% aqueous NaOH, toluene, catalytic NBu4/HS04,
25 "C) of the less polar alcohol lla with concomitant sa-
ponification of the benzoate group followed by acidic
(HOAc, H20) cleavage of the ketal moiety afforded the
ketone 12 (73% from lla): oil; 'H NMR (CD2C12) 6 1.06
(3 H, d, J = 6.8 Hz, CHCH,), 1.10 (3 H, t, J = 7.5 Hz,
CH,CH,), 4.22 (1 H, m, H-llb), 4.38 (1 H, m, H-158); IR
(neat) 1730 (C=O) cm-'. After silylation of the hydroxyl
groups in 12 (C1SiMe2-t-Bu, DMF, imidazole), the ketone
13 was subjected to a Horner-Wittig reaction with triethyl
phosphonoacetate (KO-t-Bu, THF, 0 "C). Reduction of
the 1:l mixture of the isomeric a,p-unsaturated esters 14
with diisobutylaluminum hydride (toluene, 0 "C) gave after
chromatographic separation the E isomer 15a (32% from
12) and the less polar 2 isomer 15b.11
the a,P-unsaturated bromo ketone 10 in 60% yield: oil;
(3 H, d, J = 7 Hz, CHCH,), 3.91 (4 H, m, OCH2CH20), 5.21
(1 H, m, H-llp), 7.09 (1 H, d, J = 10 Hz, H-13), 7.42-7.92
(5 H, m, COPh); IR (neat) 1720 (COPh), 1690 (COC=C)
cm-'. Reduction of 10 (NaBH,, CH,OH, -40 "C) gave a
ca. 1:l mixture of the allylic alcohols lla and llb, which
was separated chromatographically.'O Dehydrobromina-
tion (50% aqueous NaOH, toluene, catalytic NBu4/HS04,
25 "C) of the less polar alcohol lla with concomitant sa-
ponification of the benzoate group followed by acidic
(HOAc, H20) cleavage of the ketal moiety afforded the
ketone 12 (73% from lla): oil; 'H NMR (CD2C12) 6 1.06
(3 H, d, J = 6.8 Hz, CHCH,), 1.10 (3 H, t, J = 7.5 Hz,
CH,CH,), 4.22 (1 H, m, H-llb), 4.38 (1 H, m, H-158); IR
(neat) 1730 (C=O) cm-'. After silylation of the hydroxyl
groups in 12 (C1SiMe2-t-Bu, DMF, imidazole), the ketone
13 was subjected to a Horner-Wittig reaction with triethyl
phosphonoacetate (KO-t-Bu, THF, 0 "C). Reduction of
the 1:l mixture of the isomeric a,p-unsaturated esters 14
with diisobutylaluminum hydride (toluene, 0 "C) gave after
chromatographic separation the E isomer 15a (32% from
12) and the less polar 2 isomer 15b.11
Etherification of 15a under phase-transfer conditions
with tert-butyl bromoacetate (50% aqueous NaOH, tolu-
ene, catalytic Bu4NHS04, 25 "C) was accompanied by
simultaneous cleavage of the tert-butyl ester to give 16
(87%). Finally, removal of the silyl ether groups (tetra-
n-butylammonium fluoride, THF, 25 "C) afforded 2 cicaprost, in
86% yield: oil;
with tert-butyl bromoacetate (50% aqueous NaOH, tolu-
ene, catalytic Bu4NHS04, 25 "C) was accompanied by
simultaneous cleavage of the tert-butyl ester to give 16
(87%). Finally, removal of the silyl ether groups (tetra-
n-butylammonium fluoride, THF, 25 "C) afforded 2 cicaprost, in
86% yield: oil;
'H NMR (CD,Cl,) 6= delta 1.07 (3 H, d, J = 6.8
Hz), 16@-CH3), 1.11 (3 H, t, J = 7.5 Hz, CH2CH3), 3.97 (1
H, m, H-llP), 4.06 (2 H, m, OCH,CO), 4.12 (2 H, m, =
H, m, H-5); IR (neat) 1730 (COOH) cm-'.
Hz), 16@-CH3), 1.11 (3 H, t, J = 7.5 Hz, CH2CH3), 3.97 (1
H, m, H-llP), 4.06 (2 H, m, OCH,CO), 4.12 (2 H, m, =
H, m, H-5); IR (neat) 1730 (COOH) cm-'.
..................
J. Am. Chem. Soc., 2003, 125 (32), pp 9653–9667
DOI: 10.1021/ja030200l
An asymmetric synthesis of the anti-metastatic prostacyclin analogue cicaprost and a formal one of its isomer isocicaprost by a new route are described. A key step of these syntheses is the coupling of a chiral bicyclic C6−C14 ethynyl building block with a chiral C15−C21 ω-side chain amide building block with formation of the C14−C15 bond of the target molecules.
A highly stereoselective reduction of the thereby obtained C6−C21 intermediate carrying a carbonyl group at C15 of the side chain was accomplished by the chiral oxazaborolidine method. The chiral phosphono acetate method was used for the highly stereoselective attachment of the α-side chain to the bicyclic C6−C21 intermediate carrying a carbonyl group at C6.
Asymmetric syntheses of the bicyclic C6−C14 ethynyl building blocks were carried out starting from achiral bicyclic C6−C12 ketones by using the chiral lithium amide method. In the course of these syntheses, a new method for the introduction of an ethynyl group at the α-position of the carbonyl group of a ketone with formation of the corresponding homopropargylic alcohol was devised.
Its key steps are an aldol reaction of the corresponding silyl enol ether with chloral and the elimination of a trichlorocarbinol derivative with formation of the ethynyl group. In addition, a new aldehyde to terminal alkyne transformation has been realized. Its key steps are the conversion of an aldehyde to the corresponding 1-alkenyl dimethylaminosulfoxonium salt and the elimination of the latter with a strong base.
Two basically different routes have been followed for the synthesis of the enantiomerically pure C15−C21 ω-side chain amide building block. The first is based on the chiral oxazolidinone method and features a highly stereoselective alkylation of (4R)-N-acetyl-4-benzyloxazolidin-2-one, and the second encompasses a malonate synthesis of the racemic amide and its efficient preparative scale resolution by HPLC on a chiral stationary phase containing column
.......
(5E) -13,14,18,13,19,19-Hecadehydro-3-oxa-6a-carba-prostaglandin I 2derivatives of the general formula I
(5E) - (16S) -13,14-didehydro-16 ,20-dimezhyl-3-oxa-18 ,18,19,19-tetradehydro-6a-carbaprostaglandin 1 2
- Example 1(5E) - (16S) -13,14-didehydro-16 ,20-dimezhyl-3-oxa-18 ,18,19,19-tetradehydro-6a-carbaprostaglandin 1
2
- [0028]To a solution of 0.4 g in 12 ml of tetrahydrofuran was added to 80 mg of 55% sodium hydride (in mineral oil) and cook for 1 hour reflux. Is added to a solution of 127 mg of bromoacetic in 4 ml of tetrahydrofuran, boiled under reflux for 18 hours, diluted with ether and extracted four times with 30 ml of 5% sodium hydroxide. This extract is adjusted with 10% sulfuric acid at 0 ° C to pH 3 and extracted with methylene chloride. The organic extract is shaken with brine, dried over magnesium sulfate and evaporated under vacuum. Obtained 220 mg hydropyranyläther), which are for the elimination of the protective groups is stirred for 18 hours with 15 ml of acetic acid / water / tetrahydrofuran (65/35/10) at 25 ° C. It is evaporated to the addition of toluene, and the residue is chromatographed on silica gel with ethyl acetate / 0.1 - 1% acetic acid. This gives 145 mg of the title compound as a colorless Ö1.
- [0029]IR (CHC1 3): 3600, 3400 (broad), 2 93 0 222 3, 1730, 1600, 1425, 1380/cm.
- [0030]The starting material for the above title compound is prepared as follows:
1 a)
- [0031]To a suspension of 3.57 g of sodium hydride (55% in mineral oil) in 360 ml of dimethoxyethane was added dropwise at O ° C, a solution of 21.9 g of 3-methyl-2-oxo-oct-5-in-phosphonsäuredimethyl esters in 140 ml of dimethoxyethane was stirred for 1 hour at 0 ° C and then add 14.56 g of finely powdered N-bromosuccinimide. It is stirred for 1 hour at O ° C, treated with a solution of 22.5 g of (lR, 5S, 6R, 7R) -3,3 - ethylenedioxy-7-benzoyloxy-6-formyl-bicyclo [3.3.0] octane in 180 ml of dimethoxyethane and 4 hours the mixture is stirred at 0 ° C. The reaction mixture is diluted with 3 1 ether, washed neutral with brine, dried with sodium sulfate and evaporated in vacuo. The residue is chromatographed with hexane / ether as eluent on silica gel. Following three chromatography of the respective diastereomeric mixed fractions obtained as polar compound 8.1 g and a polar compound 7.4 g of the title compound as colorless oils.
- [0032]IR: 2935, 2878, 17 15, 1690, 1601, 1595, 1450, 1270, 948/cm.
1 b)
- [0033]To a solution of 7.4. G of produced according to Example 1 a) ketone in 140 ml of methanol is added at -20 ° C. 3 g of sodium borohydride in portions and stirred for 30 minutes at -20 ° C. Then diluted with ether, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum.
- [0034]The crude product (15-epimer) is dissolved in 300 ml of methanol, added to 2.95 g of potassium carbonate and stirred for 21 hours at 23 ° C under argon. Then concentrated in vacuo, diluted with ether and washed neutral with brine. It is dried over magnesium sulfate and evaporated under vacuum. By column chromatography on silica gel with ether / methylene chloride (7 +3) first obtained 2.6 g of the 15SS-configured alcohol as well as 2.1 g of the more polar component 15a-configured alcohol (PG nomenclature) as colorless oils.
- [0035]A solution of 2.1 g of the above prepared alcohol 15a, 20 mg of p-toluenesulfonic acid and 1.4 g of dihydropyran in 50 ml of methylene chloride is stirred for 30 minutes at 0 ° C. Then it is poured into dilute sodium bicarbonate solution, extracted with ether, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum. Chromatography of the residue on silica gel, using hexane / ether (6 +4), 2.6 g of the title compound as a colorless oil.
- [0036]IR: 2939, 2877, 1450, 969, 948 / cm.
1 c) bicyclo [3.3.0] octane-3-one
- [0037]A solution of 290 mg of the of Example 1 b) the compound prepared in 2.5 ml of dimethyl sulfoxide and 1 ml of tetrahydrofuran is mixed with 112 mg of potassium tert-butoxide and stirred for 2 hours at 23 ° C. It is diluted with 10 ml of water and extracted three times with 10 ml of ether / hexane (7 +3), wash the extract with water until neutral, dried over brine and evaporated under vacuum.
- [0038]It is stirred for 22 hours with the residue 15 ml of acetic acid / water / tetrahydrofuran (65/35/10) evaporated in a vacuum with the addition of toluene, and the residue is purified by chromatography on silica gel. With ether eluted 150 mg oily substance, which is reacted in 5 ml of dichloromethane with 140 mg of dihydropyran and 1 mg of p-toluenesulfonic acid at 0 ° C.. After 30 minutes, diluted with ether, extracted with 5% sodium bicarbonate solution and brine, dried over magnesium sulfate and evaporated under vacuum. Chromatography of the residue on silica gel with hexane / ether (1 +1), 185 mg of the title compound as a colorless oil.
- [0039]IR: 2940, 2876, 2216, 1738, 1020, 970 / cm.
1 d)
- [0040]To a solution of 529 mg Phosphonoessigsäuretri acid ethyl ester in 10 ml of tetrahydrofuran is added at 0 C 225 mg of potassium tert-butoxide, stirred for 10 minutes, treated with a solution of 0.6 g of the product of Example 1 c) ketone in 6 ml of toluene and stirred for 22 hours at 23 ° C. It is diluted with 150 mL of ether, shake once with water, once with 20% sodium hydroxide, washed neutral with water, dried over magnesium sulfate and evaporated under vacuum. The residue is filtered using hexane / ether (6 +4) over silica gel. Thereby obtain 0.58 g of the unsaturated ester as a colorless oil.
- [0041]IR: 2940, 2870, 2212, 1704, 1655, 970 / cm.
- [0042]It adds 150 mg of lithium aluminum hydride in portions at 0 ° C to a stirred solution of 570 mg of the ester prepared in 25 ml of ether and stirred for 30 minutes at 0 ° C. Destroying the excess reagent by dropwise addition of ethyl acetate, added to 1 ml of water, stirred for 3 hours at 20 ° C, filtered and evaporated under vacuum. The residue is chromatographed with ether / hexane (3 +2) on silica gel. Thereby obtained as a non-polar compound 140 mg of 2 - {(Z) - (1S, 5S, 6S, 7R) -7 - (tetrahydropyran-2-yloxy) -6 - / R3S, 4S)-4-methyl-3-( tetrahydropyran-2-yloxy)-nona-1 ,6-diinyl]-bicyclo [3.3.0] octane-3-ylidene} - ethane-1-ol and 180 mg of the title compound as a colorless oil.
- [0043]IR: 3620, 3450 (broad), 2940, 2860, 2212, 970/cm.
3 BIMATOPROST
Bimatoprost
BIMATOPROST
155206-00-1
Lumigan, Latisse, AGN 192024, bimatoprostum, UNII-QXS94885MZ, Lumigan (TN), CHEBI:51230, AC1NSJUW, AGN-192024
Molecular Formula: C25H37NO4
Molecular Weight: 415.56558
Bimatoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a prostaglandin analogue that works by increasing the outflow of aqueous fluid from the eyes. It binds to the prostanoid FP receptor.
Allergan reported Lumigan® sales of US$625 million and Latisse® sales of US$100 million in 2013
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 7-[3,5-dihydroxy-2- (3-hydroxy-5-phenyl-pent-1-enyl)- cyclopentyl]-N-ethyl-hept-5-enamide | |
| CLINICAL DATA | |
| TRADE NAMES | Lumigan |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a602030 |
| LICENCE DATA | US Daily Med:link |
| PREGNANCY CAT. | C (US) |
| LEGAL STATUS | ℞-only (US) |
| ROUTES | Topical (eye drops) |
| IDENTIFIERS | |
| CAS NUMBER | 155206-00-1 |
| ATC CODE | S01EE03 |
| PUBCHEM | CID 5311027 |
| IUPHAR LIGAND | 1958 |
| DRUGBANK | DB00905 |
| CHEMSPIDER | 4470565 |
| UNII | QXS94885MZ |
| CHEMICAL DATA | |
| FORMULA | C25H37NO4 |
| MOL. MASS | 415.566 g/mol |
Bimatoprost (marketed in the U.S., Canada and Europe by Allergan, under the trade name Lumigan) is aprostaglandinanalog/prodrug used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces intraocular pressure (IOP) by increasing the outflow of aqueous fluid from the eyes.[1] In December 2008, the indication to lengthen eyelashes was approved by the U.S. Food and Drug Administration (FDA); the cosmetic formulation of bimatoprost is sold as Latisse /ləˈtiːs/.[2] In 2008-2011, at least three case series suggested that bimatoprost has the ability to reduce adipose (fat) tissue.[3][4][5]
Allergen originally developed Bimatoprost and marketed it as Lumigan® for the treatment of elevated intraocular pressure (IOP), with open-angle glaucoma or ocular hypertension. Bimatoprost was later reformulated as a topical formulation and marketed as Latisse® for use in the treatment of hypotrichosis of the eyelashes.
Cosmetic use
In patients using ophthalmic prostaglandins such as travoprost and latanoprost, it has been anecdotally noted that there had been an increase in diameter, density and length of eyelashes. Allergan has initiatedclinical trials investigating the usage of Lumigan as a cosmetic drug.[6] On December 5, 2008, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee voted to approve bimatoprost for the cosmetic use of darkening and lengthening eyelashes.The medical term for this is treatment of hypotrichosis, however, the FDA approval is for purely cosmetic purposes.[7]
For cosmetic purposes, it is administered once daily by applying the solution to the skin at the base of the eyelash using an applicator device “Application Guide”, where it has its effect upon the hair follicle.
Bimatoprost activates prostamide alpha F2 receptors found in the hair follicle to stimulate its growth rate. Research led by Professor Randall and the University of Bradford found that it may also offer a treatment for scalp hair regrowth in trials conducted on samples taken from men undergoing hair transplants.[8]
According to Allergan’s package labeling, users of its Latisse cosmetic product didn’t develop darker irises in clinical studies; however, “patients should be advised about the potential for increased brown iris pigmentation which is likely to be permanent.”[9]
Several cosmetics companies have released products based on prostaglandin analogs, as non-drug cosmetics.
- Age Intervention Eyelash by Jan Marini Skin Research
- RevitaLash by Athena Cosmetics Corp.
These companies have been sued by Allergan for patent infringement.[6] The FDA has seized Age Intervention Eyelash as an “unapproved and misbranded drug” because Jan Marini Skin Research promoted it as something that increases eyelash growth[10]and because it is “adulterated” with bimatoprost.[11]
Fat-reducing properties
Reductions in orbital fat (i.e., fat around the eye) have been observed in patients using bimatoprost as glaucoma therapy.[12] Of particular interest, the loss of orbital fat was unilateral in patients who used bimatoprost on only one eye.[13] The effect appears reversible upon cessation of bimatoprost use. The effect is likely to explain deepening of the lid sulcus described in a series of three patients on bimatoprost.[14] The mechanism for the apparent fat reduction remains unclear. However, bimatoprost is chemically analogous to prostaglandin F2α (PGF2α), a compound which is known to reduce fat by inhibition of adipocyte differentiation and survival.[15]
Formulations
Lumigan is a 0.03% solution of bimatoprost, and contains benzalkonium chloride as a preservative. Contact lenses should therefore be removed before use, and replaced no less than 15 minutes later;[1] other eye drops or ointments should be given no less than five minutes before or after bimatoprost.[1]
Efficacy
Studies have shown once-daily bimatoprost 0.03% ophthalmic solution to be more effective than timolol twice daily in reduction of intraocular pressure (IOP) and as effective as or more effective than the prostaglandin analogues latanoprost and travoprost in reducing IOP.[16]
Side effects
Possible side effects of this medication are:
- May cause blurred vision.
- May cause eyelid redness.
- May permanently darken eyelashes.
- May cause eye discomfort.
- May eventually cause permanent darkening of the iris to brown.
- May cause a temporary burning sensation during use.
- May cause thickening of the eyelashes.
- It may cause unexpected growth of hair if applied inappropriately, on the cheek, for example.
- It may cause infection if the one-time applicators which come with the genuine product are reused.
- Lashes may grow so long that they become ingrown and scratch the cornea.
- May cause darkening of the eyelid or of the area beneath the eye.[17]
On November 19, 2007, the FDA issued a warning during the seizure of a bimatoprost-containing cosmetic.[18] The warning stated that “the extra dose of bimatoprost may decrease the prescription drug’s effectiveness. Damage to the optic nerve may lead to decreased vision and possibly blindness.”
PATENTS
| COUNTRY | PATENT NUMBER | APPROVED | EXPIRES (ESTIMATED) |
|---|---|---|---|
| Canada | 2585691 | 2009-05-19 | 2026-03-14 |
| Canada | 2144967 | 2003-11-11 | 2013-09-09 |
| United States | 7351404 | 2004-05-25 | 2024-05-25 |
| United States | 6403649 | 1992-09-21 | 2012-09-21 |
Jiang Xing Chen, “Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost.” U.S. Patent US20090287003, issued November 19, 2009.
US20090287003
LUMIGAN® 0.01% and 0.03% (bimatoprost ophthalmic solution) is a synthetic prostamide analog with ocular hypotensive activity. Its chemical name is (Z)-7-[(1R,2R,3R,5S)-3,5Dihydroxy-2-[(1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and Its molecular weight is 415.58. Its molecular formula is C24H37NO4. Its chemical structure is:
 |
Bimatoprost is a powder, which is very soluble in ethyl alcohol and methyl alcohol and slightly soluble in water. LUMIGAN® 0.01% and 0.03% is a clear, isotonic, colorless, sterile ophthalmic solution with an osmolality of approximately 290 mOsmol/kg.
LUMIGAN® 0.01% contains Active: bimatoprost 0.1 mg/mL; Preservative: benzalkonium chloride 0.2 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.
LUMIGAN® 0.03% contains Active: bimatoprost 0.3 mg/mL; Preservative: benzalkonium chloride 0.05 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.
SYNTHESIS
……………………………….
- PGF2α prostaglandin synthetic routes so far reported may comprise the following two major steps:

- The starting compound is generally protected on the secondary hydroxyl group:
 where Y stands for p-phenylbenzoyl (PPB) (Ia) or benzoyl (Bz) (Ib) or an analogous substituted aryl group.
where Y stands for p-phenylbenzoyl (PPB) (Ia) or benzoyl (Bz) (Ib) or an analogous substituted aryl group. - The product is obtained as a mixture of epimers where the 15-OH can be in α-position or β-position. The removal of unwanted β-isomer and of other impurities represents one of the main difficulties in the preparation of prostaglandins and many methods have been proposed to reduce the amount of the undesired diastereoisomers formed during the preparation.
- In EP0544899B1 (Pharmacia AB) the reduction of the α,β-unsaturated ketone is performed with lithium-tri(sec-butyl)borohydride at ―130°C.
- In this kind of processes the low selectivity in the reduction of the keto group leads to a tedious separation of the diastereomers and generally decreases the global yield of the synthesis.
- A different method of stereoselective reduction of the ketone is proposed in US7674921 B1 (Cayman Chemical Co.) where the reaction with lithium aluminium hydride in the presence of (S)-binaphtol in tetrahydrofuran at -78°C is reported.
- An alternative is presented in US6927300B2 and in US 7157590B2(Finetech Lab. Ltd.) where the critical step of the reduction of the α,β-unsaturated keto-group is performed on the compound reported below
 where R1 is an aryl carbonyl group in the presence of (-)β-chlorodiisopinocampheylborane in tetrahydrofuran at ―25°C which allows to obtain a ratio of 95/5 of the two diastereoisomers (R)-(IV)/(S)-(IV).
where R1 is an aryl carbonyl group in the presence of (-)β-chlorodiisopinocampheylborane in tetrahydrofuran at ―25°C which allows to obtain a ratio of 95/5 of the two diastereoisomers (R)-(IV)/(S)-(IV).
- A further improvement in the preparation of PGF2α is directed to the reduction of lactone to lactol in order to introduce the α-chain by subsequent Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide.
- The reaction was usually carried out using diisobutylaluminum hydride at very low temperatures, namely between -80°C and ―40°C. Unfortunately, in these conditions the partial removal of the protecting group PPB or benzoyl (Bz) is possible.
- To avoid the inconvenient of working with mixtures of products, the p-phenylbenzoyl group or the benzoyl group R is removed by basic hydrolysis and a suitable protecting group is introduced at the two hydroxyl groups of the so obtained compound according to the following scheme:

- In this way two additional steps of protection and deprotection are introduced possibly leading to a decrease of the global yield of the process at an advanced stage of the synthesis.
- Moreover when P’ is a silylated group as reported in US7268239B2(Resolution Chemical Ltd.), a mixture of products is obtained because of the migration of the silyl group as shown in scheme 3

- The same approach of hydrolysis of the aroyl group R followed by diprotection of the two hydroxyl groups with trialkylsilyl group or triaryl silyl group or tetrahydropiranyl group is disclosed in US7642370B2(Daichii Fine Chemical Co.). The protection of the two hydroxyl groups coming before the reduction of the lactone ring to lactol is described also in US7674921B1 (Cayman Chemical Co.) and in US6689901B2(Pharmacia and Upjohn Company).
- It follows that one important issue in the synthesis of PGF2α is the involvement of intermediates with the most appropriate hydroxyl protecting group. As a consequence the use of the starting Corey lactone carrying a protection able to survive to the conditions of the subsequent reactions allows to form intermediates easy to isolate and to purify. Moreover the protection should be selectively removed in mild conditions.
- In US5359095 (Pharmacia AB) the preparation of PGF2α prostaglandins is described starting from the commercially available (-)-Corey lactone (Ia) which is oxidized to the corresponding aldehyde (IIa)

- The reaction is carried out in the presence of dicyclohexylcarbodiimide in dimethylsulfoxide and 1,2-dimethoxyethane; after quenching with orthophosphoric acid the aldehyde is obtained. In the same application it is reported that the crude aldehyde (II) is particularly unstable and must be used within a short period after preparation.
- In US2010/0010239A1 (Sandoz AG) compound (I) is preferably oxidized in dichloromethane with oxalyl chloride and DMSO; the aldehyde (II) is not isolated and is processed directly in the solution where it is obtained or, when necessary, stored in solution at a temperature between ―20 and 0°C.
- In US7268239B2 (Resolution Chem. Ltd.) the aldehyde (II)
 where Z is (C6-C10)-aryl optionally substituted with one to three substituents independently selected from the group consisting of halo, C1 to C6 alkyl and unsubstituted C6 to C10 aryl, is formed by oxidation of the alcohol with sodium hypochlorite and 2,2,6,6-tetramethtyl-1-piperidinyloxy free radical (TEMPO); in order to avoid the risk of degradation, the aldehyde is directly used in the organic solution where it is synthesized.
where Z is (C6-C10)-aryl optionally substituted with one to three substituents independently selected from the group consisting of halo, C1 to C6 alkyl and unsubstituted C6 to C10 aryl, is formed by oxidation of the alcohol with sodium hypochlorite and 2,2,6,6-tetramethtyl-1-piperidinyloxy free radical (TEMPO); in order to avoid the risk of degradation, the aldehyde is directly used in the organic solution where it is synthesized. - According to US2009/0287003A1 (Eastar Chem. Corp.) five steps can be performed to prepare the aldehyde to be used as starting material for the synthesis of latanoprost. The five steps are reported in the scheme below:

- In WO2010/097672 (Sifavitor) the starting material is the Corey lactone where the hydroxyl group is protected astert-butyldimethyl silyl derivative and during the synthesis a second protection is introduced according the scheme reported below referred to the preparation of Bimatoprost:

- In case Bimatoprost is the end product, compound 9 with A=benzyl (in the following scheme, compound 9a) is directly converted by Wittig reaction to the acid 10 which is then converted in one step to ethylamide affording Bimatoprost, according to the following scheme:
- In a preferred embodiment, the conversion of compound 10a to Bimatoprost is performed by reaction with ethylamine in an organic solvent, which is chosen among amides, ethers, ketones and chlorinated solvents, more preferably N,N-dimethylformamide, at a temperature between ―35 and 25°C, more preferably between -20°C and -10°C, in the presence of triethylamine and a suitable coupling reagent, which is preferably 1-(methylsulfonyloxy)benzotriazole. The molar ratio between compound 10a and ethylamine is comprised between 1 and 5, more preferably is 3.5, while the molar ratio between compound 10a and the coupling reagent is comprised between 1 and 2.5, more preferably is 1.8.
- When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:
- In the case of Travoprost, compound 9 with A=3-(trifluoromethyl)phenoxy (in the following scheme, compound 9b) is converted into 10b, which in turn is converted into Travoprost by esterification of the carboxylic acid by reaction with 2-iodopropane, according to scheme 10:


EXAMPLE 13
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((S,E)-3-hydroxy-5-phenylpent-1-enyl)cyclopentyl)hept-5-enoic acid (Bimatoprost free acid)

- 4-Carboxybutyltriphenylphosphonium bromide 15 (65.4 g, 0.148 mol) was suspended in tetrahydrofuran (150.0 mL) at 0°C under nitrogen atmosphere. A solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((S,E)-3-hydroxy-5-phenylpent-1-enyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (10.0 g, 0.033 mol) in tetrahydrofuran (46.0 mL) was added dropwise at a temperature lower than – 10°C. After three hours at -15°C no more starting material was visible on TLC and water (200 mL) was added. The mixture was extracted with diisopropyl ether (144 mL) and the layers were separated. The aqueous phase was treated with 0.6 N HCl to pH 6.0. Two extractions with ethyl acetate (2x 250 mL) were then performed and the combined organic layers were concentrated under vacuum at 40°C. An oil (26.80 g) was obtained which was purified on silica gel (eluent: dichloromethane:methanol from 97.5:2.5 to 85:15). The fractions of interest were combined and concentrated at 35°C under reduced pressure affording a colorless oil (11.7 g, 0.030 mol, 91%).
- 1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.29-7.25 (m, 2H, Ph), 7.19-7.15 (m, 3H, Ph), 5.60 (dd, J=7.2, 15.2 Hz, 1H, vinyl), 5.51-5.41 (m, 2H, H vinyl), 5.38-5.31 (m, 1H, vinyl), 4.5-3.8 (m, 7H), 2.67 (m, 2H, -CH 2-Ph), 2.37-1.43 (m, 14H).
- 13C-NMR {400 MHz, CDCl3, δ (ppm)}: 177.2 (C), 141.9 (C), 134.9 (CH), 133.0 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH, arom.), 128.3 (2xCH arom.),125.8 (CH), 77.4 (CH), 72.3 (CH), 72.2 (CH), 55.1 (CH), 50.5 (CH), 42.7 (CH2), 38.5 (CH2), 32.9 (CH2), 31.8 (CH2), 26.3 (CH2), 25.2 (CH2), 24.4 (CH2).
- HPLC-MS (ESI): [M-H2O+1]+= 371; [M+Na]+ = 411; [2M+Na]+ = 799.
EXAMPLE 14
(5Z)-7-[(2R)-3,5-Dihydroxy-2-[(1E)-3-hydroxy-5-phenylpent-1-en-1-yl]cyclopentyl]-N-ethylhept-5-enamide (Bimatoprost)

- Bimatoprost acid (11.50 g, 0.030 mol) was dissolved in dimethylformamide (92.0 mL), and stirred at -15°C. Triethylamine (7.25 mL, 5.26 g, 0.052 mol) was then added over 5 minutes followed by the portionwise addition of 1-(methylsulfonyloxy)benzotriazole 16 (prepared according to Bulletin of the Chemical Society of Japan, 1978, 51(11), 3320-3329) (11.27 g, 0.053 mol). The mixture was then stirred for one hour at -15°C and an aqueous solution of ethylamine (70% weight, 8.4 mL, 0.104 mol) was added dropwise over 5 minutes. The temperature was allowed to reach 0°C and the reaction was checked by TLC. The mixture was washed with water (172.0 mL) and extracted four times with ethyl acetate (4x 230.0 mL). The combined organic layers were washed with 5% sodium bisulfate solution (100 mL, 50 mL, 50 mL). The bisulfate aqueous phases were extracted with ethyl acetate (50.0 mL). The organic layers were concentrated at 40°C under reduced pressure affording the crude product as an oil (16.98 g). Treatment with dichloromethane and diisopropylether at 0°C for one hour followed by filtration afforded a solid which was then recrystallized from ethyl acetate (6.79 g, 0.016 mol, 55%).
- 1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.26 (m, 2H, Ph), 7.17 (m, 3H, Ph), 6.07 (t, J=5.6Hz, 1H, -NH-), 5.57 (dd, J=7.6, 15.2 Hz, 1H, H-14), 5.45 (dd, J=8.8, 15.2 Hz, 1H, H-13), 5.35 (m, 2H, H-5+H-6), 4.32 (d, J=4.8 Hz, 1H, OH-11), 4.08 (m, 2H, H-9+H-15), 3.90 (m, 1H, H-11), 3.73 (m, 2H, OH-9+OH-15), 3.20 (m, 2H, -N-CH 2-CH3), 2.64 (m, 2H, -CH2-17), 2.30 (m, 1H, H-12), 2.18 (m, 2H, H-7+H-10), 2.11 (m, 3H, CH2-2+H-7), 2.03 (m, 2H, H-4), 1.89 (m, 1H, H-16), 1.77 (m, 2H, H-10+H-16), 1.64 (m, 2H, H-3), 1.45 (m, 1H, H-8), 1.09 (t, J=6.8Hz, 3H, -N-CH2-CH3).
- 13C-NMR {400 MHz, CDCl3, δ (ppm)}: 173.4 (C), 142.0 (C), 135.0 (CH), 133.2 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH arom), 128.3 (2xCH arom), 125.7 (CH arom), 77.5 (CH), 72.22 (CH), 72.20 (CH), 55.4 (CH), 50.1 (CH), 42.8 (CH2), 38.7 (CH2), 35.8 (CH2), 34.3 (-N-CH2), 31.8 (CH2), 26.6 (CH2), 25.6 (CH2), 25.3 (CH2), 14.7 (CH3).
- HPLC-MS (ESI): [M+Na]+ = 438, [(M-H2O) +H]+=398, [(M-2H2O) +H]+=380.
………………………………..
Bimatoprost refers to (Z)-7-[(1R,2R,3R,5S)-3,5-Dihydroxy-2-[1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and its molecular weight is 415.58. Its molecular formula is C25H37NO4. Its chemical structure is:
References
- “Bimatoprost Ophthalmic”. MedlinePlus. January 1, 2003. Archived from the original on 2007-10-05. Retrieved 2007-11-19.
- “Allergan gets FDA approval for eyelash treatment”. BusinessWeek. Associated Press. December 26, 2008. Retrieved December 26, 2008.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27.doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit29 (6): 357–359. doi:10.3109/01676830.2010.527028.PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df.PMID 18645437.
- Rundle, Rhonda L. (2007-11-19). “Drug That Lengthens Eyelashes Sets Off Flutter”. The Wall Street Journal. Retrieved 2007-11-19.
- The Pink Sheet: [1] Lauren Smith December 15, 2008; Volume 70, Number 050,Page
- Federation of American Societies for Experimental Biology Journal “The prostamide-related glaucoma therapy, bimatoprost, offers a novel approach for treating scalp alopecias” Randall et all. October 26, 2012.
- Latisse prescribing information: “Important Safety Information”
- MSNBC: FDA Seizes $2 Million Of Potentially Harmful SJ Eye Product KNTV-TV November 17, 2007
- Reuters: “U.S. seizes discontinued eyelash product”. Jim Wolf. November 16, 2007.
- Tappeiner C, Perren B, Iliev ME, Frueh BE, Goldblum D (May 2008). “Orbitale Fettgewebsatrophie bei lokaler Bimatoprost-Therapie – Kann Bimatoprost einen Enophthalmus verursachen?” [Orbital fat atrophy in glaucoma patients treated with topical bimatoprost--can bimatoprost cause enophthalmos?]. Klinische Monatsblätter für Augenheilkunde (in German)225 (5): 443–5.doi:10.1055/s-2008-1027362. PMID 18454393.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL (2008). “Periorbital changes associated with topical bimatoprost”. Ophthalmic Plastic and Reconstructive Surgery 24 (4): 302–7. doi:10.1097/IOP.0b013e31817d81df.PMID 18645437.
- Peplinski LS, Albiani Smith K (August 2004). “Deepening of lid sulcus from topical bimatoprost therapy”. Optometry and Vision Science 81 (8): 574–7.doi:10.1097/01.opx.0000141791.16683.4a. PMID 15300114.
- Serrero G, Lepak NM (April 1997). “Prostaglandin F2alpha receptor (FP receptor) agonists are potent adipose differentiation inhibitors for primary culture of adipocyte precursors in defined medium”. Biochemical and Biophysical Research Communications 233 (1): 200–2. doi:10.1006/bbrc.1997.6433. PMID 9144422.
- Curran MP (2009). “Bimatoprost: a review of its use in open-angle glaucoma and ocular hypertension”. Drugs Aging 26 (12): 1049–71. doi:10.2165/11203210-000000000-00000.PMID 19929032.
- “Long Lashes Without Prescription, but With Risks”. Catherine Saint Louis. The New York Times. May 1, 2010
- “Potentially Harmful “Cosmetic” Eye Product Seized” (Press release). U.S. Food and Drug Administration (FDA). November 19, 2007. Retrieved 2007-12-05.
Citations
- Chen M, Cheng C, Chen Y, Chou C, Hsu W (2006). “Effects of bimatoprost 0.03% on ocular hemodynamics in normal tension glaucoma.”. J Ocul Pharmacol Ther 22 (3): 188–93. doi:10.1089/jop.2006.22.188. PMID 16808680.
- Kruse P, Rieck P, Sherif Z, Liekfeld A (2006). “Cystoid macular edema in a pseudophakic patient after several glaucoma procedures. Is local therapy with bimatoprost the reason?”. Klinische Monatsblätter für Augenheilkunde 223 (6): 534–7.doi:10.1055/s-2005-858992. PMID 16804825.
- Steinhäuser S (2006). “Decreased high-density lipoprotein serum levels associated with topical bimatoprost therapy.”.Optometry 77 (4): 177–9.doi:10.1016/j.optm.2006.02.001. PMID 16567279.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27. doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit29 (6): 357–359.doi:10.3109/01676830.2010.527028. PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df.PMID 18645437.
External links
- Medical News Today: FDA Seizes $2 Million Of Cosmetic Eye Product Which Contains Drug Ingredient And Makes Unapproved Drug Claims. Christian Nordqvist. 18 November 2007
- Wired Science: FDA Seizes Cosmetic That Can Blind. Brandon Keim. November 19, 2007
- Eye Drops: [The generic name of the Latisse eye drop is Bimatoprost Ophthalmic Solution 0.03%]. Crazzy Paul. Aug 01, 2013
1-25-2012
|
Method of enhancing hair growth
| |
1-25-2012
|
NITRIC OXIDE DONATING PROSTAMIDES
| |
11-25-2011
|
BIMATOPROST CRYSTALLINE FORM I
| |
10-19-2011
|
Method of Enhancing Hair Growth
| |
7-22-2011
|
IMPROVED PROCESS FOR THE PRODUCTION OF BIMATOPROST
| |
6-3-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
| |
5-27-2011
|
COMPOSITIONS AND METHODS FOR STIMULATING HAIR GROWTH
| |
5-27-2011
|
ENHANCED BIMATOPROST OPHTHALMIC SOLUTION
| |
5-25-2011
|
Bimatoprost crystalline form I
| |
5-13-2011
|
COMPOSITIONS FOR ENHANCING HAIR GROWTH
|
3-2-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
| |
12-24-2010
|
METHOD FOR THE PURIFICATION OF PROSTAGLANDINS
| |
12-15-2010
|
Enhanced bimatoprost ophthalmic solution
| |
9-17-2010
|
Compositions and Methods for Reducing Body Fat
| |
5-28-2010
|
COMPLEXES OF PROSTAGLANDIN DERIVATIVES AND MONOSUBSTITUTED, CHARGED BETA-CYCLODEXTRINS
| |
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
| |
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
| |
2-24-2010
|
Compositions and methods for reducing body fat
| |
1-27-2010
|
10-HYDROXY-11-DIHYDROPROSTAGLANDIN ANALOGS AS SELECTIVE EP4 AGONISTS
| |
1-15-2010
|
Process for the Production of Prostaglandins and Prostaglandin Analogs
|
11-20-2009
|
Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost
| |
11-4-2009
|
Enzymatic transformation of a prostaglandin (bimatoprost) intermediate
| |
10-16-2009
|
Method for preparing prostaglandin F analogue
| |
8-14-2009
|
METHOD OF ENHANCING HAIR GROWTH
| |
6-12-2009
|
Enhanced Bimatoprost Ophthalmic Solution
| |
4-3-2009
|
METHOD FOR SCREENING OF PROSTAGLANDIN COMPOUNDS COMPRISING AN OPTIMAL FORMULATION FOR THE ENHANCEMENT OF HAIR GROWTH AND THE STIMULATION OF FOLLICULAR ANAGEN AND FORMULATIONS RESULTING THEREFROM
| |
10-15-2008
|
5-Thiopiperdinyl prostaglandin e analogs
| |
2-20-2008
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
| |
2-15-2008
|
Novel Prostamides For The Treatment Of Glaucoma And Related Diseases
| |
11-23-2007
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
|
7-18-2007
|
Compositions and methods comprising prostaglandin related compounds and trefoil factor family peptides for the treatment of glaucoma with reduced hyperemia
| |
5-18-2007
|
NOVEL PROSTAMIDES FOR THE TREATMENT OF GLAUCOMA AND RELATED DISEASES
| |
4-27-2007
|
Compositions comprising benzo (g) quinoline derivatives and prostaglandin derivatives
| |
3-7-2007
|
Prostamides for the treatment of glaucoma and related diseases
| |
1-31-2007
|
10-Hydroxy-11-dihydroprostaglandin analogs as selective EP4 agonists
| |
1-24-2007
|
Process for the preparation of prostaglandin derivatives
| |
1-10-2007
|
Protected diols for prostaglandin synthesis
| |
1-3-2007
|
Process for the preparation of 17-phenyl-18,19,20-thinor-pgf 2a and its derivatives
| |
11-29-2006
|
Protected and unprotected triols for prostaglandin synthesis
| |
9-20-2006
|
Prostaglandin synthesis
|
9-6-2006
|
Cyclopentane heptan(ENE)OIC acid, 2-heteroarylalkenyl derivatives as therapeutic agents
| |
7-5-2006
|
Method for imparting artificial tan to human skin
| |
8-24-2005
|
Inhibition of irritating side effects associated with use of a topical ophthalmic medication
| |
3-18-2005
|
Methods for the treatment of gray hair using cyclopentane(ene) heptan(en)oic acid amides
| |
3-9-2005
|
9,11-cycloendoperoxide pro-drugs of prostaglandin analogues for treatment of ocular hypertension and glaucoma
| |
2-11-2005
|
Compositions for delivery of therapeutics into the eyes and methods for making and using same
| |
5-29-2002
|
Ocular hypotensive lipids
|
| EP0364417A1 | Sep 6, 1989 | Apr 18, 1990 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0364417B1 | Sep 6, 1989 | Feb 9, 1994 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0544899B1 | Jun 19, 1992 | Sep 6, 1995 | CHINOIN Gyogyszer és Vegyészeti Termékek Gyára RT. | Prostaglandins |
| US5359095 | Feb 8, 1994 | Oct 25, 1994 | Pharmacia Ab | Hydrogenation of doulbe bond in intermediate compound without deoxygenatio of allytic alcohol |
| US6689901 | Jun 25, 2002 | Feb 10, 2004 | Pharmacia & Upjohn Company | Process and intermediates to prepare latanoprost |
| US6927300 | Jan 26, 2001 | Aug 9, 2005 | Finetech Laboratories Ltd | Process for the preparation of Latanoprost |
| US7157590 | May 3, 2002 | Jan 2, 2007 | Finetech Laboratories Ltd. | Chemical intermediate for vision defect drug |
| US7268239 | Jul 27, 2005 | Sep 11, 2007 | Resolution Chemicals Limited | Process for the preparation of prostaglandins and analogues thereof |
| US7642370 | Mar 20, 2007 | Jan 5, 2010 | Daiichi Fine Chemical Co., Ltd. | Method for preparing prostaglandin derivative |
| US7674921 | Feb 25, 2008 | Mar 9, 2010 | Cayman Chemical Company, Inc. | cloprostenol or latanoprost; topical use; highly crystalline structures that are easy to formulate into ophthalmic solutions; hydrolysis of these analogs releases only the active PGF2 alpha analog free acid, without the production of toxic and irritant small aliphatic alcohol coproducts |
| US20090259066 * | Jul 3, 2008 | Oct 15, 2009 | Everlight Usa, Inc. | Method for preparing prostaglandin F analogue |
| US20090287003 | Sep 29, 2006 | Nov 19, 2009 | Jiang Xing Chen | Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost |
| US20100010239 | Jul 10, 2009 | Jan 14, 2010 | Sandoz Ag | Process for the Production of Prostaglandins and Prostaglandin Analogs |
| WO2010097672A1 | Feb 18, 2010 | Sep 2, 2010 | Sifavitor S.R.L. | Process for the preparation of prostaglandin derivatives |
''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
5 LATANOPROST
Latanoprost
isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate.
130209-82-4
XA41, PhXA34 [as 15 (R, S) -isomer], PhXA41, Xalatan
Latanoprost (pronounced la-TA-noe-prost) ophthalmic solution is a medication administered into the eyes to control the progression of glaucoma or ocular hypertension by reducing intraocular pressure. It is a prostaglandin analogue (more specifically an analogue ofprostaglandin F2α[1]) that lowers the pressure by increasing the outflow of aqueous fluid from the eyes through the uvealsclearal tract.[2] Latanoprost is an isopropyl ester prodrug, meaning it is inactive until it is hydrolyzed by esterases in the cornea to the biologically active acid.[3]
It is also known by the brand name of Xalatan manufactured by Pfizer. Annual sales are approximately $1.6 billion. The patent for latanoprost expired in March 2011, and at least one generic version (manufactured by Mylan Inc.) is now widely available in the U.S. The Veterans Health Administration, part of the U.S. Department of Veterans Affairs, uses generic Latanoprost manufactured by Alcon Laboratories of Fort Worth, Texas distributed by Novartis generic brand Sandoz Pharmaceuticals.
Latanoprost was invented by Johan W. Stjernschantz and Bahram Resul, employees of the Pharmacia Corporation of Upsalla, Sweden.[4]
It is on the World Health Organization's List of Essential Medicines, a list of the most important medication needed in a basic health system.[5]
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| isopropyl (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2- [(3R)3-hydroxy-5-phenylpentyl]-cyclopentyl] hept-5-enoate | |
| Clinical data | |
| Trade names | Xalatan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697003 |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Pharmacokinetic data | |
| Half-life | 17 minutes |
| Identifiers | |
| CAS number | 130209-82-4 |
| ATC code | S01EE01 |
| PubChem | CID 5311221 |
| IUPHAR ligand | 1961 |
| DrugBank | DB00654 |
| ChemSpider | 4470740 |
| UNII | 6Z5B6HVF6O |
| KEGG | D00356 |
| ChEBI | CHEBI:6384 |
| ChEMBL | CHEMBL1051 |
| Chemical data | |
| Formula | C26H40O5 |
| Mol. mass | 432.593 g/mol |
Medical uses
Ocular hypertension
- In well-controlled clinical trials including patients with open-angle glaucoma or ocular hypertension (IOP ≥21 mm Hg), monotherapy with latanoprost reduced IOP levels by 22 to 39% over 1 to 12 months’ treatment. Latanoprost was significantly more effective than timolol 0.5% twice daily in 3 of 4 large (n = 163 to 267) randomised, double-blind trials. Latanoprost demonstrated a stable long-term IOP-lowering effect in 1- or 2-year continuations of these trials, with no sign of diminishing effect during prolonged treatment.[6]
- Meta analysis suggests that latanoprost is more effective than timolol in lowering IOP. However, it often causes iris pigmentation. While current evidence suggests that this pigmentation is benign, careful lifetime evaluation of patients is still justified.[7]
Closed-angle glaucoma
- Patients who had elevated IOP despite iridotomy and/or iridectomy (including patients of Asian descent), latanoprost was significantly more effective than timolol in two double-blind, monotherapy trials (8.2 and 8.8 mm Hg vs 5.2 and 5.7 mm Hg for latanoprost vs timolol at 12 and 2 weeks, respectively).[8]
Method of administration
One drop in the affected eye(s) once daily in the evening; do not exceed the once daily dosage because it has been shown that more frequent administration may decrease the intraocular-pressure (IOP) lowering effect[2]
Adverse effects[
Listed from most to least common:
- >5% to 15%: Blurred vision, burning and stinging, conjunctival hyperemia, foreign body sensation, itching, increased pigmentation of the iris causing (heterochromia), punctate epithelial keratopathy
- 4%: Cold or upper respiratory tract infections, flu-like syndrome
- 1-4%: Dry eyes, excessive tearing, eye pain, lid crusting, lid edema, lid erythema (hyperemia), lid pain, photophobia (light intolerance)
- 1 % - 2%: Chest pain, allergic skin reactions, arthralgia, back pain, myalgia, thickening of the eyelashes.(used,also bimatoprost,in cosmetic industry as eyelash growth enhancers)
- <1% (Limited to important or life-threatening): Asthma, herpes keratitis, iritis, keratitis, retinal artery embolus, retinal detachment, toxic epidermal necrolysis, uveitis, vitreous hemorrhage from diabetic retinopathy
- A single case report links latanoprost use to the progression of keratoconus.[9]
Concerns related to adverse effects:
- Bacterial keratitis: Inadvertent contamination of multiple-dose ophthalmic solutions, has caused bacterial keratitis.
- Ocular effects: May permanently change/increase brown pigmentation of the iris, the eyelid skin, and eyelashes. In addition, may increase the length and/or number of eyelashes (may vary between eyes); changes occur slowly and may not be noticeable for months or years. Long-term consequences and potential injury to eye are not known.
- Ocular disease: Use with caution in patients with intraocular inflammation, aphakic patients, pseudophakic patients with a torn posterior lens capsule, or patients with risk factors for macular edema. Safety and efficacy have not been determined for use in patients with angle-closure-, inflammatory-, or neovascular glaucoma.
Special populations
Contact lens wearers: Contains benzalkonium chloride which may be absorbed by contact lenses; remove contacts prior to administration and wait 15 minutes before reinserting
Contraindications
Hypersensitivity to latanoprost, benzalkonium chloride, or any component of the formulation
Drug Interactions
Bimatoprost: The concomitant use of Latanoprost and Bimatoprost may result in increased intraocular pressure. Risk D: Consider therapy modification
Nonsteroidal Anti-Inflammatory Agents: May diminish the therapeutic effect of Prostaglandins (Ophthalmic). Nonsteroidal Anti-Inflammatory Agents may also enhance the therapeutic effects of Prostaglandins (Ophthalmic). Risk C: Monitor therapy
Pregnancy
Prescription of Latanoprost is limited in human studies due to high incidence of abortion shown in animal experiments. Because of this, Latanoprost is classified as Risk factor C (Adverse events were observed in animal reproduction studies at maternally toxic doses)according to United States Food and Drug Administration's use-in-pregnancy ratings.[10]Lactation Excretion in breast milk unknown/use caution. Breast-Feeding Considerations It is not known if latanoprost is excreted in breast milk. The manufacturer recommends that caution be exercised when administering latanoprost to nursing women.[2]
Storage
Latanoprost is a substance exhibiting thermal and solar instability. Concentration of latanoprost will decrease by 10% when stored at 50 and 70 degrees Celsius every 8.25 and 1.32 days respectively. Reaction with ultraviolet radiation will cause rapid degradation of Latanoprost. It is therefor important to store Latanoprost ideally in temperature below room temperature and free from sunlight in order to attain acceptable drug quality. [11]
Latanoprost is a prostaglandin F2α analogue. Its chemical name is isopropyl-(Z)7[(1R,2R,3R,5S)3,5-dihydroxy-2-[(3R)-3-hydroxy-5-phenylpentyl]cyclopentyl]-5-heptenoate. Its molecular formula is C26H40O5and its chemical structure is:
XALATAN Sterile Ophthalmic Solution (latanoprost ophthalmic solution) is supplied as a sterile, isotonic, buffered aqueous solution of latanoprost with a pH of approximately 6.7 and an osmolality of approximately 267 mOsmol/kg. Each mL of XALATAN contains 50 micrograms of latanoprost. Benzalkonium chloride, 0.02% is added as a preservative. The inactive ingredients are: sodium chloride, sodium dihydrogen phosphate monohydrate, disodium hydrogen phosphate anhydrous, and water for injection. One drop contains approximately 1.5 μg of latanoprostLatanoprost is a colorless to slightly yellow oil that is very soluble in acetonitrile and freely soluble in acetone, ethanol, ethyl acetate, isopropanol, methanol, and octanol. It is practically insoluble in water.
...............................
When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:
EXAMPLE 16
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((R)-3-hydroxy-5-phenylpentyl)cyclopentyl)hept-5-enoic acid (Latanoprost acid)

- 4-Carboxybutyltriphenylphosphonium bromide 15 (32.7 g, 0.074 mol) was suspended in tetrahydrofuran (75.0 mL) at 0°C under nitrogen atmosphere. A 1M solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((R)-3-hydroxy-5-phenylpentyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (5.0 g, 0.016 mol) in tetrahydrofuran (23.0 mL) was added dropwise at a temperature lower than -10°C. After stirring overnight at -15°C no more starting was visible on TLC and water (100 mL) was added. The mixture was extracted with diisopropyl ether (70 mL) and after separation the aqueous phase was treated with 0.6 N HCl to pH 6.0. Three extractions with ethyl acetate (3x 125 mL) were then performed, each time adjusting the pH of the aqueous phase to 6.0. The combined organic layers were concentrated under vacuum at 35°C. An oil (13.87 g) was obtained which was used in the subsequent step without further purification.
- 1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.71 (m, 1H, Ph), 7.49 (m, 1H, Ph), 7.30-7.17 (m, 3H, Ph), 5.52-5.35 (m, 2H, -CH=CH-), 4.34 (bs, 4H, OH), 4.17 (m, 1H, -CH-OH(C-9)), 3.96 (m, 1H, -CH-OH (C-11)), 2.78 (m, 1H, -CH-OH (C-15)), 2.78 (m, 1H, -CH2Ph), 2.66 (m, 1H, -CH2Ph), 2.36-1.27 (m, 18H).
- 13C-NMR {400 MHz, CDCl3, δ (ppm)}: 176.5 (C), 142.2 (C), 130.8 (CH), 130.7 (CH), 129.4 (CH), 128.8 (CH), 128.7 (CH), 128.4 (CH), 125.7 (CH), 78.3 (CH), 74.2 (CH), 71.4 (CH), 52.2 (CH), 51.6 (CH), 42.4 (CH2), 38.8 (CH2), 35.2 (CH2), 33.4 (CH2), 32.0 (CH2), 29.1 (CH2), 26.6 (CH2), 26.4 (CH2), 24.7 (CH2).
- HPLC-MS (ESI): [M+Na]+ = 413, [M+H]+ = 391.

- Latanoprost acid (6.78 g, corresponding to 0.008 mol) was dissolved in N,N-dimethylformamide (108 mL) and cesium carbonate (8.48 g, 0.026 mol) was added at room temperature. 2-Iodopropane (3.46 mL, 0.035 mol) was added and the suspension was stirred at 40°C for 3 hours, checking the conversion on TLC. The mixture was then allowed to reach 25°C and a mixture of ice (184 g), water (40 mL), sodium thiosulfate (1M, 18 mL), was added stirring at -5/0°C for 15 minutes. The mixture was extracted with tert-butylmethylether (285 mL) and the phases were separated. The aqueous phase was extracted twice with tert-butylmethyl ether (2x 200 mL) and the combined organic layers were washed with brine (176 mL, 130 mL). The organic phase was concentrated under reduced pressure at 25°C and the crude product was obtained as a yellow oil (6.60 g). Purification by column chromatography on silica gel was performed eluting with dichloromethane:methanol increasing the percentage of methanol from 0 to 5%. A second purification on silica gel afforded Latanoprost (2.36 g, 0.005 mol, 68% over two steps).
- 1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.32-7.19 (m, 5H, Ph), 5.45-5.51 (m, 2H, H-5 e H-6 vinyl), 5.0 (hept, J=6.3 Hz, 1H, CH3CHCH3), 4,18 (bs, 1H, CHOH), 3.95 (bs, 1H, CHOH), 3.67 (bs, 1H, CHOH), 2.76 (m, 2H, CH 2Ph), 1.23 (d, J=6.3Hz, 6H, C(CH3)2), 2.55-1.3, 21H).
- HPLC-MS (ESI): [M+Na]+ = 455, [M+H]+ = 432.
.....................................
Example 3
Synthesis of Latanoprost
ether MTBE
8c-iso
Latanoprost
Scheme 5. Synthesis of Latanoprost
Synthesis of Latanoprost from 12c:
As shown in Scheme 5 in Example 3, a 250 ml.3-necked round-bottom flask equipped with a magnetic bar, a temperature probe, rubber septa, and a nitrogen gas inlet was charged at room temperature with 7.3 g (19.6 mmol) of deprotected lactone 12c in 70 mL of 2-propanol and 1.6 g (39.2 mmol) of sodium hydride, 60%, in mineral oil. The reaction mixture was heated at 35 0C for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of water and the pH was adjusted to 6 with 1 N HCI. The layers were separated and the aqueous layer was back extracted with 40 mL of 2-propanol four times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was dissolved in 60 mL of THF, 5.0 mL (33.3 mmol) of DBU and 3.3 mL (33.3 mmol) of iodopropane. The reaction mixture was stirred at room temperature for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 60 mL of ethyl acetate and 60 mL of water. The layers were separated and the aqueous layer was back extracted with 40 mL of ethyl acetate for two times. The combined organic layers were washed with 50 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The material was purified by using reverse phase biotage, 70 : 30 ACN :
H2O to obtain 4.1 g (49% yield) of Latanoprost, confirmed by 1H NMR.
......................
Wittig condensation of lactol (XIII) with (carboxybutyl) triphenylphosphonium bromide (XV) in the presence of potassium tert-butoxide produced the Z-olefin (XVI). Conversion of carboxylic acid (XVI) to the title isopropyl ester was then accomplished by alkylation with 2-iodopropane in the presence of DBU.
....................................
...........................
References
- Ishikawa H, Yoshitomi T, Mashimo K, Nakanishi M, Shimizu K (February 2002). "Pharmacological effects of latanoprost, prostaglandin E2, and F2alpha on isolated rabbit ciliary artery". Graefes Arch. Clin. Exp. Ophthalmol. 240 (2): 120–5. doi:10.1007/s00417-001-0412-4. PMID 11931077.
- Patel SS, Spencer CM (1996). "Latanoprost. A review of its pharmacological properties, clinical efficacy and tolerability in the management of primary open-angle glaucoma and ocular hypertension". Drugs Aging 9 (5): 363–378. doi:10.2165/00002512-199609050-00007. PMID 8922563.
- Huttunen et al. (2011) Prodrugs—from Serendipity to Rational Design. Pharmacol Rev 63:750–771
- "Patent US5296504 - Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension - Google Patents".
- "WHO Model List of EssentialMedicines". World Health Organization. October 2013. Retrieved 22 April 2014.
- Perry CM, McGavin JK, Culy CR, Ibbotson T (2003). "Latanoprost. An Update of its Use in Glaucoma and Ocular Hypertension". Drugs Aging 20 (8): 1170–2229.PMID 12795627.
- Zhang WY, Wan Po AL, Dua HS, Azuara-Blanco A (2001). "Meta-analysis of randomised controlled trials comparing latanoprost with timolol in the treatment of patients with open angle glaucoma or ocular hypertension". British Journal of Ophthalmology 85: 983–990. doi:10.1136/bjo.85.8.983. PMID 11466259.
- Aung T; Wong HT; Yip CC; et al. (2000). "Comparison of the intraocular pressure-lowering effect of latanoprost and timolol in patients with chronic angle closure glaucoma: a preliminary study.". Ophthalmology 107 (6): 1178–83. doi:10.1016/s0161-6420(00)00073-7. PMID 10857840.
- Amano S, Nakai Y, Ko A, Inoue K, Wakakura M (2008). "A case of keratoconus progression associated with the use of topical latanoprost". Japanese Journal of Ophthalmology 52 (4): 334–6. doi:10.1007/s10384-008-0554-6. PMID 18773275.
- De Santis, M., Lucchese, A., Carducci, B., Cavaliere, A., De Santis, L., & Merola, A. et al. (2004). Latanoprost exposure in pregnancy. American Journal Of Ophthalmology, 138(2), 305.pmid=15289149.1
- Morgan, P., Proniuk, S., Blanchard, J., & Noecker, R. (2001). Effect of temperature and light on the stability of latanoprost and its clinical relevance. Journal Of Glaucoma, 10(5), 401--405.
External links
- LATANOPROST solution [Greenstone LLC] (Nov 2011), Daily Med, U.S. National Library of Medicine, National Institutes of Health
''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
6 TRAVOPROST
Travoprost
cas 157283-68-6
[1R-[lα(Z),2β(lE,3R*),3α,5α]]-7-[3,5-Dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1 -butenyl]cyclopentyl]-5-heptenoic acid, 1 -methylethylester
(+)-16-m-trifluoromethylphenoxy tetranor Prostaglandin F2α isopropyl ester; (+)-Fluprostenol ispopropyl ester
(+)-(5Z,9α,1α,13E,15R)-trihydroxy-16-(3-(trifluoromethyl)phenoxy)-17,18,19,20-tetranor-prosta-5,13-dien-1-oic acid, isopropyl ester
(+) – Fluprostenol isopropyl ester,
Travatan, Travatan Z, AL-6221, Travatanz, Travatan Alcon, Travatan (TN), Travatan, Travoprost, Travoprost [USAN]
Molecular Formula: C26H35F3O6
Molecular Weight: 500.54771
Alcon (Originator)
Antiglaucoma Agents, OCULAR MEDICATIONS, Ophthalmic Drugs, Prostaglandins, Prostanoid FP Agonists
Ophthalmic solution used for the reduction of elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension who are intolerant of other intraocular pressure lowering medications or insufficiently responsive (failed to achieve target IOP determined after multiple measurements over time) to another intraocular pressure lowering medication.
Travoprost free acid is a selective FP prostanoid receptor agonist and is believed to reduce intraocular pressure by increasing the drainage of aqueous humor, which is done primarily through increased uveoscleral outflow and to a lesser extent, trabecular outflow facility.
Travoprost, an isopropyl ester prodrug, is a synthetic prostaglandin F2 alpha analogue that is rapidly hydrolyzed by esterases in the cornea to its biologically active free acid. The travoporst free acid is potent and highly selective for the FP prostanoid receptor.
Travoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a synthetic prostaglandin analog (or more specifically, an analogof prostaglandin F2α)[1][2] that works by increasing the outflow of aqueous fluidfrom the eyes.[3] It is also known by the brand names of Travatan andTravatan Z, manufactured by Alcon, and Travo-Z, manufactured by Micro Labs.
Travoprost is a synthetic prostaglandin F analogue. Its chemical name is [1R-[lα(Z),2β(lE,3R*),3α,5α]]-7-[3,5-Dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl)phenoxy]-1 -butenyl]cyclopentyl]-5-heptenoic acid, 1 -methylethylester. It has a molecular formula of C26H35F3O6 and a molecular weight of 500.55. The chemical structure of travoprost is:
 | |||
| COUNTRY | PATENT NUMBER | APPROVED | EXPIRES (ESTIMATED) |
|---|---|---|---|
| Canada | 2181172 | 2003-04-29 | 2015-12-19 |
| Canada | 2129287 | 2002-05-14 | 2014-08-02 |
| United States | 5631287 | 1994-12-22 | 2014-12-22 |
| United States | 6503497 | 1995-05-06 | 2012-05-06 |
7-25-2012
|
TOPICAL APPLICATION OF TRAVOPROST FOR COMBATING HAIR LOSS
| |
12-28-2011
|
Stable prostaglandin-containing compositions
| |
7-22-2011
|
IMPROVED PROCESS FOR THE PRODUCTION OF BIMATOPROST
| |
6-3-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
| |
9-17-2010
|
Compositions and Methods for Reducing Body Fat
| |
5-28-2010
|
COMPLEXES OF PROSTAGLANDIN DERIVATIVES AND MONOSUBSTITUTED, CHARGED BETA-CYCLODEXTRINS
| |
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
| |
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
| |
2-24-2010
|
Compositions and methods for reducing body fat
| |
1-15-2010
|
Process for the Production of Prostaglandins and Prostaglandin Analogs
|
4-3-2009
|
METHOD FOR SCREENING OF PROSTAGLANDIN COMPOUNDS COMPRISING AN OPTIMAL FORMULATION FOR THE ENHANCEMENT OF HAIR GROWTH AND THE STIMULATION OF FOLLICULAR ANAGEN AND FORMULATIONS RESULTING THEREFROM
| |
3-9-2005
|
9,11-cycloendoperoxide pro-drugs of prostaglandin analogues for treatment of ocular hypertension and glaucoma
| |
10-8-2004
|
Use of cloprostenol and fluprostenol analogues to treat glaucoma and ocular hypertension
| |
4-21-2004
|
Use of cloprostenol and fluprostenol analogues to treat glaucoma and ocular hypertension
|
Side effects
Possible side effects of this medication are:
- May cause blurred vision
- May cause eyelid redness
- May permanently darken eyelashes
- May cause eye discomfort
- May eventually cause permanent darkening of the iris to brown (heterochromia)
- May cause a temporary burning sensation during use
- May cause thickening of the eyelashes
- May cause inflammation of the prostate gland, restricting urine flow (BPH)
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| propan-2-yl 7-[3,5-dihydroxy-2-[3-hydroxy-4-[3-(trifluoromethyl) phenoxy]-but-1-enyl]-cyclopentyl]hept-5-enoate | |
| CLINICAL DATA | |
| TRADE NAMES | Travatan |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a602027 |
| PREGNANCY CAT. | C US |
| LEGAL STATUS | Rx only (US) |
| ROUTES | Topical (eye drops) |
| IDENTIFIERS | |
| CAS NUMBER | 157283-68-6 |
| ATC CODE | S01EE04 |
| PUBCHEM | CID 5282226 |
| DRUGBANK | DB00287 |
| CHEMSPIDER | 4445407 |
| UNII | WJ68R08KX9 |
| CHEMICAL DATA | |
| FORMULA | C26H35F3O6 |
| MOL. MASS | 500.548 g/mol |
The condensation of 2- [3- (trifluoromethyl) phenoxy] acetyl chloride (I) with methylphosphonic acid dimethyl ester (II) by means of BuLi in THF gives 2-oxo-3- [3- (trifluoromethyl) phenoxy] propylphosphonic acid dimethyl ester (III), which is condensed with the known bicyclic aldehyde (IV) by means of BuLi in dimethoxyethane, yielding the unsaturated ketone (V). The reduction of (V) with zinc borohydride in dimethoxyethane affords the unsaturated alcohol (VI), which is treated with K2CO3 to give a diastereomeric mixture of unsaturated diols, resolved by chromatography to yield the chiral unsaturated diol (VII). The protection of (VII) with dihydropyran and TsOH in dichloromethane provides the bis (tetrahydropyranyl) ether (VIII), which by reduction of the lactone ring with diisobutylaluminum hydride in THF gives the lactol (IX). The condensation of (IX) with the phosphonium bromide (X) by means of NaH in DMSO yields the prostenoic acid (XI), which is esterified with isopropyl iodide and 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) in acetone to afford the corresponding isopropyl ester (XII). Finally, this compound is deprotected with acetic acid in hot THF / water.
…………………………………………..
Org Process Res Dev2002,6, (2): 138
A commercial synthesis of the antiglaucoma agent, travoprost 2, is described. A total of 22 synthetic steps are required to provide the single enantiomer prostanoid, with the longest linear sequence being 16 steps from 3-hydroxybenzotrifluoride. The route is based upon a cuprate-mediated coupling of the single enantiomer vinyl iodide 13 and the tricyclic ketone 5, of high stereochemical purity, to yield the single isomer bicyclic ketone 15. A Baeyer−Villiger oxidation provides the lactone 16 as a crystalline solid, thus limiting the need for chromatographic purification. DIBAL-H reduction, Wittig reaction, esterification, and silyl group deprotection complete the synthesis of travoprost.
…………………………………………
- In the case of Travoprost, compound 9 with A=3-(trifluoromethyl)phenoxy (in the following scheme, compound 9b) is converted into 10b, which in turn is converted into Travoprost by esterification of the carboxylic acid by reaction with 2-iodopropane, according to scheme 10:
……………………………
Example 2
Synthesis of Travoprost MTBE MTBE
C
7b
8b
9b-iso
Travoprost Scheme 4. Synthesis of Travoprost
References
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN – travoprost solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- Alcon Laboratories, Inc. (September 2011). “TRAVATAN Z (travoprost) solution”. DailyMed. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- AHFS Consumer Medication Information (2011-01-01). “Travoprost Ophthalmic”. MedlinePlus. Bethesda, MD: U.S. National Library of Medicine. Retrieved 2011-09-30.
- Ota T, Aihara M, Narumiya S, Araie M: The effects of prostaglandin analogues on IOP in prostanoid FP-receptor-deficient mice. Invest Ophthalmol Vis Sci. 2005 Nov;46(11):4159-63. PubMed: 16249494
- Thieme H, Schimmat C, Munzer G, Boxberger M, Fromm M, Pfeiffer N, Rosenthal R: Endothelin antagonism: effects of FP receptor agonists prostaglandin F2alpha and fluprostenol on trabecular meshwork contractility. Invest Ophthalmol Vis Sci. 2006 Mar;47(3):938-45. PubMed: 16505027
- Lim KS, Nau CB, O’Byrne MM, Hodge DO, Toris CB, McLaren JW, Johnson DH: Mechanism of action of bimatoprost, latanoprost, and travoprost in healthy subjects. A crossover study. Ophthalmology. 2008 May;115(5):790-795.e4. PubMed: 18452763
- Neacsu AM: [Receptors involved in the mechanism of action of topical prostaglandines] Oftalmologia. 2009;53(2):3-7. PubMed: 19697832
- Costagliola C, dell’Omo R, Romano MR, Rinaldi M, Zeppa L, Parmeggiani F: Pharmacotherapy of intraocular pressure – part II. Carbonic anhydrase inhibitors, prostaglandin analogues and prostamides. Expert Opin Pharmacother. 2009 Dec;10(17):2859-70.PubMed: 19929706
- Ferrari G, Scagliotti GV: Serum and urinary vascular endothelial growth factor levels in non-small cell lung cancer patients. Eur J Cancer. 1996 Dec;32A(13):2368-9. PubMed: 9038626
- Toris CB, Gabelt BT, Kaufman PL: Update on the mechanism of action of topical prostaglandins for intraocular pressure reduction. Surv Ophthalmol. 2008 Nov;53 Suppl1:S107-20. PubMed: 19038618
- Arranz-Marquez E, Teus MA: Prostanoids for the management of glaucoma. Expert Opin Drug Saf. 2008 Nov;7(6):801-8. PubMed: 18983226
- Chen X, Ji ZL, Chen YZ: TTD: Therapeutic Target Database. Nucleic Acids Res. 2002 Jan 1;30(1):412-5. PubMed: 11752352
..............................
''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
 |
|---|
isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate,
propan-2-yl (E)-7-[2-[(E)-3,3-difluoro-4-phenoxybut-1-enyl]-3,5-dihydroxycyclopentyl]hept-5-enoate
Molecular Formula: C25H34F2O5
Molecular Weight: 452.531266
Drug: Zioptan
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
Generic molecule: tafluprost
Company: Merck
Approval date: Feb. 10, 2012
The scoop: Merck says this is the first (get ready for a mouthful) preservative-free prostaglandin analog ophthalmic solution and is for treating elevated eye pressure in some patients with the most common form of glaucoma. Merck sells the ointment in the U.S. and most of Europe, while it licensed it to Japanese drugmaker Santen in Japan, Germany and northern Europe.
Tafluprost (trade names Taflotan, marketed by Santen Pharmaceutical Co. and Zioptan, by Merck (U.S.)) is a prostaglandin analogue used topically (aseye drops) to control the progression of glaucoma and in the management ofocular hypertension. It reduces aqueous fluid from the eyes.[1][2]
Taflotan contains 15 µg/ml Tafluprost. Taflotan sine is a preservative-free, single-dose formulation containing 0.3 ml per dose.[3]
| |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| isopropyl (5Z)-7-{(1R,2R,3R,5S)-2-[(1E)-3,3-difluoro-4-phenoxybut-1-en-1-yl]-3,5-dihydroxycyclopentyl}hept-5-enoate | |
| CLINICAL DATA | |
| TRADE NAMES | Saflutan, Taflotan, Tapros, Zioptan |
| AHFS/DRUGS.COM | International Drug Names |
| PREGNANCY CAT. | C (US) |
| LEGAL STATUS | ℞-only (US) |
| ROUTES | Topical (eye drops) |
| IDENTIFIERS | |
| CAS NUMBER | 209860-87-7 |
| ATC CODE | S01EE05 |
| PUBCHEM | CID 6433101 |
| CHEMSPIDER | 8044182 |
| UNII | 1O6WQ6T7G3 |
| CHEBI | CHEBI:66899 |
| CHEMBL | CHEMBL1963683 |
| CHEMICAL DATA | |
| FORMULA | C25H34F2O5 |
| MOL. MASS | 452.531266 g/mol |
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Santen Home Page
- Gelbe Liste (in German)
Its synthesis from compound Corey aldehyde and HWE (Hormer-Wadsworth-Emmons) reagent 1 generates trans olefin 2 , 2 and fluorination reagent 3 hydrolysis reaction 4 , lactone 4 with DIBAL reduction to give the ring hemiacetal 5 , 5 and Wittig reagent 6 cis olefin reaction after esterification with isopropyl iodide Tafluprost.
……………………….
European Patent No. 8509621 discloses a process for the preparation of tafluprost. In the first step, (3afl,4fl,5fl,6aS)-4-formyl-2-oxohexahydro-2 — cyclopenta[b]furan-5-ylbenzoate (CTAF 1 (i)) is condensed with dimethyl (2-oxo-3- phenoxypropyl)-phosphonate in the presence of lithium chloride and triethylamine, to provide (3aft,4F?,5F?,6aS)-2-oxo-4-((£)-3-oxo-4-phenoxybut-1 -en-1 -yl)hexahydro-2H- cyclopenta[b]-furan-5-ylbenzoate (CTAF1 ). In the second step, CTAF 1 is reacted with morpholinosulfurtrifluoride to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-2-oxohexahydro-2 –cyclopenta-[b]furan-5-yl benzoate (CTAF2). CTAF 2 is debenzoylated by potassium carbonate in methanol, to provide (3aH,4H,5H,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-5-hydroxyhexahydro-2H- cyclopenta[b]furan-2-one(CTAF 3), which is further reduced by diisobutyl aluminum hydride (DIBALH) to provide (3af?,4f?,5f?,6aS)-4-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 – yl) hexahydro-2H-cyclopenta[b]furan-2,5-diol (CTAF 4). CTAF 4 is then treated with (4- carboxybutyl)triphenylphosphonium bromide, in the presence of potassium bis(trimethylsilyl)amide in THF, to provide (Z)-7-((1 f?,2f?,3f?,5S)-2-((£)-3,3-difluoro-4- phenoxybut-1 -en-1 -yl)-3,5-dihydroxycyclopentyl)hept-5-enoic acid (“tafluprost free acid,” CTAF5), which is reacted with isopropyl iodide in the presence of DBU to provide (Z)- isopropyl 7-((1 F?,2F?,3F?,5S)-2-((£)-3,3-difluoro-4-phenoxybut-1 -en-1 -yl)-3,5-dihydroxy- cyclopentyl)hept-5-enoate (“tafluprost,” CTAF 6). The reaction sequence is summarized in Scheme 1 .
CTAF 1(i)
CTAF 1 CTAF 2
U.S. Patent Application Publication No. 2010/0105775A1 discloses amino acid salts of prostaglandins. The application also discloses a process for the preparation of prostaglandins, comprising forming an amino acid salt of a prostaglandin and converting the amino acid salt to the prostaglandin.
EXAMPLE 1 : Preparation of CTAF 1
CTAF1(i)
CTAF1
To a stirred suspension of sodium hydride (60% dispersion in mineral oil, 0.217 g, 5.429 mmol) in THF (5 ml_) was added a solution of dimethyl (2-oxo-3- phenoxypropyl)phosphonate(1 .21 g, 4.705 mmol) in THF (2 ml_), over 15 minutes at 0- 5°C under a nitrogen atmosphere. The mixture was warmed to 25-35 , 0.5 M zinc chloride solution in THF (9.4 ml_, 4.705 mmol) was added over 10 minutes, and then the mixture was stirred for 15 minutes at 25-35<€. CTAF1 (i) (3af?,4F?,5F?,6aS)-4-formyl-2- oxohexahydro-2 –cyclopenta[b]furan-5-yl benzoate (1 g) in dichloromethane (10 ml_) was added over 5 minutes at 25-35 °C. The temperature was raised to 35-40 °C and the mixture was stirred for 2hours under a nitrogen atmosphere. The mixture was cooled to 15°C and the reaction was quenched by adding acetic acid (0.2 mL), followed by adding saturated ammonium chloride solution (10 mL), and further stirring for 15 minutes. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (5 mL). The combined organic layers were evaporated under reduced pressure below 50°C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (0.9 g, 61 %yield).
EXAMPLE 2: Preparation of CTAF 2
CTAF1 CTAF2
To a stirred solution of CTAF1 (5 g, 0.0123 mol) in dichloromethane (100 mL) was added diethylaminosulfurtrifluoride (13 mL, 0.09841 mol) at 0-5 °C under a nitrogen atmosphere. The temperature was raised to 25-35 °C and maintained for 24 hours under a nitrogen atmosphere at the same temperature. The mass was slowly added into a saturated sodium bicarbonate solution (75 mL) at 0-5 °C. Temperature was raised to 25- 35 °C, the layers were separated, and the aqueous layer was extracted with dichloromethane (2×25 mL). The combined organic layer was washed with water (25mL) and dried over sodium sulfate (5 g). The organic layer was evaporated to dryness under reduced pressure below 40 °C. The crude product was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane, to afford the title compound (4.2 g, 79% yield). EXAMPLE 3: Preparation of CTAF 4
CTAF 2 CTAF 4
CTAF 2 (2.30 g, 5.37 mmol) was dissolved in toluene (25 mL) and the solution was cooled to -65 °C under nitrogen. Diisobutyl aluminum hydride (1 .5 M in toluene, 1 1 .8 mL, 17.7mmol) was added over 15 minutes at -61 to -65 . The mixture was stirred for 3hours and then the reaction was quenched by adding methanol (1 .5 mL). Sulfuric acid (1 M, 25 mL) was added and the temperature rose to -20°C during the addition. Methyl t-butyl ether (MTBE) (10 mL) was added and the mixture was allowed to warm to room temperature. The organic phase was separated and the aqueous phase was extracted with MTBE (2x 10 mL). The combined organic phase was washed with water (10 mL), saturated aqueous sodium bicarbonate (10 mL), and then brine (10 mL). The washes were back-extracted with MTBE (10 mL). The combined organic phases were dried with magnesium sulfate, filtered, and evaporated to give a colourless oil (2.20 g). The crude product was chromatographed on silica (60 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), and then with ethyl acetate, to give CTAF 4 as a colourless oil (1 .71 g, 97% yield).
EXAMPLE 4: Preparation of CTAF 2
CTAF1 CTAF2
To a stirred solution of CTAF1 (20 g, 0.0492 mol) in dichloromethane(400 mL) was added diethylaminosulfurtrifluoride (52 mL, 0.393 mol) at 0-10°C under a nitrogen atmosphere. The temperature was raised to 25-35 and maintained for 96hours under a nitrogen atmosphere at that temperature. The mass was slowly added to a saturated NaHCOs solution (600 mL) at 0-10°C. The mixture was heated to 25-35 <€ and filtered through aCelite bed. The layers were separated and the aqueous layer was extracted with DCM (2×100 mL). The combined organic layer was washed with 10% NaCI solution (100 mL) and evaporated to dryness under reduced pressure below 40°C. The residue was purified by column chromatography on silica gel (100-200 mesh) with 30% ethyl acetate in hexane.
Column purified material was dissolved in MTBE (80 mL) at 40°C and stirred for 30 minutes at that temperature. Diisopropyl ether (160 mL) was added at 35-40 and stirring continued for 30 minutes at 35-40 . Cooled the mass to 5-15°C and stirred for 30 minutes at that temperature. The solid was filtered, washed with a mixture of MTBE and diisopropyl ether (DIPE) (1 :2 by volume, 60 mL), and dried at 40°C under vacuum, to afford pure CTAF2 (12.0 g, 57% yield).
EXAMPLE 5: Preparation of CTAF 5
(4-Carboxybutyl)triphenylphosphonium bromide (10.32 g, 23.3 mmol, 4 eq) was suspended in THF (20 mL) under a nitrogen atmosphere and cooled to 5°C. NaHMDS solution (1 M in THF, 46.6 mL, 46.6 mmol, 8 eq) was added over 10 minutes. The red/orange mixture was stirred for 30 minutes. A solution of CTAF 4 (1 .90 g, 5.82 mmol) in THF (10 mL) was added over 30 minutes at 0-3 . The mixture was stirred for 1 .5hours and then the reaction was quenched by adding water (30 mL) and the masswas warmed to room temperature. The aqueous phase was separated and the organic phase was washed with water (20 mL). The combined aqueous phases were washed with MTBE (30 mL). The organic phases up to this point were discarded. The aqueous phase was acidified with 2M hydrochloric acid (14 mL, to pH 3-4) and extracted with ethyl acetate (2×30 mL). The combined ethyl acetate layers were washed with brine (20 mL), dried with magnesium sulfate, filtered, and evaporated under reduced pressure to give CTAF 5 asa yellow oil (8.60 g).
A 2.96 g sample was removed and the remainder (5.64 g) was chromatographed on silica (30 g) eluting with ethyl acetate to give purified CTAF 5 (1 .41 g) asa yellow oil. NMR analysis showed approximately 90% purity, remainder triphenyl phosphine oxide.
EXAMPLE 6: Preparation of CTAF 5 DCHA salt
CTAF 5 CTAF 5 DCHA sa t
CTAF5 (1 1 .72 g, 90% purity, 25.7 mmol, containing 1 .4% trans isomer) was dissolved in acetone (60 mL). Dicyclohexylamine (4.66 g, 25.7 mmol) was added and the mixture was stirred at room temperature overnight. The solid was filtered and washed with acetone (6 mL), then dried to give the DCHA salt (12.93 g, 85% yield, 0.29% trans-isomer).
A sample (7.03 g) was further purified by recrystallisation. It was dissolved in hot acetone (30 mL) and cooled to room temperature with stirring. The mixture was stirred for 3 hours, filtered and the solid was washed with acetone (3 mL) and dried to give a white solid (6.41 g, 91 % recovery, 0.1 1 % trans-isomer).
A PXRD pattern of the product is shown as Fig. 1 , obtained using copper Ka radiation. In the drawing, the y-axis is intensity units and the x-axis is the 2-theta angle, in degrees. EXAMPLE 7: Pre aration of CTAF 6
CTAF 5 DCH A sa l ^ I AI- O
CTAF 5 DCHA salt (5.80 g, 9.80 mmol) was suspended in ethyl acetate (20 mL). Sulfuric acid (1 M, 20 mL) was added and the mixture was stirred until a clear solution was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2×20 mL). The combined organic layers were washed with water (15 mL) and brine (15 mL), dried with magnesium sulfate, filtered, and evaporated. The residue was dissolved in acetone (40 mL) and charged into a jacketed vessel at 30°C. 1 ,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (8.95 g, 58.8 mmol) was added, then 2- iodopropane (10.0 g, 58.8 mmol) was added, and the mixture was stirred for 20hours. The mixture was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (30 mL) and aqueous potassium dihydrogen orthophosphate (8 g) in water (50 mL). The organic phase was separated and the aqueous was extracted with ethyl acetate (30 mL). The combined organic phases were washed with brine (20 mL), dried with magnesium sulfate, filtered and evaporated to give a yellow oil (4.83 g). The crude product was chromatographed on silica (130 g), eluting with a mixture of ethyl acetate and heptane (2:1 by volume), to give CTAF 6 (3.98 g, 90% yield) as a colorless oil.
............................................
''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
BERAPROST
2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta(b)benzofuran-5-butanoic acid
(±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid
rac-4-{(1R,2R,3aS,8bS)-2-hydroxy-1-[(1E,3S,4RS)-3-hydroxy-4-methyloct-1-en-6-ynyl]-2,3,3a,8b-tetrahydro-1H-cyclopenta[b][1]benzofuran-5-yl}butanoic acid
- Beraprost
- Beraprostum
- Beraprostum [INN-Latin]
- MDL 201229
- MDL-201229
- ML 1229
- ML-1229
- UNII-35E3NJJ4O6
Beraprostum, Beraprostum [INN-Latin], ML 1229, MDL 201229, 88430-50-6
Molecular Formula: C24H30O5
Molecular Weight: 398.492
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=Beraprost
Beraprost is a synthetic analogue of prostacyclin, under clinical trials for the treatment of pulmonary hypertension. It is also being studied for use in avoiding reperfusion injury.
As an analogue of prostacyclin PGI2, beraprost effects vasodilation, which in turn lowers the blood pressure. Beraprost also inhibits plateletaggregation, though the role this phenomenon may play in relation to pulmonary hypertension has yet to be determined.
Beraprost …sodium salt
ML 1129; Procyclin; TRK 100 (CAS 88475-69-8)
Beraprost is an analog of prostacyclin in which the unstable enol-ether has been replaced by a benzofuran ether function. This modification increases the plasma half-life from 30 seconds to several hours, and permits the compound to be taken orally. Doses of 20-100 µg in humans, given 1 to 3 times per day, have been demonstrated to improve clinical end points in diseases responsive to prostacyclin. Oral beraprost therapy improved the survival and pulmonary hemodynamics of patients with primary pulmonary hypertension.1 Beraprost inhibits platelet aggregation in healthy subjects and in diabetic patients at similar doses.2,3
| SYNONYMS |
|
|---|---|
| FORMAL NAME | 2,3,3a,8b-tetrahydro-2-hydroxy-1-(3-hydroxy-4-methyl-1-octen-6-ynyl)-1H-cyclopenta[b]benzofuran-5-butanoic acid, monosodium salt |
| CAS NUMBER | 88475-69-8 |
| MOLECULAR FORMULA | C24H29O5 · Na |
| FORMULA WEIGHT | 420.5 |
- Beraprost sodium is a prostacyclin analog and an NOS3 expression enhancer that was first launched in 1992 in Japan pursuant to a collaboration between Astellas Pharma and Toray for the oral treatment of peripheral vascular disease (PVD), including Raynaud’s syndrome and Buerger’s disease. In 2000, the drug was commercialized for the treatment of pulmonary hypertension. Development for the oral treatment of intermittent claudication associated with arteriosclerosis obliterans (ASO) was discontinued at Kaken and United Therapeutics after the product failed to demonstrate statistically significant results in a phase III efficacy trial.
- In terms of clinical development, beraprost sodium is currently in phase II clinical trials at Kaken for the treatment of lumbar spinal canal stenosis and at Astellas Pharma for the oral treatment of primary chronic renal failure. The company is also conducting phase III trials for the treatment of nephrosclerosis. The drug has also been studied through phase II clinical trials at Kaken for the oral treatment of diabetic neuropathy, but recent progress reports for this indication have not been made available.
- Beraprost is an oral form of prostacyclin, a member of the family of lipid molecules known as eicosanoids. Prostacyclin is produced in the endothelial cells from prostaglandin H2 by the action of the enzyme prostacyclin synthase. It has been shown to keep blood vessels dilated and free of platelet aggregation.
- Beraprost sodium was originally developed at Toray in Japan, and rights to the drug were subsequently acquired by Astellas Pharma. A 1972 alliance between Toray and Kaken Pharmaceutical to develop and commercialize prostaglandin led to a later collaboration agreement for the development of beraprost. In 1990, Toray granted the right to market the drug to Sanofi (formerly known as sanofi-aventis), a licensing agreement that was later expanded to include Canada, the U.S., South America, Africa, Southeast Asia, South Asia, Korea and China. In September 1996, Bristol-Myers Squibb entered into separate agreements with Sanofi and Toray to acquire all development and marketing rights to beraprost in the U.S. and Canada. In January 1999, United Therapeutics and Toray agreed to cooperatively test the drug in North America, and in July 2000, a new agreement was signed pursuant to which United Therapeutics gained exclusive North American rights to develop and commercialize sustained-release formulations of beraprost for all vascular and cardiovascular diseases. In 1999, orphan drug designation was received in the U.S. for the treatment of pulmonary arterial hypertension associated with any New York Heart Association classification (Class I, II, III, or IV). In 2011, orphan drug designation was assigned in the U.S. for the treatment of pulmonary arterial hypertension.
- The compound name of beraprost which is used as an antimetastasis agent of malignant tumors according to the present invention is (±)-(IR*,2R*,3aS*,8bS*)-2,3,3a,8b-tetrahydro-2-hydroxy-1-[(E)-(3S*)-3-hydroxy-4-methyl-1-octene-6-inyl]-1H-cyclopenta[b]benzofuran-5-butyric acid. This compound has the following structure.
 Beraprost is described in Japanese Laid-open Patent Application (Kokai) Nos. 58-32277, 57-144276 and 58-124778 and the like as a PGI₂ derivative having a structure in which the exoenol moiety characteristic to beraprost is converted to inter-m-phenylene structure. However, it is not known that beraprost has an activity to inhibit metastasis of malignant tumors.
Beraprost is described in Japanese Laid-open Patent Application (Kokai) Nos. 58-32277, 57-144276 and 58-124778 and the like as a PGI₂ derivative having a structure in which the exoenol moiety characteristic to beraprost is converted to inter-m-phenylene structure. However, it is not known that beraprost has an activity to inhibit metastasis of malignant tumors. - The beraprost which is an effective ingredient of the agent of the present invention includes not only racemic body, but also d-body and l-body. Beraprost can be produced by, for example, the method described in the above-mentioned Japanese Laid-open Patent Application (Kokai) No. 58-124778. The salts of beraprost include any pharmaceutically acceptable salts including alkaline metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as magnesium salt and calcium salt; ammonium salt; primary, secondary and tertiary amine salts; and basic amino acid salts.
…………………..
EXAMPLE 6 Beraprost of the Formula (I)
0.246 g (0.6 mmol) of compound of the general formula (II) obtained in Example 5 is dissolved in 1 ml of methanol and 1 ml of 1 M aqueous sodium hydroxide solution is added dropwise slowly thereto. After stirring for an hour the methanol is distilled off from the reaction mixture in vacuum. The aqueous residue is diluted with 10 ml of water extracted with methyl-tert.butyl-ether and the combined organic phase is washed with saturated NaCl solution, dried on Na2SO4 and evaporated. The residue of evaporation is crystallized from ethylacetate-hexane mixture and the pure above mentioned title compound is obtained as colourless crystals.
Yield: 0.21 g (87%)
TLC-Rf (toluene-dioxan-acetic acid 20:10:1)=0.41
Melting point: 98–112° C.
1H NMR (400 MHz, CDCl3), δH (ppm): 1.00d, 1.03d [3H; J=6.8 Hz; 21-H3]; 1.79m [1H; 16-H]; 1.80t, 1.81t [3H, J=2.5,2.4 Hz; 20-H3]; 2.3–1.9m [5H, 3-H2, 10Hb, 17-H2]; 2.34t [1H; J=7.4 Hz; 2-H2]; 2.43m [1H; 12-H]; 2.64m [3H; 10-Ha, 4-H2]; 3.43t, 3.44t [1H, J=8.7,8.5 Hz; 8-H]; 3.92m [1H; 11-H]; 4.07t, 4.17t [1H, J=7.3,5.6 Hz; 15-H]; 4.3b [2H; OH]; 5.09m [1H, 9-H]; 5.58dd, 5.61dd [1H; J=15.3,6.5 Hz; 14-H]; 5.67dd, 5.68dd [1H; J=15.3,8.0 Hz; 13-H]; 6.77m [1H; 2′-H]; 6.95m [2H; 1′-H,3′-H]13C NMR (100 MHz, CDCl3), δC (ppm): 3.5, 3.6 [C-20]; 14.7, 15.8 [C-21]; 22.3, 22.6 [C-17]; 24.6 [C-2]; 29.1 [C-4]; 33.1 [C-3]; 38.2, 38.3 [C-16]; 41.2 [C-10]; 50.4 [C-8]; 58.8 [C-12]; 75.8, 76.3, 76.4 [C-11, C-15]; 77.2, 77.4 [C-18, C-19]; 84.5, 84.6 [C-9]; 120.6 [C-2′]; 121.9 [C-3′]; 123.2 [C-5]; 129.0 [C-1′]; 129.7 [C-7]; 132.3, 133.0, 133.8, 134.0 [C-13, C-14]; 157.2 [C-6]; 178.3 [C-1].
EXAMPLE 7 Beraprost Sodium Salt (The Sodium Salt of the Compound of Formula (I)
0.199 g of beraprost is dissolved in 2 ml of methanol, 0.5 ml of 1 M aqueous solution of sodium hydroxide is added thereto and after their mixing the solvent is evaporated in vacuum and thus the above title salt is obtained as colourless crystals.
Yield: 0.21 g (100%)
Melting point: >205° C.
1H NMR (400 MHz, DMSO-d6), δH (ppm): 0.90d, 0.92d [3H; J=6.7 Hz; 21-H3]; 1.75–1.55m [7H; 10Hb, 16-H, 3-H2, 20-H3]; 1.89t [2H, J=7.6 Hz; 2-H2]; 1.94m [1H; 17-Hb]; 2.16q [1H, J=8.5 Hz; 12-H]; 2.25m [1H; 17-Ha]; 2.44t [2H; J=7.5 Hz; 4-H2]; 2.50o [1H; 10-Ha]; 3.39t [1H, J=8.5 Hz; 8-H]; 3.72td [1H; J=8.5,6.1 Hz; 11-H]; 3.84t 3.96t [1H, J=6.5,6.0 Hz; 15-H]; 4.85b [2H, OH]; 5.01dt [1H, J=8.5,6.6 Hz; 9-H]; 5.46dd, 5.47dd [1H; J=15.4,6.5 Hz, J=15.4,6.0 Hz; 14-H]; 5.65dd, 5.66dd [1H; J=15.4,8.5 Hz; 13-H]; 6.71m [1H; 2′-H]; 6.92m [2H; 1′-H, 3′-H] During the above thin layer chromatography (TLC) procedures we used plates MERCK Kieselgel 60 F254, thickness of layer is 0.2 mm, length of plates is 5 cm.
…………….
- Reaction Scheme A.




- The starting material of bromocarboxylic acid, Compound 1, and the process for the preparation thereof are disclosed in Japanese Patent Application No. 29637/81.
- Scheme B.
REACTION SCHEME B

- REACTION SCHEME C

 Org Lett 2012, 14(1): 299
Org Lett 2012, 14(1): 299
EP0024943A1 Sep 2, 1980 Mar 11, 1981 Toray Industries, Inc. 5,6,7-Trinor-4,8-inter-m-phenylene PGI2 derivatives and pharmaceutical compositions containing them EP0084856A1 Jan 19, 1983 Aug 3, 1983 Toray Industries, Inc. 5,6,7-Trinor-4, 8-inter-m-phenylene prostaglandin I2 derivatives JP3069909B Title not available
THANKS AND REGARD'S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
You may search for our products through the search bar on our website. If you would like to receive a copy of our product catalog, please contact us at info@alfa-chemistry.com. N-butyl,methylpyrrolidinium bis((trifluoromethyl)sulfonyl)imide
ReplyDeleteHello, I want to write a little review about the good work of doctor Ojamo, who cured me from HSV 1&2 for just 2 week with his herbal medicine, I never believe I can be cured again and have a good life like others, I always regretted the day I got diagnose with this virus, I become lost of hope when my doctor told me there is no cure for it, Well I am so grateful for my helper who get me cured with his herbal medicine, I went online in search of anything that can help me because I can’t deal with it forever so I found dr Ojamo email dr.ojamoherbalhome@gmail.com on a blog of someone who was cured by him,of the same herpes, so I quickly contacted him for help and explained all my pains to him, he told me not to worry about it there is a cure for real, I never believe until he sent me the herbs when I ordered for it and I got it within 5 working days that is how I took the medicine for 2 weeks and the following week I went for a test just to confirm I was 100% cured from this sickness what a miracle in my life, I am so happy right now, and forever greatful, you can also get in contact with him if you have such sickness through his whatsapp +2349077406037 or email address:: dr.ojamoherbalhome@gmail.com
ReplyDeletewhat a miracle i never believe there is cure because my doctor tested me HSV 2 positive and she told me there is no cure, i’m very happy today that i’m having a free life without this sickness, i can remember some months ago when i was crying all through the night and day that i can’t get cured from this sickness, i found this herbal man email on internet when i was doing research on cure for HSV 2 i contacted him to found out if i can get help from this sickness, i was so surprise when he told me that he have the herbs cure to it and he sent me the herbal in less down 4 days i was so happy when i get someone giving me hope that he can cured me i took the herbal for just 2 weeks, when i went for test after taking the herbal i found out that i am cured i was so happy and surprise, i want to use this opportunity to inform you that there is cured to HSV 2 you can also contact him for his help as soon as possible so that you can get rid of this sickness once and for all you can reach him through this email: drehiaguna@gmail.com or and you can also call or whatapp him now +2348073908953 You can also contact him on any sickness in this he all have the herbal cure to it .HIV.HEPATITIS.ANTHRAX.HPV.CANCER.ALS
ReplyDeleteI never thought i would be HIV negative again after been diagnosed in 2017, i have tried everything possible in life from one doctor to another, one hospital to another, series of tests, different kinds of medication, i had already lost hope until i meet Great Dr. OSAGIE online testimonies, a specialist in herbal medication from Africa, i contacted him (drosagiesolutiontemple@gmail.com OR DROSAGIESOULTIONTEMPLE@YAHOO.COM) and he prepared HIV herbal medication for me which i took for 14days and now i am completely cured. i want to use this medium to express my gratitude to him for saving my life and curing me from HIV, for taking away all my pains and sorrows, I''m indeed grateful and i am so happy I''m now HIV negative. i will continue to tell the good news of your great works to everyone, if you have HIV or other disease contact him, Email: his email: (drosagiesolutiontemple@gmail.com OR DROSAGIESOULTIONTEMPLE@YAHOO.COM ) or Whatsapp number: +2347030465649 DOCTOR OSAGIE CAN AS WELL HELP THE FOLLOWING PROBLEMS
ReplyDelete1. HIV/AIDS SPELL
2. HERPES CURE OF KIND
3. CANCER SPELL
4 IF YOU WANT YOUR EX LOVER BACK SPELL
5 IF YOU NEED A BABY SPELL him to solve
6 LOW SPERM COUNT SPELL get all your problem solve. No problem is too big for him to solve.