FLOXACIN SERIES
1 DELAFLOXACIN
2 AVAROFLOXACIN/ACORAFLOXACIN.
3.MOXIFLOXACIN
4.NEMONOXACIN
5.NADIFLOXACIN
6 ZABOFLOXACIN
7 DS-8587
8 OZENOXACIN
9 FINAFLOXACIN
10 CLINAFLOXACIN
11 PRULIFLOXACIN
12 FANDOFLOXACIN
13 ACH 702
14 levonadifloxacin wck 771
15 QUARFLOXACIN
16 GATIFLOXACIN
17 WCK 2349
18
WILL BE UPDATED
1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.


Ligand Partner Rib-X Initiates Phase 3 Trial of Captisol-enabled™ Delafloxacin IV


cas no1001162-01-1 February 25, 2013


 MOXIFLOXACIN
MOXIFLOXACIN
 MOXIFLOXACIN
MOXIFLOXACIN


















![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i20.png?max-width=550&background=EEEEEE)

 NADIFLOXACIN
NADIFLOXACIN

DS-8587 (Daiichi Sankyo (Japan) a new broad-spectrum antibacterial agent, is in phase I clinical trials for the treatment of bacterial infection.


Ozenoxacin in phase 3......topical formulation in the treatment of impetigo


9
FINAFLOXACIN










10
1 DELAFLOXACIN
2 AVAROFLOXACIN/ACORAFLOXACIN.
3.MOXIFLOXACIN
4.NEMONOXACIN
5.NADIFLOXACIN
6 ZABOFLOXACIN
7 DS-8587
8 OZENOXACIN
9 FINAFLOXACIN
10 CLINAFLOXACIN
11 PRULIFLOXACIN
12 FANDOFLOXACIN
13 ACH 702
14 levonadifloxacin wck 771
15 QUARFLOXACIN
16 GATIFLOXACIN
17 WCK 2349
18
WILL BE UPDATED
1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.
Ligand Partner Rib-X Initiates Phase 3 Trial of Captisol-enabled™ Delafloxacin IV

delafloxacin
Ligand Pharmaceuticals Incorporated (NASDAQ: LGND) announced today that its partner Rib-X Pharmaceuticals, Inc. has initiated a Phase 3 clinical trial of a Captisol-enabled™ intravenous (IV) formulation of delafloxacin for the first-line treatment of acute bacterial skin and skin structure infections (ABSSSI), including infections caused by MRSA. Under the terms of a license and supply agreement, Ligand has earned a $500,000 milestone payment.
read all at
Delafloxacin (originally RX-3341) is a fluoroquinolone antibiotic being developed by Rib-X Pharmaceuticals, Inc.
It is more active (lower MIC90) than other quinolones against Gram-positive bacteria such as MRSA. In contrast to most approved fluoroquinolones, which are zwitterionic, delafloxacin has an anionic character, which results in a 10-fold increase in delafloxacin accumulation in both bacteria and cells at acidic pH. This property is believed to confer to delafloxacin an advantage for the eradication of Staphylococcus aureus in acidic environments, including intracellular infections.[1]
Phase II clinical trials have been completed,[2][3][4] including a trial with tigecycline as a comparator[5] The company states Delafloxacin met its primary and secondary efficacy endpoints based on the draft guidance from the FDA. A Phase III trial for acute bacterial skin and skin structure infections (ABSSSI) is due to begin in the 2nd half of 2012.[6]
- Lemaire, S. et al. Contrasting Effects of Acidic pH on the Extracellular and Intracellular Activities of the Anti-Gram-Positive Fluoroquinolones Moxifloxacin and Delafloxacin against Staphylococcus aureus Antimicrob. Agents Chemother. February 2011; 55:649-658 doi:10.1128/AAC.01201-10
- http://www.bio-medicine.org/biology-technology-1/Rib-X-Pharmaceuticals-Announces-Positive-Phase-2-Study-Results-for-Delafloxacin-and-a–2425-Million-Financing-10093-1/
- http://clinicaltrials.gov/ct2/show/NCT00719810
- http://www.medicalnewstoday.com/articles/132200.php “Rib-X Pharmaceuticals Reports Positive Top-Line Results From Phase 2 Study Of Delafloxacin” 9 Dec 2008
- http://www.citybizlist.com/lstg/lstgDetail.aspx?id=45749 “ABS Ventures Joins $25M Series D Rib-X Pharmaceuticals Inc.” 5 Feb 2009
- Delafloxacin – Pipeline – Rib-X Pharmaceuticals. Accessed: 6/27/2012
2 Phase 3
FDA -Avarofloxacin / Acorafloxacin Granted QIDP and Fast Track Designation
Avarofloxacin .HCl/Acorafloxacin.HCl
Avarofloxacin Granted QIDP and Fast Track Designation
JNJ-Q2, JNJ-32729463-AAA
CAS NO
878592-87-1 of base
Furiex Pharmaceuticals Inc. announced that the FDA has granted Qualified Infectious Disease Product (QIDP) and Fast Track designation for avarofloxacin (JNJ-Q2). Avarofloxacin is a Phase 3-ready broad-spectrum fluoroquinolone antibiotic for the treatment of acute bacterial skin and skin-structure (ABSSSI) infections, community-acquired pneumonia and has proven to be effective in treating methicillin-resistant Staphylococcus aureus (MRSA) infections.
Avarofloxacin is an investigational novel fluoroquinolone antibiotic that has been shown to be effective in a Phase 2 study of ABSSSI infections. In this study, avarofloxacin demonstrated favorable efficacy for both early clinical response endpoints as well as all clinical cure endpoints for the intent to treat population.
Avarofloxacin has a low tendency for development of drug resistance and exhibits a broad range of antibacterial activities in vitro, including MRSA, fluoroquinolone-resistant Staphylococcus aureus, Streptococcus pneumoniae (including multi-drug resistant strains), gram positive, gram negative, atypical respiratory pathogens (such as legionella and mycoplasma) and anaerobic bacteria, which are often associated with abscesses of the skin and other organs.
The availability of IV and oral formulations for avarofloxacin differentiates it from a number of other products for MRSA infections which are only available for intravenous administration.
For more information call (919) 456-7800or visit http://www.furiex.com/
About Methicillin-Resistant Staphylococcus aureus (MRSA)
MRSA is a strain of the bacteria Staphylococcus aureus (staph) which commonly causes skin and soft tissue infections and is resistant to many antibiotics. Although MRSA had previously been primarily a hospital-acquired pathogen, its incidence has been rising in the community, and it has become the most frequent cause of skin and soft tissue infections presenting to emergency departments in the United States. There are a limited number of antibiotics approved to treat MRSA, and their frequent usage has led to emergence of multi-drug resistant bacteria. Thus, we believe there is significant unmet medical need for new antibiotics such as avarofloxacin that provide flexible (hospital and outpatient) treatment options for MRSA.
Avarofloxacin nonproprietary drug name
November 28, 2012. N12/130. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (ZZ-145). AVAROFLOXACIN.- [PDF]
Avarofloxacin hydrochloride nonproprietary drug name
December 26, 2012. M12/158. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (zz-146). AVAROFLOXACIN …
3 MOXIFLOXACIN
MOXIFLOXACIN
Bay-12-8039
CAS 354812-41-2
186826-86-8 HYDROCHLORIDE
| 1-cyclopropyl-7-[(1S,6S)-2,8-diazabicyclo[4.3.0]non-8-yl]-6-fluoro-8-methoxy-4-oxo- quinoline-3-carboxylic acid |
(4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acid
ALSO AS….
Vigamox, Avelox I.V., 151096-09-2, Moxifloxacin [INN:BAN], MXFX, CHEMBL32, Actira (*Hydrochloride*), Avelox (*Hydrochloride*)
Molecular Formula: C21H24FN3O4
Molecular Weight: 401.431363
Moxifloxacin is a synthetic fluoroquinolone antibiotic agent. Bayer AG developed the drug (initially called BAY 12-8039) and it is marketed worldwide (as the hydrochloride) under the brand name Avelox (in some countries also Avalox) for oral treatment
For the treatment of sinus and lung infections such as sinusitis, pneumonia, and secondary infections in chronic bronchitis. Also for the treatment of bacterial conjunctivitis (pinkeye).
Moxifloxacin and its salts are antibacterial agents, which were disclosed in EP 550,903. Moxifloxacin, chemically 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS, 7aS)-octahydro-6H-pyrrolo[3,4-b]pyridine-6-yl]-4-oxo-3-quinolinecarboxylic acid
Moxifloxacin is a fourth-generation synthetic fluoroquinolone antibacterial agent developed by Bayer AG (initially called BAY 12-8039). It is marketed worldwide (as the hydrochloride) under the brand names Avelox, Avalox, and Avelon for oral treatment. In most countries, the drug is also available in parenteral form for intravenous infusion. Moxifloxacin is also sold in an ophthalmic solution (eye drops) under the brand names Vigamox, Moxezafor the treatment of conjunctivitis (pink eye).
A United States patent application was submitted on 30 June 1989, for Avelox (moxifloxacin hydrochloride).[1] In 1999 Avelox was approved by theU.S. Food and Drug Administration (FDA) for use in the United States.[2]
In the United States, moxifloxacin is licensed for the treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, community acquired pneumonia, complicated and uncomplicated skin and skin structure infections, and complicated intra-abdominal infections.[3]
In the European Union, it is licensed for acute bacterial exacerbations of chronic bronchitis, non-severe community-acquired pneumonia, and acute bacterial sinusitis. Based on its investigation into reports of rare but severe cases of liver toxicity and skin reactions, the European Medicines Agency recommended in 2008 that the use of the oral (but not the IV) form of moxifloxacin be restricted to infections in which other antibacterial agents cannot be used or have failed.[4] In the US, the marketing approval does not contain these restrictions, though the label contains prominent warnings against skin reactions.
MOXIFLOXACIN
Avelox (moxifloxacin) was launched in the United States in 1999 and is currently marketed in more than 80 countries worldwide. In the United States, Avelox is marketed by Bayer’s partner Merck.
In 2011 the FDA added two boxed warnings for this drug in reference to spontaneous tendon ruptures and the fact that moxifloxacin may cause worsening of myasthenia gravis symptoms, including muscle weakness and life-threatening breathing problems.[5]
Moxifloxacin is used to treat a number of infections including: respiratory tract infections, cellulitis, anthrax, intraabdominal infections, endocarditis,meningitis, and tuberculosis.[6]
The initial approval by the FDA (December 1999)[7] encompassed the following indications:
- Acute Exacerbations of Chronic Bronchitis (AECB)
- Acute Bacterial Sinusitis (ABS)
- Community Acquired Pneumonia (CAP)
Additional indications were approved by the FDA as follows:
- April 2001: Uncomplicated Skin and Skin Structure Infections (uSSSI)[8]
- May 2004: Community Acquired Pneumonia caused by multi-drug resistant Streptococcus pneumoniae.[9]
- June 2005: Complicated Skin and Skin Structure Infections (cSSSI)[10]
- November 2005: Complicated Intra-Abdominal Infections (cIAI).[11]
The European Union requires that moxifloxacin only be prescribed when other antibiotics that have been initially recommended for treatment cannot be used or have failed.[12][13]
At the current time,[when?] there are no approved uses within the pediatric population for Oral and I.V. moxifloxacin. A significant number of drugs found within this class, including moxifloxacin, are not licensed by the FDA for use in children due to the risk of permanent injury to the musculoskeletal system.[14][15][16]
In ophthalmology, moxifloxacin is approved for the treatment of conjunctival infections caused by susceptible bacteria.[17]
Note: Moxifloxacin may be licensed for other uses, or restricted, by the various regulatory agencies worldwide
Marketing authorisations for the tablet and injectable forms of Moxifloxacin are held by Bayer, while Alcon (now a subsidiary of Novartis) produces ophthalmic solutions for treating conjunctivitis under the brand names of Moxeza, Vigamox, and Moxivig. Avelox generated sales of USD320 million in the first 9 months of 2013.
Moxifloxacin is available in three distinct administration forms. Formulated as a salt, Moxifloxacin hydrochloride is sold as an oral 400 mg film-coated tablet and as an injectable solution for infusion by Bayer; although in the US it is distributed by Merck Sharp and Dohme under license from Bayer.
Alcon has formulated Moxifloxacin hydrochloride as a 0.5% ophthalmic solution under license from Bayer. Moxifloxacin was first discovered in 1988 and received the first market authorisation eleven years later in 1999 in the US.
Moxifloxacin hydrochloride is a synthetic broad-spectrum antibacterial agent. The active moiety, moxifloxacin has been shown to be clinically active against most strains of microorganisms such as aerobic gram-positive microorganisms including staphylococcus aureus, streptococcus pneumonia (penicillin-susceptible strains) and streptococcus pyogenes, aerobic gram-negative microorganisms including haemophilus influenza hemophilus parainfluenzae, klebisiella pneumonia. Moxifloxacin is commercially available under the brand name of AVELOX® marketed by Bayer pharms.
VIGAMOX® (moxifloxacin hydrochloride ophthalmic solution) 0.5% is a sterile solution for topical ophthalmic use. Moxifloxacin hydrochloride is an 8-methoxy fluoroquinolone anti-infective, with a diazabicyclononyl ring at the C7 position.
C21H24FN304•HC1 Mol Wt 437.9
Chemical Name: l-Cyclopropyl-6-fluoro-l,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolol[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid, monohydrochloride. Moxifloxacin hydrochloride is a slightly yellow to yellow crystalline powder. Each mL of VIGAMOX® solution contains 5.45 mg moxifloxacin hydrochloride, equivalent to 5 mg moxifloxacin base.
Contains: Active: Moxifloxacin 0.5% (5 mg/mL); Inactives: Boric acid, sodium chloride, and purified water. May also contain hydrochloric acid/sodium hydroxide to adjust pH to approximately 6.8.
VIGAMOX® solution is an isotonic solution with an osmolality of approximately 290 mOsm/kg.
Market Considerations
Amongst the US approvals, Dr. Reddy’s, Teva, Torrent, and Aurobindo have received tentative approvals for the 400 mg oral tablet formulation. Akorn, Teva and Apotex have received tentative approvals for a Moxifloxacin hydrochloride ophthalmic solution. No 180 day period of exclusivity has been awarded since all patents were found to be valid.
In the UK, Teva, Rivopharm, and Double-E Pharma have received marketing authorisations for the 400 mg Moxifloxacin tablets, while Noridem has received a market authorisation for the equivalent 400mg/250ml solution for infusion. A similar trend of generic competition, for tablets and infusions, following molecule patent expiry is expected throughout Europe. Currently no generic market authorisations for ophthalmic formulations have been granted in major European countries. However, Sandoz and Hexal have gained market authorisations in some European markets for the ophthalmic dosage form following Novartis’ acquisition of Alcon.
In Canada the only generic manufacturer holding a marketing authorisation is Sandoz, however, this was granted as a New Drug Submission rather than as an ANDS. Following patent expiries from mid-2014, the European and North American markets are likely to have significant competition if the numbers of companies filing litigation, ANDS, ANDAs and the like in the northern hemisphere is anything to go by.
MOXIFLOXACINHistory
Moxifloxacin was first patented (United States patent) in 1991 by Bayer A.G., and again in 1997.[47] Avelox was subsequently approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999 to treat specific bacterial infections.[2] Ranking 140th within the top 200 prescribed drugs in the United States for 2007[48] moxifloxacin, in the same manner asciprofloxacin, has proven to be a blockbuster drug for Bayer A. G., generating billions of dollars in additional revenue. In 2007 alone, Avelox generated sales of $697.3 million dollars worldwide.[26]
Moxifloxacin is also manufactured by Alcon as Vigamox.[citation needed]
Patent
A United States patent application was made on 30 June 1989, for Avelox (moxifloxacin hydrochloride),(Bayer A.G. being the assignee), which was subsequently approved on 5 February 1991. This patent was scheduled to expire on 30 June 2009. However, this patent was extended for an additional two and one half years on 16 September 2004, and as such is not expected to expire until 2012.[49] Moxifloxacin was subsequently (ten years later) approved by the U.S. Food and Drug Administration (FDA) for use in the United States in 1999. There have been at least four additional United States patents filed regarding moxifloxacin hydrochloride since the 1989 United States application,[47][50] as well as patents outside of the USA.
Additional regulatory history
6/12/2002 Changes made to minimize the impact of warnings concerning adverse reactions.[51]
26 June 2003 New Zealand Pharmacovigilance warns of moxifloxacin induced respiratory insufficiency.[52]
10/6/2003 Changes made to minimize the impact of post marketing reports as well as the risk of tendon injuries.[53]
29 December 2008 Addition of numerous adverse reactions associated with the use of moxifloxacin.[54]
27 April 2009 Issuance of a Medication Guide and revisions to include new safety information including the addition of the Black Box Warning to the Medication Guide. The FDA had determined that Moxifloxacin poses a serious and significant public health concern, requiring the distribution of a Medication Guide.[55]
24 June 2009 Updating of the carton and container labels to include a statement to let dispensers know that a Medication Guide must be dispensed with the product.(emphasis added)[56]
Patent related
|
As indicated by the Key Patent Indicator (Fig. 2), patents in the families with priority DE3824072A, 15/07/1988 (‘072), and DE4200414A, 10/01/1992, (‘414) provide protection for the Moxifloxacin molecule and are considered to be the main constraint to generic entry. As patents in the ‘414 family have expired or were never granted, the only remaining constraint to generic entry is the ‘072 family. The term of the Australian patent of this family have been extended to 19 June 2014 while the Canadian member will enjoy the longer term of 17 years from grant, expiring in November 2015.
Supplementary protection certificates (SPCs) have been granted in France, Germany, Spain and the UK, and will expire in June 2014. Given that there are less than 2 years until these SPCs expire, and that as yet no applications for paediatric extension of the SPCs have been published in Europe, they are unlikely to be extended by 6 months on the basis of the approved Paediatric Investigation Plans.
The US member, 4,990,517 (‘517), protecting the general structure of the Moxifloxacin molecule, expired in June 2012, after enjoying 6 month paediatric extension on top of a 901 day s156 extension. However, the absence of generics on the US market is due to Bayer securing a divisional patent, 5,607,942 (‘942). This patent claims the Moxifloxacin molecule specifically and is due to expire in September 2014, after being awarded a 6 month paediatric extension.
Members of the ‘072 family from both Canada and the US have been the subject of litigation after generic manufacturers identified these patents in paragraph IV filings, and the equivalent in Canada, as early as 2006. After filing an infringement suit in the US against Teva in relation to US ‘517, US ‘942, Bayer enjoyed a satisfying validification of their patents when Teva agreed that it would be infringing two of the patents, while the third was decided by the court to be equally valid.
In Canada, Novopharm, Cobalt, Apotex, Mylan and Apotex have also all tested the litigation waters relating to the equivalent patent with no success noted so far.
A third patent family that promises to be a constraint for generic ophthalmic formulations is Alcon’s 1998 patent, US10250498P (Fig. 2), which identifies an ophthalmic formulation of Moxifloxacin and its use in the treatment and prevention of eye infections. Patents in this family are set to expire in August 2019. US members 6,716,830 (‘830) and 7,671,070 (‘070) have been awarded a 6 month paediatric extension, extending their expiration until March 2020. The validity of US ‘830 was upheld following Teva filing paragraph IV certifications to manufacture generic Vigamox. Teva has since appealed this ruling.
In addition, Alcon has filed patent infringement suits against Watson, Lupin and Apotex in relation to US ‘070 after they submitted Abbreviated New Drug Applications (ANDAs)with paragraph IV filings in preparation for commercialisation of a Moxeza/Vigamox generic equivalent. There has been no outcome from these suits to date. In addition, applications for Orders of Prohibition against Cobalt, Apotex and Teva have also been noted for the equivalent Canadian patent following the filing of Abbreviated New Drug Submissions (ANDS) by these companies.
The equivalent European patent 1,117,401 was revoked following opposition by Teva filed in the European patent office. Its divisional patents, 1,384,478 (granted) and 2,301,541 (accepted) have restricted claims to the use of Moxifloxacin in the topical treatment of ophthalmic infections caused by P. aeruginosa and H. influenza, respectively. This may provide a prepared generic competitor an opportunity to launch their Moxifloxacin ophthalmic equivalent in Europe soon after the expiry of patents protecting the molecule, subject to legal review of the remaining claims of the patent.
Alcon have secured additional protection for their Moxeza ophthalmic formulation by way of patents in the family with priority US5987708P (09/06/2008). Patent claims specify ratios of Moxifloxacin to inactive ingredients and additional inactive ingredients and therefore generic competitors are likely to circumvent the patent by reformulation. Lupin has filed paragraph IV certifications to US8450311, which is currently subject of a patent infringement suit.
Families with priorities DE19546249A (12/12/1995), DE19751948A (24/11/1997), DE19855758A (10/11/1998) and US36433499A (30/07/1999) are not considered to be a constraint to generic entry because the protected technologies are likely to be circumvented.
Generic equivalents
In 2007, the U.S. District Court for the District of Delaware held that two Bayer patents on Avelox (moxifloxacin hydrochloride) are valid and enforceable, and infringed by Dr. Reddy’s ANDA for a generic version of Avelox.[70][71] The district court sided with Bayer, citing the Federal Circuit’s prior decision in Takeda v. Alphapharm[72] as “affirming the district court’s finding that defendant failed to prove a prima facie case of obviousness where the prior art disclosed a broad selection of compounds, any one of which could have been selected as a lead compound for further investigation, and defendant did not prove that the prior art would have led to the selection of the particular compound singled out by defendant.” According to Bayer’s press release[70] announcing the court’s decision, it was noted that Teva had also challenged the validity of the same Bayer patents at issue in the Dr. Reddy’s case. Within Bayer’s first quarter 2008 stockholder’s newsletter[73]
Bayer stated that they had reached an agreement with Teva Pharmaceuticals USA, Inc., the adverse party, to settle their patent litigation with regard to the two Bayer patents. Under the settlement terms agreed upon, Teva would obtain a license to sell its generic moxifloxacin tablet product in the U.S. shortly before the second of the two Bayer patents expires in March 2014.
- Economic impact: adverse reactions:
The advocacy group Public Citizen has lobbied for increasing safety warnings and for the removal of some fluoroquinolone drugs from clinical practice.[74][75][76][77][78][79][80][81]
| US5607942 | 3-5-1997 | 7-(1-pyrrolidinyl)-3-quinolone- and – naphthyridone-carboxylic acid derivatives as antibacterial agents and feed additives |
| WO0032196 | 6-9-2000 | INHIBITORS OF MULTIDRUG TRANSPORTERS INHIBITORS OF MULTIDRUG TRANSPORTERS |
| WO0027398 | 5-19-2000 | PHARMACEUTICAL MOXIFLOXACIN PREPARATION |
| WO0025765 | 5-12-2000 | AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION AQUEOUS DRUG FORMULATION FOR ORAL APPLICATION |
| WO0018386 | 4-7-2000 | ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE |
| WO0018388 | 4-7-2000 | ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE |
| WO0018404 | 4-7-2000 | ANTIBIOTIC COMPOSITIONS FOR TREATMENT OF THE EYE, EAR AND NOSE |
| EP0971691 | 1-20-2000 | COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS |
| WO9964037 | 12-17-1999 | NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES |
| WO9915172 | 4-2-1999 | MEDICAMENT FORMULATION WITH A CONTROLLED RELEASE OF AN ACTIVE AGENT |
| WO9836732 | 8-28-1998 | COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS COMBINATION PREPARATION FOR ORALLY ADMINISTERED ANTIBIOTICS |
| US2006252789 | 11-10-2006 | Amorphous moxifloxacin hydrochloride |
| US7115744 | 10-4-2006 | Method for producing 8-methoxy-quinolinecarboxylic acids |
| US2005152975 | 7-15-2005 | Pharmaceutical composition |
| US6916484 | 7-13-2005 | Aqueous pharmaceutical composition containing moxifloxacin or salts thereof |
| US6897315 | 5-25-2005 | Method for producing 8-methoxy-quinolinecarboxylic acids |
| US2004022848 | 2-6-2004 | Medicinal composition |
| US6566523 | 5-21-2003 | Method for the enantiomer separation of cis-8-benzyl-7,9-dioxo-2,8-diazabicyclo[4.3.0]nonane |
| WO0071541 | 11-31-2000 | OPTICALLY ACTIVE QUINOLINE CARBOXYLIC ACID DERIVATIVES HAVING 7-PYRROLIDINE SUBSTITUTES CAUSING OPTICAL ACTIVITY AND A PROCESS FOR PREPARING THEREOF |
| WO0068229 | 11-17-2000 | ANTIBACTERIAL OPTICALLY PURE BENZOQUINOLIZINE CARBOXYLIC ACIDS, PROCESSES, COMPOSITIONS AND METHODS OF TREATMENT (S)-BENZOQUINOLIZINE CARBOXYLIC ACIDS AND THEIR USE AS ANTIBACTERIAL AGENTS |
| WO0050007 | 8-32-2000 | COMPOSITIONS AND METHODS FOR IMPROVED DELIVERY OF HYDROPHOBIC THERAPEUTIC AGENTS |
| US2010152414 | 6-18-2010 | MULTI-ARM POLYMER PRODRUGS |
| US2010087409 | 4-9-2010 | COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID |
| US7563805 | 7-22-2009 | Tri-, tetra-substituted-3-aminopyrrolidine derivative |
| US2009170893 | 7-3-2009 | NOVEL HYDRATE FORM |
| US2009074704 | 3-20-2009 | Multi-Arm Polymer Prodrugs |
| US2009018169 | 1-16-2009 | Sulfonamide Derivatives for the Treatment of Bacterial Infections |
| US2008287396 | 11-21-2008 | Phosphonated Fluoroquinolones, Antibacterial Analogs Thereof, and Methods for the Prevention and Treatment of Bone and Joint Infections |
| US2008194612 | 8-15-2008 | Multi-arm polymer prodrugs |
| US2007196504 | 8-24-2007 | PHARMACEUTICAL COMPOSITION |
| US2007148235 | 6-29-2007 | PHARMACEUTICAL COMPOSITION |
| US8211910 | 7-4-2012 | TRI-, TETRA-SUBSTITUTED-3-AMINOPYRROLIDINE DERIVATIVE |
| US8198451 | 6-13-2012 | Process for the Synthesis of Moxifloxacin Hydrochloride |
| US2011293717 | 12-2-2011 | COMPACTED MOXIFLOXACIN |
| US8034817 | 10-12-2011 | Treatment of bacterial diseases of the respiratory organs |
| US2011224249 | 9-16-2011 | Novel Hydrate Form |
| US2011212990 | 9-2-2011 | NOVEL POLYMORPH OF MOXIFLOXACIN HYDROCHLORIDE |
| US2011159049 | 6-31-2011 | PHARMACEUTICAL COMPOSITION |
| US2011137036 | 6-10-2011 | SYNTHESIS OF (4aS,7aS)-OCTAHYDRO-1H-PYRROLO[3,4-b]PYRIDINE |
| US2010190933 | 7-30-2010 | MULTI-ARM POLYMER PRODRUGS |
| US7744861 | 6-30-2010 | Multi-arm polymer prodrugs |
DESCRIPTION
- Moxifloxacin is a therapeutic agent that shows a broad spectrum antibacterial action. Moxifloxacin is the international non-proprietary name (INN) for 1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid.
- The structure of moxifloxacin corresponds to formula (I):

- Racemic moxifloxacin was firstly described in EP-A-350733 and, particularly, moxifloxacin having a (S,S)-configuration is described inEP-A-550903 .
- In experimental example 19 of EP-A-550903 and example Z19 of EP-A-591808 , a method for preparing and isolating moxifloxacin base is described. The same method is described in patent document EP-A-592868 ). These published patent applications neither describe nor suggest the possible existence of a crystalline form of moxifloxacin base. In WO-A-2008059521 it is disclosed that by performing the mentioned examples an acetonitrile solvated form of moxifloxacin with low purity is obtained, and so the obtained solvate form can not be used as such in pharmaceutical formulations. In the above mentioned examples, the obtained moxifloxacin base crude is purified and isolated by chromatography using methylene chloride/methanol/17% aqueous ammonia as the solvent system. The purification process disclosed in said documents has been reproduced by the authors of the present invention, but only an amorphous form of moxifloxacin was obtained. This process for the purification and isolation of moxifloxacin base is complex and difficult to perform on industrial scale due to the need for purifying the product by column chromatography.
- WO-A-9926940 discloses a process for the preparation of moxifloxacin from a difluoro precursor comprising the step of adjusting the pH to 6.8- 7.0. However, the reproduction of this example shows that at this pH moxifloxacin hydrochloride or a mixture of moxifloxacin hydrochloride and moxifloxacin base is obtained, since the X-ray diffractogram of the isolated compound corresponds with the hydrochloride moxifloxacin. This fact has also been confirmed by the reproduction of the subsequent crystallization described in the international patent application of the previous compound in ethanol/water. The behaviour of this solid was very different regarding its solubility in respect what was described in the international patent application.
- WO-A-2004091619 and WO-A-2007010555 disclose the preparation of moxifloxacin base as a solid. Nevertheless, they do not describe nor suggest the preparation of a crystalline form of moxifloxacin base. The authors of the present invention have proved that the X-ray diffractogram of the product obtained by reproducing the reference example disclosed in WO-A-2004091619 , based on a pH adjustment to 7.0-7.2, corresponds to the X-ray diffractogram of the monohydrate of moxifloxacin hydrochloride disclosed in US 5849752 . Similarly, by reproducing the reference example of WO-A-2007010555 , also for obtaining moxifloxacin base but based on a pH adjustment to 5.0-6.0, it is neither expected to obtain moxifloxacin base due to the adjustment of the solution within a range of acidic pH values.
- [0008]WO-A-2008059521 discloses a process for the preparation of a crystalline form of moxifloxacin base, designated as Form I, by recrystallization in a ketosolvent, such as acetone. This form is characterized by its X-ray diffraction pattern corresponding to a hemihydrate. The best yield obtained is a 78.2% starting from moxifloxacin hydrochloride in example 18. This crystalline form has a tendency to occlude solvent molecules within the crystalline network in amounts very superiors to the allowed ones by for Guidelines Residual Solvents (CPMP/ICH/283/95) and can be difficult to impossible to remove by drying, what forces to carry out laborious treatments, either physical, or chemical to reach allowed solvent levels. The presence of any non-aqueous solvents in amounts over the allowed ones would not make this crystalline form suitable for the preparation of pharmaceutical formulations.
- The existence of polymorphs is unpredictable and there is no a priori established procedure to prepare an unknown polymorph. The difference in the physical properties of different morphological forms results from the orientation and intermolecular interactions of adjacent molecules are complexes in the bulk solid. Furthermore, the different solid forms of a pharmaceutically active ingredient can have different characteristics, and offer certain advantages in methods of manufacture and also in pharmacology. Thus, the discovery of new solid forms can contribute to clear improvements in the efficiency of methods of production and/or improvements in the characteristics of the pharmaceutical formulations of the active ingredients, since some forms are more adequate for one type of formulation, and other forms for other different formulations.
Moxifloxacin Hydrochloride namely (4aS-Cis) -l-cyclopropyl-7- (2, 8- diazabicyclo [4.3.0] non-8-yl) -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylic acidhydrochloride has the formula
Moxifloxacin Hydrochloride
Moxifloxacin is a fluoroquinolone broad spectrum antibacterial particularly against Gram-positive bacteria significantly better than those of Sparfloxacin and Ciprofloxacin that was disclosed in EP No 350,733 and EP No 550,903. Moxifloxacin has activity against Gram- negative and Gram-positive organisms, including Streptococcus pneumonia, Staphylococcus aureus, Pseudomonas aeruginosa, particularly against the respiratory disease-causing pathogens like Mycoplasma pneumonia, Mycobacterium tuberculosis, Chlamydia pneumoniae and the activity shown to be unaffected by B-lactamases .
US Patent No 5,157,117 discloses (l-cyclopropyl-6, 7-difluoro-8-methoxy- 4-oxo-l, 4-dihydro-3-quinoline carboxylic acid-O3, 04)bis (acyloxy-O) borate and process for its preparation by reacting ethyl-1- cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylate with Boric acid and acetic anhydride in presence of zinc chloride and its conversion to Gatifloxacin hydrochloride.
WO 2005/012285 discloses the process for the preparation of moxifloxacin hydrochloride using a novel intermediate namely (4aS-Cis)-(1-cyclopropyl-7-(2,8-diazabicyclo[4,3,0]non-8-yl)-6-fluoro-8-methoxy-4-oxo-1,4-dihydro-3-quinoline carboxylic acid-O3,O4)bis(acycloxy-O)borate.
Hydrates of Moxifloxacin hydrochloride known are the anhydrous and monohydrate. US Patent No. 5,849,752 discloses the monohydrate of Moxifloxacin hydrochloride and its preparation by treating the anhydrous crystalline form with ethanol/ water mixtures.
The prior art disclosed in European Patent No’s EP 350,733, EP 550,903 and EP 657,448 discloses the preparation of Moxifloxacin hydrochloride involving the condensation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid or its esters with (S,S) 2,8-Diaza bicyclo [4.3.0] nonane in presence of a base and its conversion to hydrochloride at higher temperatures leading to the desired Moxifloxacin along with its positional isomer namely (4aS-Cis)-l- cyclopropyl-6- (2, 8-diazabicyclo [4.3.0] non-8-yl) -7-fluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid as a major impurity. As the impurity and the Moxifloxacin are positional isomers they are difficult to separate. Purification of Moxifloxacin to remove this isomer results in lower yields thereby increasing the product cost. Similarly methods described in the prior art involves the preparation of Moxifloxacin and then its conversion to its hydrochloride thereby incorporating an additional step in the manufacturing process also leading to lowering of yields.
Moxifloxacin and its pharmacologically acceptable salts are disclosed in European patents EP 350733, EP 550903 and EP 657,448. The disclosed process for the preparation of moxifloxacin hydrochloride comprises of condensing l-cyclopropyl-6,7- difluoro-8-methoxy-4-oxo-l,4-dihydro-3-quinoline carboxylic acid or its esters with (S,S)2,8-diazobicyclo[4.3.0]nonane, in presence of a base at high temperature followed by conversion into hydrochloride salt .
This process not only produces desired moxifloxacin hydrochloride but also its positional isomer namely l-cyclopropyl-7-fluoro- 1,4- dihydro -8- methoxy -6- (4aS,7aS)- octahydro- 6H -pyrrolo [3,4-b] pyridine-6-yl] -4- oxo-quinolinecarboxylic acid as a major impurity which is difficult to separate. The purification of moxifloaxcin to remove this isomer results in lower yields thereby increasing the product cost.
The International publication WO 2005/012285 discloses an improved process for the preparation of moxifloxacin hydrochloride incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin hydrochloride from the ethyl 1 -cyclopropyl-6,7-difluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3-quinolme carboxylate through a novel intermediate (4aS-cis)-l-cyclopropyl-7-(2,8-diazabicyclo[4.3.0]non-8- yl)-6-fluoro-8-methoxy-4-oxo- 1 ,4-dihydro-3 -quinolinecarboxylicacid-03,04)bis(acyloxy -0)-borate.
US patent application 6897315 discloses a process for the preparation of 8-methoxy-3-quinoline carboxylic acid especially moxifloxacin incorporated herein by reference. The disclosed process involves the preparation of moxifloxacin from 8-halo moxifloxacin derivative using methanol and potassium tertiary butoxide. US patent 5639886 discloses one-pot process for the preparation of 3-quinoline carboxylic acid derivatives including moxifloxacin.
WO 2004 091619 claims anhydrous crystalline form-Ill of moxifloxacin hydrochloride and WO 2004/039804 claims amorphous form of moxifloxacin hydrochloride. US Pat.No.5, 849,752 discloses specific crystalline forms of anhydrous moxifloxacin mono hydrochloride and monohydrated moxifloxacin mono hydrochloride. Anhydrous moxifloxacin mono hydrochloride disclosed in US Pat. No.5, 849,752 has been designated as “Form-I” and the hydrated form as “Form-II” in US Pat. No.7,230,006. It also discloses a novel crystalline Form-Ill of anhydrous moxifloxacin mono hydrochloride.
US patent US 5,480,879 discloses the melting range of moxifloxacin in example part as 203-208°C and does not speaks about polymorphism of moxifloxacin. Experiment executed as per the procedure given in example Zl 9 of US 5,480,879 and resulted in acetonitrile solvated form of moxifloxacin with low purity and the obtained solvated form can not used for formulations
U.S. Pat. No. 5,849, 752 (“the ‘752 patent”), incorporated by reference, described two crystalline forms of moxifloxacin hydrochloride namely, anhydrous moxifloxacin hydrochloride and monohydrated moxifloxacin hydrochloride. For convenience, the anhydrous crystalline form described in the 752 patent is designated as “Form I”, and the hydrated form as “Form II”. According to U.S. Pat. No. ‘752’, moxifloxacin hydrochloride monohydrate Form II was obtained by stirring a suspension of the anhydrous moxifloxacin hydrochloride in aqueous media until hydration. Moxifloxacin hydrochloride monohydrate of ‘752’ was also prepared by crystallizing moxifloxacin hydrochloride from a media having a water content which is stoichiometrically sufficient but limited to 10%.
WO patent application publication No. 04/091619 disclosed anhydrous Form III of moxifloxacin hydrochloride.
WO patent application publication No. 04/039804 disclosed amorphous form of moxifloxacin hydrochloride.
WO 2005/054240 disclosed two novel crystalline forms which were designated as Form A and Form B of moxifloxacin hydrochloride.
WO patent application publication No. 07/010555 disclosed two crystalline forms which were Form X and Form Y of moxifloxacin hydrochloride. According to WO Publication No. 2007/010555, Form Y was obtained by crystallization of moxifloxacin hydrochloride from the mixture of methanol and water in the ratio of about 8:1 by volume.
WO patent application publication No. 07/148137 disclosed hydrate form of moxifloxacin hydrochloride. According to WO Publication No. 2007/148137, moxifloxacin hydrochloride monohydrate was obtained by crystallization moxifloxacin hydrochloride by humidification of moxifloxacin hydrochloride at 50-90% relative humidity at 25-60° C. for 8 to 24 hours.
WO patent application publication No. 08/028959 disclosed crystalline form of moxifloxacin hydrochloride. According to WO Publication No. 2008/028959, moxifloxacin hydrochloride was obtained by dissolving moxifloxacin hydrochloride in a mixture of methanol and water and adding acetone and recovering moxifloxacin hydrochloride crystalline form.
WO patent application publication No. 08/059521 disclosed process for the preparation of anhydrous crystalline form I of moxifloxacin hydrochloride.
WO patent application publication No. 08/095964 disclosed crystalline form of moxifloxacin base.
……………………
SYNTHESIS
Drugs Fut 1997,22(2),109
36th Intersci Conf Antimicrob Agents Chemother (Sept 15-18, New Orleans) 1996,Abst. F1.
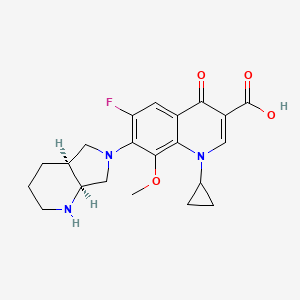
The anhydrization of pyridine-2,3-dicarboxylic acid (I) with acetic anhydride gives the corresponding anhydride (II), which by treatment with benzylamine (III) is converted into the benzylimide (IV). The hydrogenation of (IV) with H2 over Pd/C yields 8-benzyl-2,8-diazabicyclo[4.3.0]nonane-7,9-dione (V), which is further hydrogenated with LiAlH4, affording (?-cis-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VI) (1). The optical resolution of (VI) by separation of the cis-(R,R)-isomer as crystalline L-(+)-tartrate and further purification of the cis-(S,S)-isomer (VII) as the D-(-)-tartrate affords enantiomerically pure (S,S)-8-benzyl-2,8-diazabicyclo[4.3.0]nonane (VII). The debenzylation of (VII) by hydrogenolysis with H2 over Pd/C gives (S,S)-2,8-diazabicyclo[4.3.0]nonane (VIII), which is condensed with 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) in basic medium and finally salified with HCl. The 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (IX) has been obtained as follows: The reaction of 2,4,5-trifluoro-3-methoxybenzoyl chloride (X) with malonic acid monoethyl ester monopotassium salt (XI) by means of triethylamine gives 2-(2,4,5-trifluoro-3-methoxybenzoyl)acetic acid ethyl ester (XII), which is condensed with triethyl orthoformate yielding the corresponding ethoxymethylene derivative (XIII). The reaction of (XIII) with cyclopropylamine affords the cyclopropylaminomethylene derivative (XIV), which is finally cyclized to (IX) by means of NaF in DMF.
………………………..
J Label Compd Radiopharm 2000,43(8),795
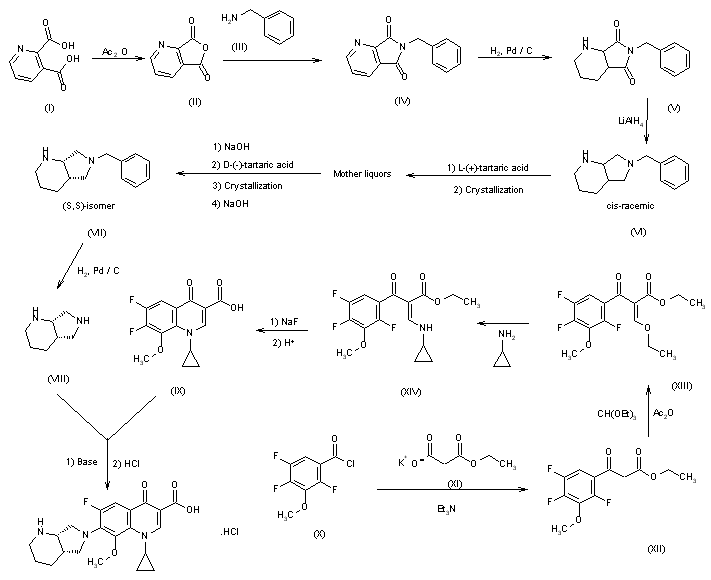
The condensation of 2,4,5-trifluoro-3-methoxybenzoyl chloride (I) with 14C-labeled diethyl malonate (II) by means of MgCl2 and TEA gives the benzoylmalonate (III), which is monodecarboxylated with TsOH in refluxing water, yielding the benzoylacetate (IV). The reaction of (IV) with triethyl orthoformate and Ac2O at 140 C affords the benzoylacrylate (V), which is treated with cyclopropylamine (VI) in cyclohexane to provide ethyl 3-(cyclopropylamino)-2-(2,4,5-trifluoro-3-methoxybenzoyl)acrylate (VII). The cyclization of (VII) by means of K2CO3 in hot N-methylpyrrolidone gives the quinolone carboxylate (VIII), which is hydrolyzed with NaOH in hot methanol, affording the carboxylic acid (IX). Finally, this compound is condensed with (S,S)-2,8-diazabicyclo[4.3.0]octane (X) by means of 1,4-diazabicyclo[2.2.2]octane (DABCO) in refluxing acetonitrile.
…………………….
……………………
The reaction scheme is given below: Stage-I
Acetic anhydride
Ethyl-l-cyclopropyl-6, 7- ( l-cyclopropyl-6, 7-difluoro-l, 4- difluoro-1, 4-dihydro-8- dihydro-8- methoxy-4-oxo-3- methoxy-4-oxo-3-quinoline quinoline carboxylic acid-03, 04) carboxylate . Bis ( acetate-O) -borate (Borate complex)
Stage-II
Triet yl amine
Acetonitrile
l-cyclo propyl-6, 7-difluoro- [S, S] -2, 8-diazabicyclo- (1- cyclo propyl-6, fluoro-7 (2, 8- 1, 4-dihydro-8- methoxy-4-oxo [4 ,3.0]nonane Diazabicyclo-nonane) 1,4- -3-quinoline carboxylic acid- dihydro-8-methoxy-4-oxo-3 03,04)Bis ( acetate-O) -borate- quinoline carboxylic acid- (Borate complex) (03,04) bis (acetate-O) -borate itage-III
(1- cyclo propyl-6, fluoro- (2, 8- Moxifloxacin HCI pseudohydrate Diazabicyclo-nonane) 1,4- dihydro-8-methoxy-4-oxo-3 -quinolinecarboxylicacid- (O^O4) bis (acetate-O) -borate
Stage-TV
Moxifloxacin HCI pseudohydrate Moxifloxacin HCI monohydrate
EXAMPLE – I
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo- l,4-dihydro-3-quinoline carboxylic acid-O3,O*)bis (acyloxy-O)borate
Acetic anhydride (175 g) is heated to 70°C and boric acid (30 g) is slowly added lot wise in a temperature range of 70°C to 90°C. The temperature is then raised, maintained under reflux for 1 hr followed by cooling to about 70°C. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylate (100 g) is added under stirring. The temperature is then raised and maintained for 1 hr in the range of 100°C to 105°C. The reaction mass is cooled to 0°C, chilled water (400 ml) is added slowly followed by cold water (600 ml) at temperature 0°C to 5°C and maintained for 2 hrs at 0°C to 5°C. The product which is a boron acetate complex is filtered, washed with water (500 ml) and dried at 55°C to 60°C under vacuum to constant weight. The dry wt is 130.0 g corresponding to yield of 95.2%.
Stage-2: Preparation of (4aS-Cis) -l-Cyclopropyl-7- (2, 8-diazabicyclo [4.3.0]non-8-yl) -6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3-quinoline carboxylicacid-03,0*)bis (acyloxy-O)borate
The boron acetate complex (130 g) prepared in stage 1 is suspended in acetonitrile (650 ml), and [S, S] -2, 8-diazabicyclo [4.3.0] nonane (47 g) and triethyl amine (72.9 g) are added. The temperature is raised to reflux and maintained for 1 hr. at reflux, followed by cooling to about 40°C. The solvent is removed under vacuum at temperature below 40°C, and n-hexane (200 ml) is added. After maintaining the reaction mass for 1 hr at room temperature the product is isolated by filtration followed by washing of the wet cake with n-hexane . The product is dried at about 45°C to about 50°C to constant weight.
Dry wt of the novel intermediate is 117.0 g corresponding to yield of 71.5%.
Elemental analysis: C: 56.42%, H: 5.62%, N: 7.76% and the calculated values for the intermediate, formula C25H29BFN308C: 56.6%, H: 5.47%, N: 7.92%
IR Spectrum (KBr, cm-1) : 3415, 3332, 2936, 1718, 1630, 1573, 1526, 1445, 1273, 1042, 935, 860, 798, 682
^ NMR (200 MHz, CDC13, ppm) : 9.00 (1H), 7.82 (1H), 4.12 (4H), 3.57 (3H), 3.43 (4H), 3.07 (2H) , 2.75 (2H), 2.4 (1H),’ 2.1 (6H), 1.84 (2H) , 1.6 (1H), 1.31 (2H)
Mass Spectrum (MJ : 530.3 [M+H] , 470.2 [M+ - CH3COOH] , 428.2 [M+- (CH3CO)20, 100%], 402.2, 388.2
Stage -3: Preparation of Moxifloxacin Hydrochloride pseudohydrate
The intermediate (117 g) prepared stage-2 is dissolved in ethanol (600 ml) by stirring for about 30 min. at room temperature and the insolubles if any are filtered off. pH of the filtrate is adjusted to about 0.5 by addition of hydrochloric acid at room temperature and maintained for 2 hrs. The reaction mass is cooled, and maintained for two hrs, at about 0°C to about 5°C. The product is filtered, washed with chilled ethanol (50 ml) and dried at about 50°C to about 55°C till constant weight.
The dry weight of the Moxifloxacin hydrochloride pseudohydrate is 87.5g corresponding to yield of 91.0%. Water content of the product by KF is 0.64% w/w.
X-ray diffraction pattern data are given in Table-1 EXAMPLE – II
Stage- 2 : Preparation of Moxifloxacin pseudohydrate with out isolating (4aS-Cis) -l-Cyclopropyl-7- (2 , 8-diazabicyclo [4.3.0] on-8-yl) -6-fluoro- 8-methoxy-4-oxo-l,4-dihydro-3-quinolinecarboxylicacid-03,04)bis (acyloxy-O) borate
The boron acetate complex (130 g) prepared in stage-1 of Example-1 is suspended in acetonitrile (650ml) and [S, S] -2, 8-Diazabicyclo [4.3.0]nonane (47 g) & triethyl amine (72.9 g) are added. Temperature of the reaction mass is raised to reflux, maintained for 1 hr. at reflux and cooled to room temperature. Methanol (600 ml) is added and maintained for 30 min at room temperature to obtain a clear solution. The solution is filtered to remove insolubles if any and pH of the filtrate is adjusted to about 0.5 with hydrochloric acid (57.5 g) . The reaction mass is maintained for 2 hrs at temperature in the range of about 20°C to about 25°C, cooled to 0°C followed by maintaining the reaction mass at about 0°C to about 5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at about 50°C to 55°C until constant weight.
Dry wt of the Moxifloxacin hydrochloride pseudohydrate is 88g corresponding to yield of 68.7%.
EXAMPLE – III : Preparation of Moxifloxacin Hydrochloride monohydrate
Moxifloxacin hydrochloride (50 g) prepared as above is suspended in a mixture of ethanol (250 ml) and hydrochloric acid (25 ml) . Raised the temperature, maintained for two hrs at 40°C to 45°C followed by cooling to about 25°C. The product is filtered and dried under vacuum at 50-55°C until become constant weight.
Dry wt of Moxifloxacin hydrochloride monohydrate is 46 g corresponding to yield of 90.5%.
The IR spectral data and XRD pattern are identical with available Moxifloxacin hydrochloride monohydrate.
……………………….
Synthesis
The preferred embodiments of the present invention is represented as follows
Formula-2a Formula-2b
Formula-3a Formula-3b Formula-3c Formula-3d
Formula-4b
Formula-4c
SCHEME-5:
SCHEME-6:
Example-1: Preparation of (S,S)-2-benzyl-8-trityI-2,8-diazabicyclo (4.3.0) nonane:
A mixture of (S5S)-2-benzyl-2,8-diazabicyclo-(4.3.0) nonane 50 grams, dichloromethane 300 ml and triethylamine 28 ml was stirred at 25-35°C. Trityl chloride 71 grams was added to the reaction mixture. The reaction mixture was stirred at 25-350C for 5 hrs. The organic layer was washed with 5% sodium bicarbonate solution followed by water. The organic layer was distilled off completely to provide the title compound as a residue.
Yield: 96 grams
Example-2:
Preparation of (S,S)-8-trityI-2,8-diazabicyclo (4.3.0) nonane:
A mixture of (S,S)-2-benzyl-8-trityl-2,8-diazabicyclo (4.3.0) nonane 130 grams,
2- butanol 1600 ml and 5% Pd/C was taken in an autoclave and heated to 45-50°C at 4.0-4.5 Kg/cm2 of hydrogen pressure. The reaction mixture was stirred at this condition for 45-50 hrs. The reaction mixture was cooled to 25-300C and filtered through hyflow bed. The solvent was distilled off completely to provide the title compound as a residue.
Yield: 95 grams.
Example-3:
Preparation of condensed compouad of formula-4a:
A mixture of 20 grams of l-cyclopropyl-NjN-diethyl-όjT-difluoro-l^-dihydro-δ- methoxy-4-oxoquinoline-3-carboxamide, 11.8 grams of (S,S)2,8-diazabicyclo (4.3.0) nonane, 100 ml of acetonitrile and 2 grams of l,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 800C. The reaction mixture was stirred for 35 hours at  0C. The reaction mixture was cooled to 320C and stirred for 45 minutes at 32°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-350C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue. Yield: 12 grams.
0C. The reaction mixture was cooled to 320C and stirred for 45 minutes at 32°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-350C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue. Yield: 12 grams.
Example-4:
Preparation of condensed compound of formula-4b:
A mixture of 5 grams of l-cyclopropyl-NjN-diethyl-όJ-difluoro-M-dihydro-δ- methoxy-4-oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-8-phenyloxy carbonyl-2,8- diazabicyclo (4.3.0) nonane, 25 ml of acetonitrile and 0.5 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 800C. The reaction mixture was stirred for 35 hours at 75-8O0C. The reaction mixture was cooled to 25-35°C. The reaction mixture was stirred for 45 minutes at 25-35°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-35°C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue. Yield: 1 gram.
Example-5: Preparation of condensed compound of formula-4c:
A mixture of 5 grams of l-cyclopropyl-NjN-diethyl-βJ-difluoro-l^-dihydro-S- methoxy-4-oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-8-tertiary butyloxycarbonyl- 2,8-diazabicyclo (4.3.0) nonane, 25 ml of acetonitrile and 0.5 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 800C. The reaction mixture was stirred for 35 hours at 75-800C. The reaction mixture was cooled to 25-350C and stirred for 45 minutes at 25-35°C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-350C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to get the title compound as a residue Yield: lgram. Example-6:
Preparation of condensed compound of formula-4d:
A mixture of 20 grams of l-cyclopropyl-6,7-difluoro-8-methoxy-l,4-dihydro-4- oxoquinoline-3-carboxamide, 2.9 grams of (S,S)-2,8-diazabicyclo (4.3.0) nonane , 100 ml of acetonitrile and 2 grams of diazabicyclo[5.4.0]undec-7-ene (DBU) was heated to 75-80°C. The reaction mixture was stirred for 35 hours at 75-80°C. The reaction mixture was cooled to 25-350C. The reaction mixture was stirred for 45 minutes at 25-350C. The solid obtained was filtered and washed with acetonitrile. The material was dried at 40-450C to get the title compound. Yield: 12 grams; M.R: 210-2120C.
Example-7:
Preparation of condensed compound of formula-4e:
A mixture of (S,S)-8-trityl-2,8-diazabicyclo (4.3.0) nonane (42 grams), l-cyclopropyl-N,N-diethyl-6,7-difluoro-l,4-dihydro-8-methoxy-4-oxoquinoline-3- carboxamide (10 grams), potassium carbonate ( 7.9 grams) and dimethyl formamide (80 ml) was heated to 110-1200C and stirred for 20 hours at 110-1200C. The reaction mixture was cooled to 800C. The solvent was distilled off under reduced pressure. Water (100 ml) was added to the obtained residue. The reaction mixture was cooled to 25-35°C. The reaction mixture was extracted with ethyl acetate. The solvent was distilled off to obtain the title compound as a residue. Yield: 14.5 grams
Example-8: Preparation of moxifloxacin from 4d:
A mixture of 165 ml of water, 35 grams of sodium hydroxide, 150 ml of ethylene glycol and 12 grams of condensed compound of formula-4d was heated to 115°C. The reaction mixture was stirred for 15 hours at 115°C. The reaction mixture was cooled to 5°C. The pH of the reaction mixture was adjusted to 5.5 using hydrochloric acid and stirred for 30 minutes at 5°C. The obtained solid was filtered and washed with water. The solid was dried at 45°C to get the title compound. Yield: 8 grams; M.R: 203-2050C Example-9:
Preparation of moxifloxacin from 4b:
Moxifloxacin can be prepared from 4b (3grams), by a method which is analogous to the method illustrated in Example-8 Yield: 0.85 grams; M.R: 203-205°C
Example-10:
Preparation of moxifloxacin from 4c:
Moxifloxacin can be prepared from 4c (3 grams), by a method which is analogous to the method illustrated in Example-8 Yield: 1.0 grams; M.R: 203-205°C
Example-11:
Preparation of moxifloxacin from 4a: Moxifloxacin can be prepared from 4a (10 grams), by a method which is analogous to the method illustrated in Example-8 Yield: 7.5 grams; M.R: 203-2050C
Example-12: Preparation of moxifloxacin from 4e:
The condensed compound of formula-4e (14.5 grams) was dissolved in ethyl acetate 100ml. Aqueous hydrochloric acid (5 ml in 20 ml of water) was added to the above reaction mixture. The reaction mixture was stirred at 25-30°C for 30-45 min. 20ml of water was added to the reaction mixture. The two layers were separated. The aqueous layer was washed twice with ethyl acetate. The pH of the aqueous layer was adjusted to 10.8 using sodium hydroxide solution. The reaction mixture was extracted with methylene chloride. The solvent distilled off to get a residue. The residue was suspended in 165 ml of water, 35 grams of sodium hydroxide; 150 ml of ethylene glycol was heated to 115°C. The reaction mixture was stirred for 15 hours at 115°C. The reaction mixture was cooled to 5°C. The pH of the reaction mixture was adjusted to 5.5 using hydrochloric acid and stirred for 30 minutes at 5°C.The obtained solid was filtered and washed with water and dried at 450C to get the title compound. Yield: 4.5 grams; M.R: 203-2050C
Example-13:
Preparation of moxifloxacin hydrochloride compound of formula-1:
A mixture of 8 grams of moxifloxacin, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35°C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The solid obtained was filtered and washed with methanol and dried at 40-450C to get crystalline Form-Y of moxifloxacin hydrochloride monohydrate. Yield: 5.5 grams
Example- 14:
Preparation of moxifloxacin hydrochloride monohydrate:
A mixture of 25 grams of moxifloxacin, 16 ml of water and 64 ml of methanol was heated to reflux temperature of 700C. The reaction mixture was filtered to remove undissolved material. Filtrate was cooled to 35°C. The pH of the filtrate was adjusted to 1.6 using hydrochloric acid. The reaction mixture was cooled to 150C. The reaction mixture was stirred for 45 minutes at 150C. The obtained solid was taken into a mixture of 22 ml of water and 0.5ml of hydrochloric acid and stirred 30 min at 5°C. The compound was filtered and washed with water, dried at 40-450C to get moxifloxacin hydrochloride monohydrate particles having oval shape. Yield: 20 grams
Example-15:
Preparation of anhydrous crystalline Form-I of moxifloxacin hydrochloride: Moxifloxacin hydrochloride (Form-Y, 10 grams) suspended in 50 ml of methanol and the reaction mixture was heated to 45-5O0C. 18ml of dichloromethane was added slowly to the reaction mixture. The reaction mixture was stirred at 45-50°C for 15-20 minutes. The reaction mixture was cooled slowly to around 0-5°C and stirred for 1-1.5 hours. The precipitated solid was filtered under nitrogen atmosphere and washed with 5 ml of methanol. The solid obtained was dried at 110-120°C till the moisture content reached below 0.5%. Yield: 8.0 grams.
Bulk density: 0.28 g/ml; Tapped density: 0.52 g/ml Particle size distribution : D( v,0.1) :1.6 μm ; D( vs0.5) : 5.5 μm ; D( v,0.9) :25.0 μm
……………………….
SYNTHESIS
Preparation of Moxifloxacin
A solution of lOOg of 1-Cyclopropyl -6,7difluoro-l,4-dihydro-4-oxo-methoxyquinolone- 3-carboxilic acid), 52gr of (S,S)2,8 diazabicyclo[4,3,0]nonane, lOgr of 1,8- diazabycyclo[5,4,0]undec-7-ene and 300ml of acetonitrile is heated to 65 -70°C temperature and stirred till the reaction get completed and evaporated the solvent. Added 5 %aqueous isopropylalcohol to the reaction mass and adjusted the pH to basic with caustic solution and then filtered the reaction mass. Combined the total filtrate and adjusted the pH to 5.0 to 6.0 with aqueous HCl.and isolated the separated solid at a temperature of 10-150C to afford Moxifloxacin.
………………….
SYNTHESIS REVIEW
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
While isolated pyridines are a vital component in numerous drugs the related quinoline/quinolone scaffolds are becoming increasingly common. One such example is nalidixic acid (1.101, in fact a naphthyridone, Figure 2). Nalidixic acid is a prototype quinolone antibiotic that has been used extensively as an effective treatment against both gram positive and gram negative bacteria. In general, quinolone antibiotics act by interfering with the enzymes DNA-gyrase and/or topoisomerase of bacteria [55]. Moxifloxacin (1.102, Avelox) and levofloxacin (1.103, Levaquin) are two third-generation fluoroquinolone antibiotics which also appear amongst the top selling drugs. These compounds display similar SAR data [56]. For instance, in the case of moxifloxacin the fluorine atom in the C6 position enhances microbial activity while a methoxy group in the C8 position is reported to increase potency and decrease toxicity. Furthermore, it was found that a cyclopropyl group was beneficial for the enzyme–DNA binding complex while the bulky nitrogen-based appendage at C7 helps to bind to DNA gyrase and hinders the efflux of the drug from the bacterial cell.
![[1860-5397-9-265-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of nalidixic acid, levofloxacin and moxifloxacin.
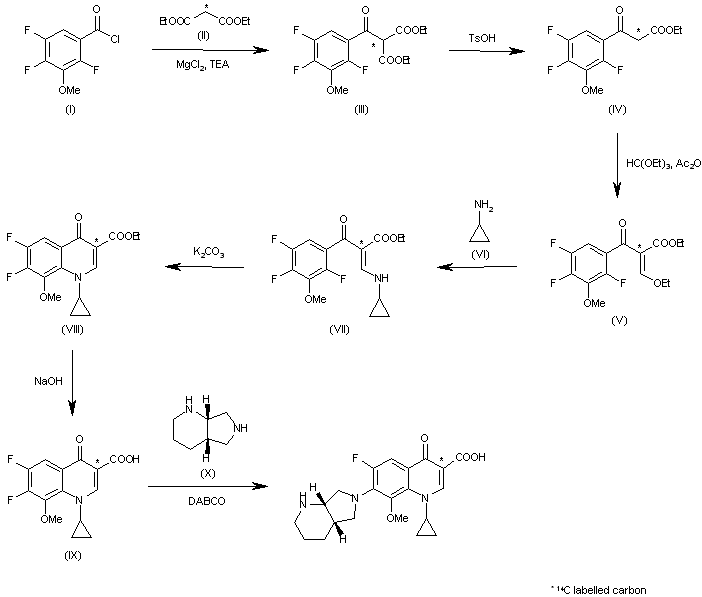
Due to the elaborate substitution pattern of the parent quinolone ring systems these compounds are usually prepared via a linear consecutive sequence. In the case of moxifloxacin, an intramolecular base catalysed nucleophilic aromatic substitution is used to prepare the bicyclic ring system of the highly substituted aromatic1.104 (Scheme 19). A SNAr reaction is then used to introduce the saturated piperidinopyrrolidine appendage 1.105to furnish the desired structure [57-60]. In order to obtain a high yield for the substitution reaction a one-pot procedure was developed, initial masking of the acid (1.104) is achieved by silylation with subsequent borane chelate formation. Addition of the amine nucleophile 1.105 under basic conditions then renders the desired product in high yield. The available patent literature however does not comment on regioselectivity issues of the SNAr reaction due to the presence of the second fluoride substituent in the substrate, although not necessarily as electronically favourable for displacement it is certainly more accessible.
![[1860-5397-9-265-i19]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i19.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of moxifloxacin.
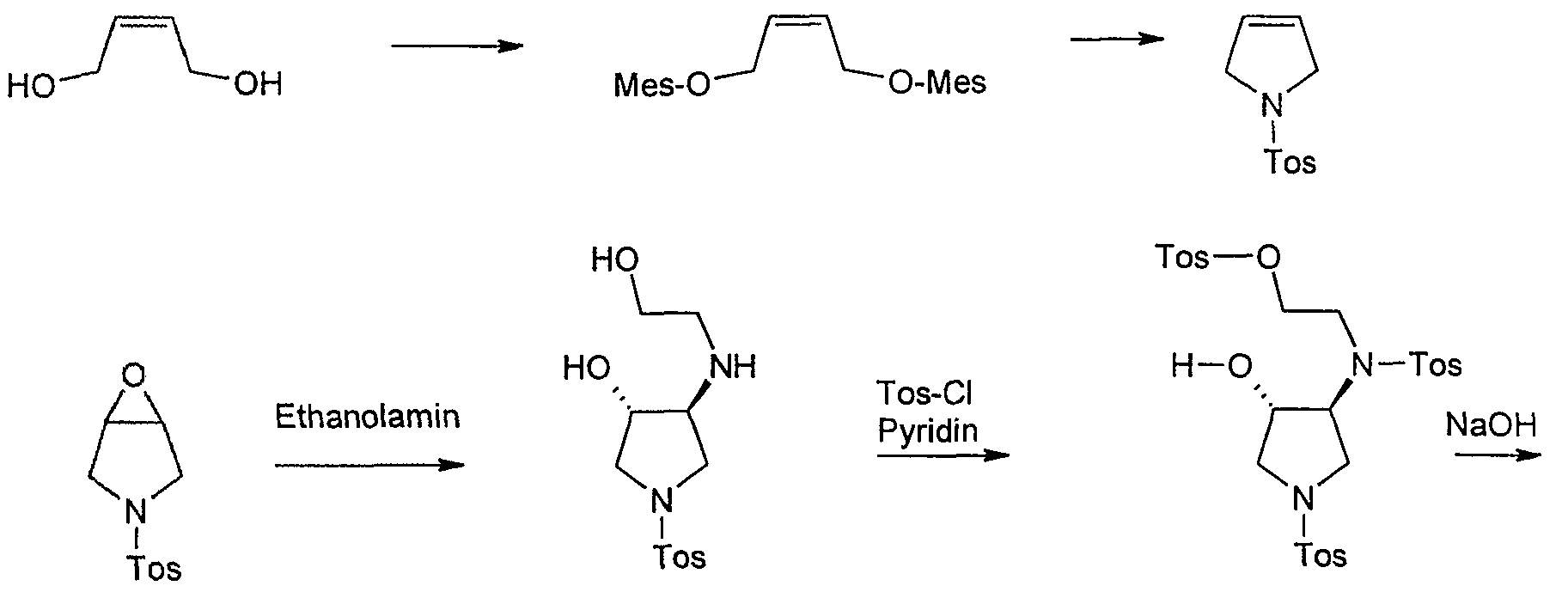
The saturated (S,S)-2,8-diazabicyclo[4.3.0]nonane (1.105) used in the final step can be prepared by a double nucleophilic substitution between tosylamine and 2,3-bis-chloromethylpyridine (1.112) followed by catalytic reduction of the resulting bicycle using palladium on carbon in acetic acid (Scheme 20). As the corresponding sulfonamide 1.113 was found to be a crystalline solid a resolution using (D)-(+)-O,O-dibenzoyltartaric acid was reported to separate the enantiomers [Petersen, U.; Schenke, T.; Krebs, A.; Schenke, T.; Philipps, T.; Grohe, K.; Bremm, K.-D.; Endermann, R.; Metzger, K. G. New Quinoline and Naphthyridinonecarboxylic Acid Derivatives. Ger. Patent DE 4 208 792 A1, March 23, 1993.].
![[1860-5397-9-265-i20]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i20.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of (S,S)-2,8-diazabicyclo[4.3.0]nonane 1.105.
…………………………………
PAPER
| First way of enantioselective synthesis of moxifloxacin intermediate LI GuangXun, WU Lei, FU QingQuan, TANG Zhuo, ZHANG XiaoMei 
A new method of enantioselective synthesis of (S,S)-2,8-diazobicyclo [4.3.0] nonane was found by using (R)-2-amino-2-phenyl-ethanol as chiral induction reagent. The entire synthetic process included 8 steps which were easy to operate with high yield. The purification method was only simple recrystallization or even used directly in the next step without further purification. The total yield was 29%.
| |
| 2013 Vol. 56 (3): 307-311 [Abstract] ( 22 ) [ PDF (518 KB) ] ( 92 ) [Supporting Information] DOI: 10.1007/s11426-012-4803-7 |
- Drugwatch. “Avelox Patent Family”. Retrieved 16 September 2012.
- “Details for NDA:021085″. DrugPatentWatch. Retrieved 17 July 2009.
- “www.accessdata.fda.gov”.
- “www.emea.europa.eu”.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021085s047,021277s041lbl.pdf
- “Avelox”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/1999/21085ltr.pdf
- http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21-085S010_Avelox_Approv.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2004/21085se1-022,21277se1-017ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s026,021277s022ltr.pdf.
- http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2005/021085s027,029,021277s024,025ltr.pdf.
- “Moxifloxacin: restricted use : MHRA”.
- European Medicines Agency (24 July 2008). “European Medicines Agency recommends restricting the use of oral moxifloxacin-containing medicines”. Retrieved 20 July 2009.
- “SYNOPSIS”. Retrieved 29 January 2009.
- Karande SC, Kshirsagar NA (February 1992). “Adverse drug reaction monitoring of ciprofloxacin in pediatric practice”. Indian Pediatr 29 (2): 181–8. PMID 1592498.
- Dolui SK, Das M, Hazra A (2007). “Ofloxacin-induced reversible arthropathy in a child”. Journal of Postgraduate Medicine 53 (2): 144–5. doi:10.4103/0022-3859.32220 .PMID 17495385.
- “Center for drug evaluation and research Application number 21-598″. Food and Drug Administration (FDA). 15 April 2005. Retrieved 21 July 2009.
- Jump up^ Babar, S. (October 2013). “SIADH Associated With Ciprofloxacin.”. The Annals of Pharmacotherapy (Sage Publiahing) 47 (10): 1359–1363.doi:10.1177/1060028013502457 . ISSN 1060-0280.PMID 24259701. Retrieved November 139,2013.
- Renata Albrecht (28 July 2004). “NDA 21-085/S-024, NDA 21-277/S-019″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (31 May 2007). “NDA 21-085/S-036, NDA 21-277/S-030″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Renata Albrecht (15 February 2008). “NDA 21-085/S-038, NDA 21-277/S-031″. Division of Special Pathogen and Transplant Products. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- DEPARTMENT OF HEALTH & HUMAN SERVICES (28 February 2008). “NDA 21-085/S-014, S-015, S-017″. Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Nea Zealand Government (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 30 January 2009.
- http://www.mhra.gov.uk/Safetyinformation/DrugSafetyUpdate/CON087781, retrieved 2012-11-10 Missing or empty
|title=(help) - http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - “EU agency recommends restricting moxifloxacin use”. Reuters. 24 July 2008. Retrieved 21 July 2009.
- Renata Albrecht (3 October 2008). “NDA 21-085/S-040, NDA 21-277/S-034″. Center for Drug Evaluation and Research. Food and Drug Administration (FDA). Retrieved 31 July 2009.
- Updated Labeling for Antibiotic Avelox (Moxifloxacin) Regarding Risk of Severe Liver Injury / Information Update: 2010-42 For immediate release 22 March 2010http://www.hc-sc.gc.ca/ahc-asc/media/advisories-avis/_2010/2010_42-eng.php
- Saint F, Gueguen G, Biserte J, Fontaine C, Mazeman E (September 2000). “[Rupture of the patellar ligament one month after treatment with fluoroquinolone]“. Rev Chir Orthop Reparatrice Appar Mot (in French) 86 (5): 495–7.PMID 10970974.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf, retrieved 2012-11-10 Missing or empty
|title=(help) - Bayer (December 2008). “AVELOX (moxifloxacin hydrochloride) Tablets AVELOX I.V. (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). p. 19. Retrieved 2 November 2010. Unknown parameter
|line=ignored (help) - “Moxifloxacin”. University of Maryland Medical Center. 2009. Retrieved 22 July 2009.
- http://www.akdae.de/Arzneimittelsicherheit/RHB/Archiv/2009/20090119.pdf
- Shin HC, Kim JC, Chung MK (September 2003). “Fetal and maternal tissue distribution of the new fluoroquinolone DW-116 in pregnant rats”. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 136 (1): 95–102.doi:10.1016/j.cca.2003.08.004 . PMID 14522602.
- Dan M, Weidekamm E, Sagiv R, Portmann R, Zakut H (February 1993). “Penetration of fleroxacin into breast milk and pharmacokinetics in lactating women” (PDF).Antimicrob. Agents Chemother. 37 (2): 293–6.doi:10.1128/AAC.37.2.293 . PMC 187655.PMID 8452360.
- Noel GJ, Bradley JS, Kauffman RE (October 2007). “Comparative safety profile of levofloxacin in 2523 children with a focus on four specific musculoskeletal disorders”.Pediatr. Infect. Dis. J. 26 (10): 879–91.doi:10.1097/INF.0b013e3180cbd382 . PMID 17901792.
- Joette Meyer; Renata Albrecht (16 March 2004).“Division of Special Pathogen and Immunologic Drug Products Summary of Clinical Review of Studies Submitted in Response to a Pediatric Written Request”. Food and Drug Administration (FDA). Retrieved 22 July 2009.
- Elbe DH, Chang SW (February 2005). “Moxifloxacin-warfarin interaction: a series of five case reports”. Ann Pharmacother 39 (2): 361–4. doi:10.1345/aph.1E179 .PMID 15632222.
- Karen E. Brown. “Top Ten Dangerous Drug Interactions in Long-Term Care”. Medication Management Project. Retrieved 1 August 2009.
- Stass HH (29 June – 3 July 1997). “Bay 12-8039 (BA) does not interact with theophylline (TH)”. Presented at the 20th International Congress of Chemotherapy.
- Medscape: Medscape Access
- http://69.20.19.211/cder/warn/dec99/wl121599.pdf
- Renata Albrecht (16 May 2002). “NDA 21-085/S-012″.Food and Drug Administration (FDA). Retrieved 17 July 2009.
- Drlica K, Zhao X (1 September 1997). “DNA gyrase, topoisomerase IV, and the 4-quinolones”. Microbiol Mol Biol Rev. 61 (3): 377–92. PMC 232616. PMID 9293187.
- “Drug card for Moxifloxacin (DB00218)”. Canada: DrugBank. 19 February 2009. Retrieved 3 August 2009.
- Alffenaar J. W. C., van Altena R., Bökkerink H. J (2009). “Pharmacokinetics of moxifloxacin in cerebrospinal fluid and plasma in patients with tuberculous meningitis”. Clinical Infectious Diseases 49 (7): 1080–2. doi:10.1086/605576 .PMID 19712035.
- “Inventors/Applicants”. patentlens.net. 3 October 2006. Retrieved 17 July 2009.
- Ed Lamb (1 May 2008). “Top 200 Prescription Drugs of 2007″. Pharmacy Times. Retrieved 21 July 2009.
- http://www.uspto.gov/web/offices/pac/dapp/opla/term/certs/4990517.pdf
- US 4990517 A (Feb 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+4990517#list US 5051509 A (Sep 1991) Nagano et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5051509#list US 5059597 A (Oct 1991) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5059597#list US 5395944 A (Mar 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5395944#list US 5416096 A (May 1995) Petersen et al. Seehttp://www.patentlens.net/patentlens/search_ajax.cgi?patnum=US+5416096#list
- Renata Albrecht (12 June 2002). “NDA 21-085/S-006, S-007 – NDA 21-277/S-002″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- The 114Th Medicines Adverse Reactions Committee Meeting; Professor P. Ellis, Professor D.C.G. Skegg, Dr M. Rademaker, Dr F. McClure, Dr N. Rafter, Dr M. Tatley (26 June 2003). “Adverse Reaction Reporting and IMMP”. New Zealand: Medsafe. Retrieved 12 August 2009.
- Renata Albrecht, (6 October 2003). “NDA 21-085/S-019 – NDA 21-277/S-011″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Renata Albrecht (6 October 2003). “NDA 21-085/S-039 NDA 21-277/S-033″. Food and Drug Administration(FDA). Retrieved 12 August 2009.
- Bayer HealthCare Pharmaceuticals Inc. (August 2008).“MEDICATION GUIDE AVELOX (AV-eh-locks) (moxifloxacin hydrochloride) Tablets AVELOX I.V. (AV-eh-locks) (moxifloxacin hydrochloride in sodium chloride injection)”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Ozlem Belen (24 June 2009). “NDA 21-085/S-041 NDA 21-277/S-035″. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- Bailey RR, Natale R, Linton AL (October 1972). “Nalidixic acid arthralgia”. Can Med Assoc J 107 (7): 604 passim.PMC 1940945. PMID 4541768.
- Bailey RR, Kirk JA, Peddie BA (July 1983). “Norfloxacin-induced rheumatic disease”. N Z Med J 96 (736): 590.PMID 6223241.
- Szarfman A, Chen M, Blum MD (January 1995). “More on fluoroquinolone antibiotics and tendon rupture” (PDF). N Engl J Med 332 (3): 193.doi:10.1056/NEJM199501193320319 . PMID 7800023.
- “Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399)”. Public Citizen. 1 August 1996. Retrieved on 27 December 2008.
- “Reports of adverse events with fluoroquinolones”. FDA Medical Bulletin 26 (3). October 1996.[dead link] Retrieved on 27 December 2008.
- “Madigan, Public Citizen, petition FDA for “black box” warning regarding potential adverse effects of certain popular antibiotics” (Press release). Office of the Illinois Attorney General. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen Petitions the FDA to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781)”. Public Citizen. 29 August 2006. Retrieved 27 December 2008.
- “Public Citizen v. Food and Drug Administration (FDA) (Fluoroquinolone)”. Public Citizen. 3 January 2008. Retrieved 27 December 2008.
- Ravn, Karen (18 August 2008). “Behind the FDA’s ‘black box’ warnings”. Los Angeles Times. Retrieved 27 December 2008.
- “FDA Requests Boxed Warnings on Fluoroquinolone Antimicrobial Drugs” (Press release). U.S. Food and Drug Administration. 8 July 2008. Retrieved 11 October 2008.
- “Drugs@FDA”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- “MedWatch: The FDA Safety Information and Adverse Event Reporting Program”. Food and Drug Administration (FDA). Retrieved 12 August 2009.
- MacCarthy, Paul (22 October 2008). “Important Change in the Avelox (moxifloxacin hydrochloride) and Cipro (ciprofloxacin) Complete Prescribing Information – Addition of Boxed Warning and Medication Guide Regarding Tendinitis and Tendon Rupture”. Bayer HealthCare Pharmaceuticals. Retrieved 27 December 2008.
- Bayer AG (6 November 2007). “Ruling in Bayer’s favor over Avelox patents”. Bayer. Retrieved 29 August 2009.
- http://www.orangebookblog.com/Bayer_20v._20Dr._20Reddy_27s.pdf
- “United States Court of Appeals for the Federal Circuit”. uscourts.gov. 28 June 2007. Retrieved 29 August 2009.
- Bayer AG (24 April 2008). “Risk Report”. Bayer. Retrieved 29 August 2009.
- In The United States District Court For The District Of Columbia Public Citizen, Inc. VS. Food And Drug Administration 3 January 2008
- Office of the Attorney General State of Illinois Lisa Madigan Citizen Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics 18 May 2005
- Public Citizen’s Petition to Include a Black Box Warning on Fluoroquinolone Antibiotics (HRG Publication #1781) 29 August 2006
- Public Citizen’s Petition to Require a Warning on All Fluoroquinolone Antibiotics (HRG Publication #1399) 1 August 1996
- Public Citizen’s Petition to Ban the Antibiotic Gatifloxacin (Tequin) (HRG Publication #1768)
- Public Citizen’s Petition to immediately ban the antibiotic Trovafloxacin (Trovan). (HRG Publication #1485) Date: 3 June 1999
- Public Citizen’s Petition to immediately stop the distribution of dangerous, misleading prescription drug information to the public. HRG Publication #1442 Date: 9 June 1998
- June 2004, A petition To the United States Congress to immediately take action to protect consumers from the reckless and negligent abuses of the FDA and the following Pharmaceutical Companies: Bayer, Ortho-McNeill, Pfizer, Merck, Bristol-Myers Squibb, Sanofi Winthrop, Bertek Pharmaceuticals – Rhone-Poulenc Rorer and Barr. These companies manufacture and distribute fluoroquinolone antibiotics in the United States in a manner that fails to warn of serious adverse event risks, and downplays and fails to warn physicians of the serious risks associated with fluoroquinolone therapy.
| EP0350733A2 | Jun 30, 1989 | Jan 17, 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0591808A1 | Sep 27, 1993 | Apr 13, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| EP0592868A1 | Sep 29, 1993 | Apr 20, 1994 | Bayer Ag | Quinolonecarboxylic acids |
| US5849752 | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO1999026940A2 | Nov 12, 1998 | Jun 3, 1999 | Bayer Ag | Method for producing 8-methoxy-quinoline carboxylic acids |
| WO2004091619A1 | Apr 9, 2004 | Oct 28, 2004 | Reddys Lab Ltd Dr | A crystalline form iii of anhydrous moxifloxacin hydrochloride and a process for preparation thereof |
| WO2007010555A2 | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| CN102030751B | Dec 1, 2010 | Nov 21, 2012 | 上虞京新药业有限公司 | Process for crystallizing moxifloxacin hydrochloride |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
| WO2005012285A1 * | Aug 5, 2004 | Feb 10, 2005 | Satyanarayana Chava | An improved process for the preparation of moxifloxacin hydrochloride |
| EP0443498A1 * | Feb 18, 1991 | Aug 28, 1991 | Kyorin Pharmaceutical Co., Ltd. | Isoindoline derivatives |
| EP0550903A1 * | Dec 28, 1992 | Jul 14, 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| US6323213 * | Feb 12, 1997 | Nov 27, 2001 | Bayer Aktiengesellschaft | Possibly substituted 8-cyano-1-cyclopropyl-7-(2,8-diazabicyclo-[4.3.0]-nonan-8-yl)-6-fluoro-1,4-dihydro-4-oxo-3-quinolin carboxylic acids and their derivatives |
| US5849752 * | Dec 5, 1996 | Dec 15, 1998 | Bayer Aktiengesellschaft | Crystal modification of CDCH a process for its preparation and pharmaceutical formulations comprising this modification |
| WO2007010555A2 * | Jul 13, 2006 | Jan 25, 2007 | Ramprasad Achampeta Kodanda | Novel crystalline forms of moxifloxacin hydrochloride and process for preparation thereof |
| WO2008059521A2 * | Sep 27, 2007 | May 22, 2008 | Ramprasad Achampeta Kodanda | Novel process for the preparation of moxifloxacin hydrochloride and a novel polymorph of moxifloxacin |
| WO2008138759A1 * | Apr 30, 2008 | Nov 20, 2008 | Sandoz Ag | Process for the preparation of moxifloxacin hydrochloride |
| WO2007148137A1 * | Jun 22, 2007 | Dec 27, 2007 | Generics Uk Ltd | Novel hydrate form of moxifloxacin monohydrochloride |
| WO2008028959A1 * | Sep 7, 2007 | Mar 13, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin hydrochloride |
| WO2008059223A2 * | Nov 13, 2007 | May 22, 2008 | Cipla Ltd | Process for the synthesis of moxifloxacin hydrochloride |
| WO2008095964A1 * | Feb 6, 2008 | Aug 14, 2008 | Sint Quimica Sa | Crystalline form of moxifloxacin base |
| WO2009087151A1 * | Jan 7, 2009 | Jul 16, 2009 | Chemo Iberica Sa | Polymorphic forms of moxifloxacin hydrochloride and processes for preparation thereof |
| CN102603738B | Feb 24, 2012 | Dec 11, 2013 | 天津市汉康医药生物技术有限公司 | 一种稳定的盐酸莫西沙星化合物 |
| EP2083010A1 | Jan 8, 2008 | Jul 29, 2009 | Chemo Ibérica, S.A. | Polymorphic Forms of Moxifloxacin hydrochloride and processes for preparation thereof |
| EP2154137A1 | Aug 4, 2008 | Feb 17, 2010 | Chemo Ibérica, S.A. | Crystalline form of moxifloxacin base |
| US8198451 | Nov 13, 2007 | Jun 12, 2012 | Cipla Limited | Process for the synthesis of moxifloxacin hydrochloride |
| US20110212990 * | Nov 6, 2008 | Sep 1, 2011 | Hetero Research Foundation | Novel polymorph of moxifloxacin hydrochloride |
4.NEMONOXACIN























Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
- C20-H25-N3-O4
- 371.4345
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
Taigexyn has been approved in Taiwan IN 2014
"TAIPEI, MARCH 13, 2014 /PRNEWSWIRE/ -- TAIGEN BIOTECHNOLOGY ..."
13.03.14 |
13.03.14 |
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ -- TaiGen Biotechnology Company, Limited ("TaiGen") today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
Taigexyn Granted QIDP and Fast Track Designations
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.
Malate salt
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.
..........................
isomeric compounds are:
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1.......DESIRED
(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1'....NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1') were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9'): Compound 9' was synthesized as shown in Scheme 1 below:
Scheme 1
3 4 Boc
A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck's Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9') was synthesized as shown in Scheme 2.
Scheme 2
HN Boc HN Boc
NaBH4,EtOH w - " MsCI1TEA . „ _. - - _. „ Benzyl Amine
THF EA1CoId
(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3
Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1')
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4
5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50"-65"C filter
A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1' was synthesized from Compound 9' as shown in Scheme 5 below:
Scheme 5
(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1'....NOT DESIRED
.....................
US2007/232650 A1,
malate salts of
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)
(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck's Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)
Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)
A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)
A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
....................
REF
A. ARJONA ET AL: "Nemonoxacin", DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: "TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia", INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: "TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)", 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: "TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic", 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
| US8211909 | 7-4-2012 | TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION |
| US8158798 | 4-18-2012 | Coupling Process For Preparing Quinolone Intermediates |
| US8039485 | 10-19-2011 | Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| US2010152452 | 6-18-2010 | STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES |
| US2010041697 | 2-19-2010 | PNEUMONIA TREATMENT |
| US7528264 | 5-6-2009 | Hydride reduction process for preparing quinolone intermediates |
| US2009042932 | 2-13-2009 | ANTIMICROBIAL PARENTERAL FORMULATION |
| US7456279 | 11-26-2008 | Coupling process for preparing quinolone intermediates |
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798
5.
NADIFLOXACIN
Nadifloxacin
OPC-7251, Nadixa, Nadoxin, Acuatim
CAS 124858-35-1
9-Fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo[ij]quinolizine-2-carboxylic acid
9-fluoro-8-(4-hydroxy-1-piperidyl)-5-methyl-6,7-dihydro-1-oxo-1H,5H-benzo[ij]quinolizine-2-carboxylic acid.
9-fluoro-6,7-dihydro-8-(4-hydroxy-l-pyperidinyl)-5-methyl- l-oxo-lH,5H-benzo(I,j)quinolizine-2-carboxylic acid
jinofloxacin
- (+-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidino)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylic acid
- CCRIS 4066
- Jinofloxacin
- Nadifloxacin
- Nadifloxacine
- Nadifloxacine [INN-French]
- Nadifloxacino
- Nadifloxacino [INN-Spanish]
- Nadifloxacinum
- Nadifloxacinum [INN-Latin]
- Nadixa
- OPC-7251
- S-Nadifloxacin
- UNII-6CL9Y5YZEQ
Acuatim (Otsuka)
Molecular Formula: C19H21FN2O4, 360.38
C 63.32%, H 5.87%, F 5.27%, N 7.77%, O 17.76%
Properties: Colorless prisms from EtOH-H2O, mp 245-247° (dec). LD50 male, female mice and rats (mg/kg): 376.5, 420.6, 225.7, 240.5 i.v. (Hashimoto).
mp 245-247° (dec
Antibacterial (topical).
(R)-isomer does not induce chromosomal aberrations, unlike (S)-isomer.
NOTE... LEVONADIFLOXACIN IS IN PHASE 2
LAUNCHED 1993 OTSUKA FOR ACNE
CLINICAL TRIALS.......http://clinicaltrials.gov/search/intervention=NADIFLOXACIN
Nadifloxacin, a novel topical fluoroquinolone, was initially launched in 1993 by Otsuka for the topical treatment of acne. It has since been marketed as an ointment for the treatment of bacterial infection. Originally developed at Otsuka, nadifloxacin is manufactured, distributed and marketed by the company in collaboration with Pfleger, Ferrer and Galderma.
Nadifloxacin is chemically, 9-fluoro-6,7-dihydro-8-(4-hydroxy-l-pyperidinyl)-5-methyl- l-oxo-lH,5H-benzo(I,j)quinolizine-2-carboxylic acid of Formula I provided below.
FORMULA I Nadifloxacin is a synthetic quinolone with potent broad-spectrum anti-bacterial activity. Nadifloxacin inhibits the enzyme DNA gyrase that is involved in bacterial DNA synthesis and replication, thus inhibiting the bacterial multiplication. RS-nadifloxacin and S-nadifloxacin, in particular, exhibit strong antibacterial activity against Gram-positive, Gram-negative and anaerobic bacteria, resistant Gram-positive organisms such as methicillin-resistant Staphylococcus aureus (MRSA), quinolone-resistant Staphylococcus aureus, coagulase negative staphylococci, such as methicillin-resistant Staphylococcus epidermidis (MRSE), enterococci, betahemolytic streptococci and viridans group of streptococci, mycobacteria and newly emerging nosocomial pathogens such as Chryseobacterium meninges epticum, and Gram-negative pathogens such as E.coli, Klebsiella, Proteus, Serratia, Citrobacter and Pseudomonas. Recently, it has also been shown that S-(-)-nadifloxacin, in particular exhibits potent antibacterial activity against glycopeptide intermediate S. aureus (GISA), vancomycin intermediate S. aureus (VISA) and vancomycin-resistant S. aureus (VRSA). Nadifloxacin is also active against quinolone-resistant Staphylococci.
Nadifloxacin is marketed in the form of cream for topical application for the treatment of acne vulgaris, folliculitis and sycosis vulgaris. It is also indicated for the treatment of topical bacterial infections with susceptible bacteria.
The use of quinolone antibiotics to treat infections is known art in the field of ophthalmic pharmaceutical compositions and methods of treatment. Several quinolone antibacterial agents available in the market include gatifloxacin (available as Zymar®), Levofloxacin (available as Quixin® or Iquix®), Ciprofloxacin (available as Ciloxan®), Ofloxacin (available as Ocuflox®), Lomefloxacin (available as Lomeflox®), Moxifloxacin (available as Vigamox®) and Norfloxacin (available as Chibroxin®).
U.S. Patent No. 4,844,902 discloses a topically applicable formulation comprising by weight about 0.01 to 30% of an anti-bacterially active compound, 0.01 to 10% of a corticosteroid and a carrier. U.S. Patent No. 6,333,045 discloses liquid pharmaceutical compositions of gatifloxacin or salt thereof and disodium edetate.
U.S. Patent No. 6,716,830 discloses ophthalmic dosage forms of moxifioxacin or salts thereof in a concentration of 0.1% to 1% (w/w) and pharmaceutically acceptable vehicle.
U.S. Patent No. 6,359,016 relates to topical suspension formulations containing ciprofloxacin and dexamethasone.
U.S. Patent No 4,399,134 discloses processes for the preparation of nadifloxacin or salts thereof and antibacterially effective pharmaceutical compositions of nadifloxacin. Typical dosage forms include tablets, pills, powders, liquid preparations, suspensions, emulsions, granules, capsules, suppositories, and injectable preparations (solutions, suspensions, etc).
U.S. Patent No 6,884,768 discloses solid oral pharmaceutical compositions that includes nadifloxacin, an absorbefacient and taurine compounds.
U.S. Patent Application 20060183698 describes topical ophthalmic formulation that includes serum electrolytes; an antimicrobial compound and an anti-inflammatory or steroidal compound. Several antimicrobial agents have been disclosed including nadifloxacin.
U.S. Patent Application 20040176337 discloses topical . compositions of benzoquinolizine-2-carboxylic acid antimicrobial drug.
U.S. Patent Application 20040176321 discloses injectable pharmaceutical composition for intravenous delivery of an active agent that includes RS-(±)-nadifloxacin; S-(-)- nadifloxacin and hydrates thereof; or S~(-)-nadifloxacin arginine and salts thereof. PCT Publication WO 04/00360 describes pharmaceutical compositions of several active ingredients including nadifloxacin for topical use for treatment of dermatosis.
European Patent EP 275,515 and U.S. Patent No. 4,923,862 disclose aqueous pharmaceutical compositions of levofloxacin and ofloxacin or salts thereof.
PCT application WO 02/39993 discloses a stable pharmaceutical preparation of a combination drug, comprising an anti-infective agent, selected from the group consisting of quinolone derivatives, amino-glycoside derivatives and their pharmaceutically acceptable salts; an ant-inflammatory agent which is a corticosteroid; a complexation enhancing polymer; a solubilizer exhibiting an inclusion phenomena; pharmaceutically acceptable excipients within a suitable carrier system.
Journal of Ocular Pharmacology and Therapeutics, vol 23(3): 243-256, 2007 discloses (7- [(3R)-3 -aminohexahydro- 1 H-azepine- 1 -yl]-8-chloro- 1 -cyclopropyl-6-fluoro- 1 ,4-dihydro- 4-oxo-3-quinolinecarboxylivc acid as the topical agent for the treatment of ophthalmic infections.
....................
JP 1983090511
The bromination of 5-fluoro-2-methylquinoline (I) with Br2 and Ag2SO4 in H2SO4 or with Br2 and AlCl3 gives 5-bromo-6-fluoro-2-methylquinoline (II), which is reduced with H2 over PtO2 in acetic acid, yielding 5-bromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline (III). The cyclization of (III) with diethyl ethoxymethylenemalonate (IV) and polyphosphoric acid (PPA) at 150 C affords 8-bromo-9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-benzo[i,j]quinolizine-2-carboxylic acid (V), which is finally condensed with 4-hydroxypiperidine (VI) by heating at 160 C in HMPT.
6-Fluoro-2-methylquinoline (I)
5-Bromo-6-fluoro-2-methylquinoline (II)
5-Bromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline (III)
diethyl 2-[(E)-ethoxymethylidene]succinate (IV)
8-Bromo-9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-pyrido[3,2,1-ij]quinoline-2-carboxylic acid (V)
4-Piperidinol; 4-Hydroxypiperidine
5-Bromo-6-fluoro-2-methylquinoline (II)
5-Bromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline (III)
diethyl 2-[(E)-ethoxymethylidene]succinate (IV)
8-Bromo-9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-pyrido[3,2,1-ij]quinoline-2-carboxylic acid (V)
4-Piperidinol; 4-Hydroxypiperidine
The synthesis of Ro 40-7592 is carried out as follows: Addition of 4-bromotoluene (I) to 4-(benzyloxy)-3-methoxybenzaldehyde (II) in the presence of butyllithium in THF at -78 C gives 4-(benzyloxy)-3-methoxy-4'-methylbenzhydrol (III). Oxidation of this compound with pyridinium chlorochromate in CH2Cl2 yields the corresponding 4-(benzyloxy)-3-methoxy-4'-methylbenzophenone (IV). Debenzylation of (IV) with 30% aqueous hydrobromic acid in acetic acid affords 4-hydroxy-3-methoxy-4'-methylbenzophenone (V). Regioselective nitration of (V) with 65% aqueous nitric acid in acetic acid gives 4-hydroxy-3-methoxy-4'-methyl-5-nitrobenzophenone (VI). Hydrolysis of the methoxy group in (VI) with 30% aqueous hydrobromic acid in boiling acetic acid affords 3,4-dihydroxy-4'-methyl-5-nitrobenzophenone, Ro 40-7592.
..................
EXAMPLE 1In a 100 ml flask were placed 7.5 g of 9-fluoro-8-bromo-5-methyl-6,7-dihydro-1-oxo-1H,5H-benzo-[ij]quinolizine-2-carboxylic acid, 9.5 g of 4-hydroxypiperidine and 60 ml of N-methyl-pyrrolidone and the mixture was stirred at 150 nitrogen gas atmosphere. After 6.5 hours disappearance of the starting materials was confirmed by thin layer chromatography, and N-methylpyrrolidone and 4-hydroxypiperidine were removed using an aspirator at a bath temperature of 140 residue were added dimethylformamide, ethanol and water and the mixture was allowed to stand overnight. On the next day, 1.6 g of crystals were obtained which were recrystallized twice each from ethanol-water to give 1.05 g of 9-fluoro-8-(4-hydroxy-1-piperidyl)-5-methyl-6,7-dihydro-1-oxo-1H,5H-benzo[ij]quinolizine-2-carboxylic acid. m.p. 244
______________________________________Elemental Analysis for C.sub.19 H.sub.21 N.sub.2 O.sub.4 F C H N______________________________________Calc'd (%): 63.32 5.87 7.78Found (%): 63.28 5.76 7.89______________________________________
References:
Fluorinated quinolone antibacterial. Prepn: H. Ishikawa et al., BE 891046; eidem, US 4399134 (1982, 1983 both to Otsuka); eidem, Chem. Pharm. Bull. 37, 2103 (1989).
Toxicity data: K. Hashimoto et al., Iyakuhin Kenkyu 21, 671 (1990), C.A. 114, 156625r (1991).
In vitro antibacterial activity: K. Vogt et al., Eur. J. Clin. Microbiol. Infect. Dis. 11, 943 (1992).
HPLC determn: M. Koike et al., J. Chromatogr. 526, 235 (1990).
Clinical trial in treatment of acne: I. Kurokawa et al., J. Am. Acad. Dermatol. 25, 674 (1991).
6 ZABOFLOXACIN
zabofloxacin, 219680-11-2
UNII-LV66BA6V2G, DW-224a
Molecular Formula: C19H20FN5O4
Molecular Weight: 401.391603
DONG WHA PHARMA SOUTH KOREA in phase 3
1-Cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxylic acid
Zabofloxacin is being developed as a new fluoroquinolone antibiotic that is a potent and selective inhibitor of the essential bacterial type II topoisomerases and topoisomerase IV. Zabofloxacin is indicated for community-acquired respiratory infections due to Gram-positive bacteria. The aim of this study was to compare the pharmacokinetics (PK) of the zabofloxacin hydrochloride 400 mg capsule (DW224a, 366.7 mg aszabofloxacin) with the PK of the zabofloxacin aspartate 488 mg tablet (DW224aa, 366.5 mg as zabofloxacin) in healthy Korean male volunteers to assess the bioequivalence between the two drug formulations
Zabofloxacin hydrochloride is a fluoroquinolone antibiotic with enhanced in vitro activity against Streptococcus pneumoniae, including strains resistant to other antibiotics. The spectrum of activity of zabofloxacin includes bacterial strains that are responsible for most community-acquired respiratory infections. Phase III clinical studies are currently ongoing at Dong-Wha for the treatment of patients with acute bacterial exacerbation of chronic obstructive pulmonary disease. Phase II trials had been ongoing at IASO; however no recent developments have been reported.The product candidate was originated by Dong Wha. In 2007, Dong Wha granted PB BioSciences worldwide exclusive development and marketing rights, except in Japan, Korea, China, Taiwan, Singapore, Indonesia, India, Thailand, Malaysia, Vietnam, Hong Kong, Australia and New Zealand.
Zabofloxacin was separated using an isocratic elution on a Capcell Pak C18 column using an acetonitrile–methanol–phosphate buffer (1 g of KH2PO4 and 1 g of heptane sulfonic acid sodium salt in 720 mL of purified water) and a 1 M tetrabutylammonium dihydrogenphosphate solution (18.5:8.5:72:1, by volume) as a mobile phase at a flow rate of 0.25 mL/min with UV detection at 275 nm. The lower limit of quantification (LLOQ) and the upper limit of quantification (ULOQ) were 100 ng/mL and 20000 ng/mL, respectively, with acceptable linearity in the range from 100 to 20000 ng/mL (R > 0.999). The intra- and inter-day accuracy (RE) ranged from −8.2% to 1.8% and the intra- and inter-day precision (CV) ranged from 3.8% to 10.6% for zabofloxacin. In addition, stock solution stability, recovery, freeze–thaw effects, and short-term and long-term stability met the acceptance criteria.
..............................
Example 1. l-Cyclopropyl-6-fluoro-7-[8-(methoxyimino)-2,6-diazaspiro[3,4]oct-6-yl]-4- oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid methanesulfonate
30 350mg of
7-[2-(t-buthoxycarbonyl)-8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-l- cyclopropyl-6-fluoro-4-oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid was dissolved in 5ml of dichloromethane and thereto 0.6ml of trifluoroacetic acid was dropped. The mixture was stirred for 5 hours at room temperature and thereto 10ml (if ethylether was added. It was stirred additionally for 1 hour and thus precipitated solid was filtered, dissolved in 5ml of diluted NaOH and neutralized with diluted hydrochloric acid. The precipitate thus obtained was filtered and dried. The resulting solid was added to 5ml of lN-methanesulfonic acid in ethanol and stirred for 1 hour. Thus obtained precipitate was filtered and dried to give 185g of the titled compound(yield : 47.8%). m.p. : 228- 229 °C
1H-NMR(DMSO-dG+CF3COOD, ppm): 0.97(s, 2H), 1.14(d, 2H), 2.48(s, 3H), 3.57(bs, IH), 3.88(s, 3H), 4.06-4.17(m, 411), 4.40(s, 2H), 4.49(s, 2H), 7.88(d, Hi, J=12.67Hz), 8.49(s, IH).
......................................
US20100184795
aspartate of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid comprises a step of reacting 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid with aspartic acid in a solvent. The method can be represented by Scheme 1.
 Example 1 Preparation of the D-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
Example 1 Preparation of the D-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (5.0 g) was added to 50% ethanol (80 mL), and then the mixture was stirred at 50° C. for 10 minutes. D-Aspartic acid (2.0 g) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature, and then the resulting solid was collected by filtration. Ethanol (100 mL) was added to the filtrate, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain a total of 5.55 g of the target compound (yield: 83%). Melting point: 200-201° C. 1H NMR (D2O): δ 0.97 (bs, 2H), 1.27 (d, 2H), 2.00 (dd, 1H, J=8.8, 17.6 Hz), 2.77 (dd, 1H, J=3.3, 17.0 Hz), 3.53 (bs, 1H), 3.84 (dd, 1H, J=3.3, 8.78 Hz), 4.01 (s, 3H), 4.31-4.45 (m, 8H), 7.46 (d, 1H, J=12.2 Hz), 8.42 (s, 1H).
Example 2 Preparation of L-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (500 mg) was added to 50% ethanol (20 mL), and then the mixture was stirred at 50° C. for 10 minutes. L-Aspartic acid (174 mg) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature. Ethanol (20 mL) was added to the reaction mixture, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 550 mg of the target compound (yield: 82%). Melting point: 205-206° C. 1H NMR (d6-DMSO): δ 0.93 (d, 2H, J=3.5 Hz), 1.20 (d, 2H, J=6.8 Hz), 2.42 (dd, 1H, J=9.2, 17.3 Hz), 2.59 (dd, 1H, J=3.3, 17.2 Hz), 3.50 (m, 1H), 3.59 (1H, dd, J=3.1, 9.1 Hz), 3.91 (s, 3H), 4.24 (m, 6H), 4.41 (br, 2H), 7.59 (d, 1H, J=12.4 Hz), 8.41 (s, 1H).
Example 3 Preparation of Hydrochloric Acid Salt, Phosphate Salt, and Formate Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
3-1 Hydrochloric Acid Salt
Ethanol (3 mL) was cooled to 0° C. and acetyl chloride (1.13 mL) was added, and then the mixture was stirred for 30 minutes. 1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (800 mg) was added to the reaction mixture, and then stirred at 0° C. for 30 minutes. Tetrahydrofuran (4 mL) was added, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration and dried to obtain 776 mg of the target compound (yield: 89%). Melting point: 244-245° C. 1H NMR (d6-DMSO): δ 1.07 (d, 2H, J=4.7 Hz), 1.21 (d, 2H, J=6.8 Hz), 3.68 (m, 1H), 3.94 (s, 3H), 4.17 (m, 2H), 4.40 (s, 2H), 4.53 (s, 2H), 8.03 (d, 1H, J=12.5 Hz), 8.59 (s, 1H).
30 350mg of
7-[2-(t-buthoxycarbonyl)-8-(methoxyimino)-2,6-diazaspiro[3.4]oct-6-yl]-l- cyclopropyl-6-fluoro-4-oxo-l,4-dihydro[l,8]naphthyridine-3-carboxylic acid was dissolved in 5ml of dichloromethane and thereto 0.6ml of trifluoroacetic acid was dropped. The mixture was stirred for 5 hours at room temperature and thereto 10ml (if ethylether was added. It was stirred additionally for 1 hour and thus precipitated solid was filtered, dissolved in 5ml of diluted NaOH and neutralized with diluted hydrochloric acid. The precipitate thus obtained was filtered and dried. The resulting solid was added to 5ml of lN-methanesulfonic acid in ethanol and stirred for 1 hour. Thus obtained precipitate was filtered and dried to give 185g of the titled compound(yield : 47.8%). m.p. : 228- 229 °C
1H-NMR(DMSO-dG+CF3COOD, ppm): 0.97(s, 2H), 1.14(d, 2H), 2.48(s, 3H), 3.57(bs, IH), 3.88(s, 3H), 4.06-4.17(m, 411), 4.40(s, 2H), 4.49(s, 2H), 7.88(d, Hi, J=12.67Hz), 8.49(s, IH).
......................................
US20100184795
aspartate of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid comprises a step of reacting 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid with aspartic acid in a solvent. The method can be represented by Scheme 1.
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (5.0 g) was added to 50% ethanol (80 mL), and then the mixture was stirred at 50° C. for 10 minutes. D-Aspartic acid (2.0 g) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature, and then the resulting solid was collected by filtration. Ethanol (100 mL) was added to the filtrate, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain a total of 5.55 g of the target compound (yield: 83%). Melting point: 200-201° C. 1H NMR (D2O): δ 0.97 (bs, 2H), 1.27 (d, 2H), 2.00 (dd, 1H, J=8.8, 17.6 Hz), 2.77 (dd, 1H, J=3.3, 17.0 Hz), 3.53 (bs, 1H), 3.84 (dd, 1H, J=3.3, 8.78 Hz), 4.01 (s, 3H), 4.31-4.45 (m, 8H), 7.46 (d, 1H, J=12.2 Hz), 8.42 (s, 1H).
Example 2 Preparation of L-Aspartic Acid Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (500 mg) was added to 50% ethanol (20 mL), and then the mixture was stirred at 50° C. for 10 minutes. L-Aspartic acid (174 mg) was added and then the mixture was stirred at 50° C. for 1 hour. The mixture was cooled to room temperature. Ethanol (20 mL) was added to the reaction mixture, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration to obtain 550 mg of the target compound (yield: 82%). Melting point: 205-206° C. 1H NMR (d6-DMSO): δ 0.93 (d, 2H, J=3.5 Hz), 1.20 (d, 2H, J=6.8 Hz), 2.42 (dd, 1H, J=9.2, 17.3 Hz), 2.59 (dd, 1H, J=3.3, 17.2 Hz), 3.50 (m, 1H), 3.59 (1H, dd, J=3.1, 9.1 Hz), 3.91 (s, 3H), 4.24 (m, 6H), 4.41 (br, 2H), 7.59 (d, 1H, J=12.4 Hz), 8.41 (s, 1H).
Example 3 Preparation of Hydrochloric Acid Salt, Phosphate Salt, and Formate Salt of 1-cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid
3-1 Hydrochloric Acid Salt
Ethanol (3 mL) was cooled to 0° C. and acetyl chloride (1.13 mL) was added, and then the mixture was stirred for 30 minutes. 1-Cyclopropyl-6-fluoro-7-(8-methoxyimino-2,6-diaza-spiro[3.4]oct-6-yl)-4-oxo-1,4-dihydro-[1,8]naphthyridine-3-carboxylic acid (800 mg) was added to the reaction mixture, and then stirred at 0° C. for 30 minutes. Tetrahydrofuran (4 mL) was added, and then the mixture was stirred for 30 minutes. The resulting solid was collected by filtration and dried to obtain 776 mg of the target compound (yield: 89%). Melting point: 244-245° C. 1H NMR (d6-DMSO): δ 1.07 (d, 2H, J=4.7 Hz), 1.21 (d, 2H, J=6.8 Hz), 3.68 (m, 1H), 3.94 (s, 3H), 4.17 (m, 2H), 4.40 (s, 2H), 4.53 (s, 2H), 8.03 (d, 1H, J=12.5 Hz), 8.59 (s, 1H).
ref
Han J, Kim JC, Chung MK, Kim B, Choi DR.
Biol Pharm Bull. 2003 Jun;26(6):832-9
Jin HE, Kang IH, Shim CK.
J Pharm Pharm Sci. 2011;14(3):291-305.
Jin HE, Lee KR, Kang IH, Chung SJ, Shim CK.
J Pharm Biomed Anal. 2011 Mar 25;54(4):873-7. doi: 10.1016/j.jpba.2010.11.001. Epub 2010 Nov 9.
Kosowska-Shick, K.; Credito, K.; Pankuch, G.A.; Lin, G.; Bozdogan, B.; McGhee, P.; Dewasse, B.;
Choi, D.-R.; Ryu, J.M.; Appelbaum, P.C. Antipneumococcal activity of DW-224a, a new
quinolone, compared to those of eight other agents. Antimicrob. Agents Chemother. 2006, 50,
2064–2071.
Choi, D.-R.; Ryu, J.M.; Appelbaum, P.C. Antipneumococcal activity of DW-224a, a new
quinolone, compared to those of eight other agents. Antimicrob. Agents Chemother. 2006, 50,
2064–2071.
Park, H.-S.; Kim, H.-J.; Seol, M.-J.; Choi, D.-R.; Choi, E.-C.; Kwak, J.-H. In vitro and in vivo
antibacterial activities of DW-224a, a new fluoronaphthyridone. Antimicrob. Agents Chemother.
2006, 50, 2261–2264.
antibacterial activities of DW-224a, a new fluoronaphthyridone. Antimicrob. Agents Chemother.
2006, 50, 2261–2264.
Dong Wha Pharmaceutical Co. Ltd. A study to evaluate efficacy and safety profile of
Zabofloxacin tablet 400 mg and moxifloxacin tablet 400 mg. Available online:
http://www.clinicaltrials.gov/ct2/show/NCT01658020 (accessed on 15 July 2013).
Zabofloxacin tablet 400 mg and moxifloxacin tablet 400 mg. Available online:
http://www.clinicaltrials.gov/ct2/show/NCT01658020 (accessed on 15 July 2013).
Dong Wha Pharmaceutical Co. Ltd. A new quinolone antibiotic. Available online:
http://www.dong-wha.co.kr/english/rnd/rnd02_03.asp (accessed on 15 April 2013).
http://www.dong-wha.co.kr/english/rnd/rnd02_03.asp (accessed on 15 April 2013).
| US4957922 * | Mar 29, 1989 | Sep 18, 1990 | Bayer Aktiengesellschaft | Infusion solutions of 1-cyclopropyl-6-fluoro-1,4-di-hydro-4-oxo-7-(1-piperazinyl)-quinoline-3-carboxylic acid |
| US5563149 * | Aug 8, 1994 | Oct 8, 1996 | Cheil Foods & Chemicals, Inc. | Aqueous solutions of pyridone carboxylic acids |
| US6552196 * | Sep 6, 2001 | Apr 22, 2003 | Dong Wha Pharmaceutical Industrial Co., Ltd. | Quinolone carboxylic acid derivatives |
7-23-2010
|
ASPARTATE OF 1-CYCLOPROPYL-6-FLUORO-7-(8-METHOXYIMINO-2,6-DIAZA-SPIRO[3.4]OCT-6-YL)-4-OXO-1,4-DIHYDRO-[1,8]NAPHTHYRIDINE-3-CARBOXYLIC ACID, METHOD FOR PREPARING THE SAME, AND ANTIMICROBIAL PHARMACEUTICAL COMPOSITION COMPRISING THE SAME
|
7 DS 8587
DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
- [Reference Example 71]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester


- [(3S)-3-(3-Hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (46 g) and imidazole (11.9 g) were dissolved in dimethylformamide (600 mL). After addition of tert-butyldimethylsilyl chloride (23.2 g) under ice-cooling, the mixture was stirred at room temperature for 59.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 2:1) to give 29.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.11 Hz), 3.58 (2H, t, J=6.13 Hz), 3.34 (1H, d, J=10.05 Hz), 3.12 (1H, d, J=10.05 Hz), 2.94 (1H, d, J=16.91 Hz), 2.31 (1H, d, J=17.16 Hz), 1.86-1.74 (1H, m), 1.72-1.62 (1H, m), 1.51 (3H, d, J=7.11 Hz), 1.49-1.24 (2H, m), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (30 g) was dissolved in tetrahydrofuran (280 mL), and the atmosphere was replaced with argon. Then, lithium hexamethyldisilazide (1.0 M solution in tetrahydrofuran) (78.0 mL) was added dropwise at -15°C, and the mixture was stirred at -5°C for 30 minutes. After cooling to -15°C again, a solution of N-fluorobenzenesulfonimide (26.6 g) in tetrahydrofuran (220 mL) was added dropwise, and the mixture was stirred at room temperature for 17 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2) to give 8.15 g of the title compound as a pale yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.53-5.44 (1H, m), 5.18 (1H, d, J=51.72 Hz), 3.64-3.52 (2H, m), 3.32-3.19 (2H, m), 1.92-1.65 (2H, m), 1.55 (3H, d, J=4.66 Hz), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
MS (FAB) m/z: 480 (M+H)+.
IR (ATR) ν: 3421, 2977, 2935, 2877, 1698, 1454, 1369, 1309, 1249, 1153, 1058, 1035, 1006, 842 cm-1.
[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (8.15 g) was dissolved in tetrahydrofuran (25.0 mL). Acetic acid (22.0 mL) and tetrabutylammonium fluoride (1.0 M solution in tetrahydrofuran) (25.0 mL) were added under ice-cooling, and the mixture was stirred at room temperature for 21.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 1:1) to give 5.77 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.03 Hz), 5.20 (1H, d, J=51.48 Hz), 3.69-3.59 (2H, m), 3.31-3.21 (2H, m), 1.95-1.72 (2H, m), 1.68-1.43 (2H, m), 1.56 (3H, d, J=7.11 Hz), 1.33 (9H, s).
[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester

- (3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (12.20 g) was dissolved in dichloromethane (400 mL). Benzenesulfonyl chloride (9.06 mL), triethylamine (10.7 mL), and 4-dimethylaminopyridine (2.04 g) were added under ice-cooling, and the mixture was stirred at room temperature for 12.5 hours. Saturated sodium bicarbonate water was added to the reaction solution, and the mixture was stirred for 30 minutes, followed by extraction with dichloromethane. The organic layer was sequentially washed with a 10% citric acid solution and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 8:2 -> 1:1) to give 11.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.94 - 7.87 (2H, m), 7.71-7.63 (1H, m), 7.60-7.53 (2H, m), 7.37-7.23 (5H, m), 5.46 (1H, q, J=7.11 Hz), 5.15 (1H, d, J=51.48 Hz), 4.10-3.98 (2H, m), 3.26-3.15 (2H, m), 1.88-1.50 (4H, m), 1.55 (3H, s), 1.30 (9H, s).
[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester

- (3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (10.9 g) was dissolved in tetrahydrofuran (350 mL), and the atmosphere was replaced with argon. Then, potassium hexamethyldisilazide (0.5 M solution in toluene) (86.5 mL) was added dropwise at - 15°C, and the mixture was stirred at 0°C for 1.5 hours. After cooling to -10°C, saturated aqueous ammonium chloride (100 mL) was added dropwise, and the mixture was stirred at room temperature for 30 minutes. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 7:1) to give 4.36 g of the title compound as a pale yellow solid.
1H-NMR (400 MHz, CDCl3) δ: 7.38-7.25 (5H, m), 5.58-5.49 (1H, m), 3.63 (1H, d, J=10.3 Hz), 2.91 (1H, dd, J=10.3, 3.2 Hz), 2.67-2.56 (1H, m), 2.50-2.38 (1H, m), 2.26-2.09 (1H, m), 2.06-1.94 (1H, m), 1.74-1.66 (1H, m), 1.54 (3H, d, J=7.1 Hz), 1.50-1.40 (1H, m), 1.34 (9H, s).
[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane


- (1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester (4.36 g, 12.5 mmol) was dissolved in dichloromethane (70 mL). Trifluoroacetic acid (70 mL) was added dropwise, and the mixture was stirred at room temperature for six hours. The solvent was evaporated under reduced pressure, and then the residue was azeotropically distilled with toluene to give carboxylic acid (3.70 g).
- The resulting carboxylic acid was dissolved in toluene. Triethylamine (3.51 mL, 25.2 mmol) and diphenylphosphoryl azide (2.98 ml, 13.8 mmol) were added, and the mixture was heated to reflux for five hours. The solvent was evaporated under reduced pressure. Then, 1,4-dioxane (110 ml) and 6N hydrochloric acid (110 mL) were added to the residue, and the mixture was stirred at 60°C for 2.5 hours. After extraction with water and ethyl acetate, the aqueous layer was made alkaline with a saturated sodium hydroxide solution and extracted with chloroform twice. The organic layers were combined, dried over anhydrous sodium sulfate, and filtered, and then the solvent was evaporated under reduced pressure. Di-tert-butyl dicarbonate (11.05 g) was added to the residue, and the mixture was stirred at 75°C for six hours. The reaction solution was concentrated under reduced pressure, and then the residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 1:1) to give 3.69 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.50 (1H, q, J=7.1 Hz), 5.22 (1H, brs), 3.34 (2H, s), 2.49-2.37 (1H, m), 2.32-2.03 (3H, m), 2.02-1.90 (1H, m), 1.51 (3H, d, J=7.1 Hz), 1.55-1.48 (1H, m), 1.35 (9H, s).
[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane

- (1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (3.69 g, 10.2 mmol) was dissolved in tetrahydrofuran (200 mL). A 1.20 M solution of a borane-tetrahydrofuran complex in tetrahydrofuran (42.4 mL, 50.9 mmol) was added dropwise under ice-cooling, and the mixture was stirred for two hours while gradually heating to room temperature. The solvent was evaporated under reduced pressure. Under ice-cooling, 90% aqueous ethanol (100 mL) and triethylamine (100 mL) were added to the residue, and the mixture was heated to reflux for two hours. The solvent was evaporated under reduced pressure, and then the residue was extracted with saturated sodium bicarbonate water and dichloromethane. Thereafter, the target substance was extracted from the aqueous layer with dichloromethane. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure, and the resulting residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 95:5 -> 90:10) to give 3.33 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.32 - 7.18 (5H, m), 5.38 (1H, brs), 3.22 (1H, q, J=6.37 Hz), 2.92-2.57 (4H, m), 2.12-1.86 (4H, m), 1.80-1.67 (1H, m), 1.63-1.52 (3H, m), 1.42 (9H, s), 1.32 (3H, d, J=6.37 Hz)
- [Reference Example 78]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane

- (1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (700 mg, 2.0 mmol) was dissolved in ethanol (30 mL). 10% palladium-carbon (50% wet) (1.01 g) was added, and the mixture was stirred in a hydrogen atmosphere at 50°C for 15 hours. The catalyst was removed by filtration, and then the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2 -> 95:5) to give 446 mg of the title compound as a pale yellow solid.
[α]D 23-15° (c=0.100, MeOH).
1H-NMR (400 MHz, CDCl3) δ: 5.29 (1H, brs), 3.47-3.18 (2H, m), 2.93-2.79 (2H, m), 2.15-1.71 (6H, m), 1.45 (9H, s).
- [Example 17]
7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid

- Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were added to a solution of (1R,5S)-1-(tert-butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane (250 mg, 1.02 mmol) in dimethyl sulfoxide (5.0 mL). The mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for ten days. Ethanol (6.0 mL), water (2.0 mL), and triethylamine (2.0 mL) were added to the reaction solution, and the mixture was stirred at 80°C for one hour. The solvent was evaporated under reduced pressure, and then the residue was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with water twice and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2), and the resulting fraction was concentrated under reduced pressure. Then, the residue was dissolved in concentrated hydrochloric acid (3.5 mL) under ice-cooling, and the solution was stirred at room temperature for one hour. The reaction solution was washed with chloroform five times, and then the aqueous layer was adjusted to pH 12 with a saturated sodium hydroxide solution. The basic solution was adjusted to pH 7.4 with hydrochloric acid, followed by extraction with chloroform. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the solvent was evaporated under reduced pressure. The resulting residue was purified by PTLC (developed with the lower layer of chloroform:methanol:water = 7:3:1). The resulting residue was solidified with isopropanol to give 7.1 mg of the title compound as a pale yellow solid.
- 1H-NMR (400 MHz, 0.1N NaOD) δ: 8.50 (1H, s), 7.77 (1H, d, J=13.73 Hz), 5.80-4.80 (1H, m), 4.22-4.10 (1H, m), 3.99-3.85 (1H, m), 3.68-3.47 (2H, m), 3.43-3.34 (1H, m), 2.68 (3H, s), 2.21-1.98 (3H, m), 1.97-1.56 (4H, m), 1.42-1.23 (1H, m).
MS (FAB); m/z: 422 (M+H)+.
Anal.Calcd C21H22F3N3O3·0.5H2O·0.25IPA: C, 58.65; H, 5.66; F, 12.80; N, 9.43. Found: C, 58.63; H, 5.35; F, 12.35; N, 9.22.
IR (ATR) ν: 2971, 2856, 1722, 1614, 1450, 1432, 1322, 1132, 1097, 987, 954, 929, 887, 856, 804 cm-1.
WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula
compound represented by, 7 - [(1R, 5S) -1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, . the 2S) -2 - fluoro-cyclopropane-1 - yl] -1,4 - dihydro-8 - methyl-4 - oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A - [(1R, 5S) - 1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, 2S) -2 - fluoro-cyclo-1 - yl] -1 , 4 - dihydro-8 - methyl-4 - oxo-3-quinoline - was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue - was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A - [(1R, 5S) - 1 - amino-5 - fluoro-3 - azabicyclo [3.3.0] octan-3 - yl] -6 - fluoro -1 - [(1R, 2S) -2 - fluoro-cyclo-1 - yl] -1 , 4 - dihydro-8 - methyl-4 - oxo-3-quinoline - was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue - was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A - was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A - was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
- Higuchi, S.; Onodera, Y.; Chiba, M.; Hoshino, K.; Gotoh, N. Potent in vitro antibacterial activity of DS-8587, a new generation of broad spectrum quinolone, against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, doi:10.1128/AAC.02374-12.
- Daiichi Sankyo. Major R&D pipeline as of July, 2013. Available online: http://www.daiichisankyo.com/rd/pipeline/pdf/Pipeline_EN.pdf (accessed on 28 September 2013).
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |
8 OZENOXACIN
1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster......http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H
Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.
- Ferrer Group. Key development projects. Available online: http://www.ferrergrupo.com/Innovation_Innovacion-Pipeline-de-proyectos-ENG (accessed on 15 April 2013).
- Yamakawa, T.; Mitsuyama, J.; Hayashi, K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J. Antimicrob. Chemother. 2002, 49, 455–465, doi:10.1093/jac/49.3.455.
- Ferrer Internacional. Efficacy and safety of ozenoxacin 1% cream versus placebo in the treatment of patients with impetigo. Available online: http://clinicaltrials.gov/ct2/show/NCT01397461 (accessed on 13 April 2013).
9
FINAFLOXACIN
FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)...POSTER.......http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., "The Sanford Guide to Antimicrobial Therapy 2007" 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:
The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:
An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .
The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 - (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 - diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.
HBr / AcOH
Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:
Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester
 a 3-bromo-2 ,4,5-trifluoro-benzoate
a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 - (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 - trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 - (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 - cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene
 a solvent-free
a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 - dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 - 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene
 In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid
 In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid
 In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 - 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 - 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 - 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor
 In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl
 50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 - dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 -
50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 - dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 -
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride
 76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 - cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 - (toluene-4-sulfonyl-pyrroline
 In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 - (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o "|" h "CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-('toluene-4-sulfonyl') pyrrolidine
 643.7 g (2.65 mol) 3 - (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
643.7 g (2.65 mol) 3 - (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] - ftoluol-4-sulfonyl)-amino]-ethyl ester)
 552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) - pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) - pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane
 1092 g of crude trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l-(toluene-4-sulfonyl) - pyrrolidin-3-yl] - (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
1092 g of crude trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l-(toluene-4-sulfonyl) - pyrrolidin-3-yl] - (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l - (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] - (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 - diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide
 38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid
 Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan
 1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 - oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + - form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan
 Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 - (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester
 To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 - (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate
 To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester
 To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid
 33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 - (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 - trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 - (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 - cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 - dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 - 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 - 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 - cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 - (toluene-4-sulfonyl-pyrroline
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 - (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o "|" h "CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-('toluene-4-sulfonyl') pyrrolidine
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] - ftoluol-4-sulfonyl)-amino]-ethyl ester)
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 - [[4-hydroxy-l - (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] - (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 - diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 - oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + - form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan
Example Z 19
2 - (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester
Example Z 20
2 - (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1
 A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl - 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl - 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-('(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 - diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 - diazabicyclo [4.3.0] non-8-yl) - l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
...................
Tetrahedron Lett 2009, 50(21): 2525

Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
...................

1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-('(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 - diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 - diazabicyclo [4.3.0] non-8-yl) - l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
...................
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528Jian Hong, Zonghua Zhang, Huoxing Lei, Haiying Cheng, Yufang Hu, Wanliang Yang, Yinglin Liang, Debasis Das, Shu-Hui Chen, Ge Li | ||
Graphical abstract
http://www.sciencedirect.com/science/article/pii/S0040403909005875
...................
- Durata Therapeutics, Inc. Finafloxacin for the treatment of cUTI and/or acute pyelonephritis. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01928433 (accessed on 28 September 2013).
- Merlion Pharma. A multi-dose, double-blind, double-dummy, active control, randomized clinical (Phase II) study of two dosing regimens of finafloxacin for the treatment of cUTI and/or acute pyelonephritis.Available online: http://www.clinicaltrialsregister.eu/ctr-search/trial/2011–006041–14/PL/ (accessed on 14 April 2013).
- Pharma, M. FDA Grants Qualified Infectious Disease Product Designation and Fast Track Status for MerLion Pharma’s Lead Antibacterial Candidate Finafloxacin; Merlion Pharma: Singapore, 2013; Volume 2013.
- Lemaire, S.; van Bambeke, F.; Tulkens, P.M. Activity of finafloxacin, a novel fluoroquinolone with increased activity at acid pH, towards extracellular and intracellular Staphylococcus aureus, Listeria monocytogenes and Legionella pneumophila. Int. J. Antimicrob. Agents 2011, 38, 52–59, doi:10.1016/j.ijantimicag.2011.03.002.
- Finafloxacin hydrochlorideDrugs Fut 2009, 34(6): 451
- A novel approach to finafloxacin hydrochloride (BAY35-3377)Tetrahedron Lett 2009, 50(21): 2525
- New fluoroquinolone finafloxacin HCI (FIN): Route of synthesis, physicochemical characteristics and activity under neutral and acid conditions48th Annu Intersci Conf Antimicrob Agents Chemother (ICAAC) Infect Dis Soc Am (IDSA) Annu Meet (October 25-28, Washington DC) 2008, Abst F1-2036
| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |
10
CLINAFLOXACIN



J. Med. Chem., 23, 1358 (1980)




















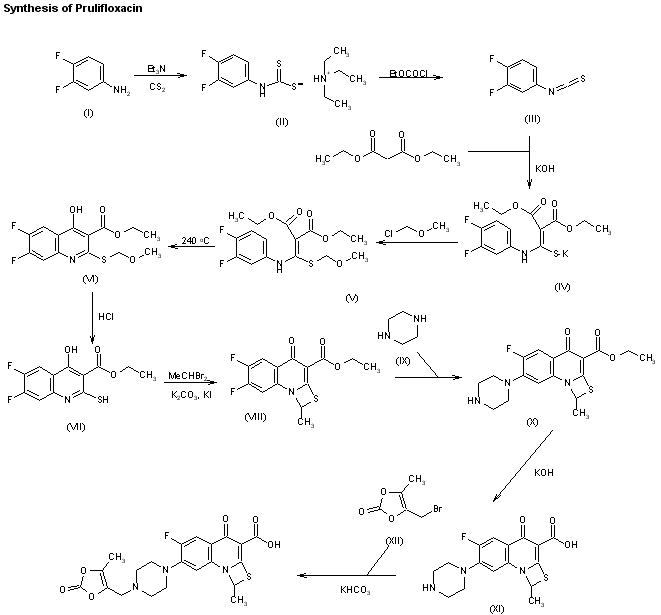
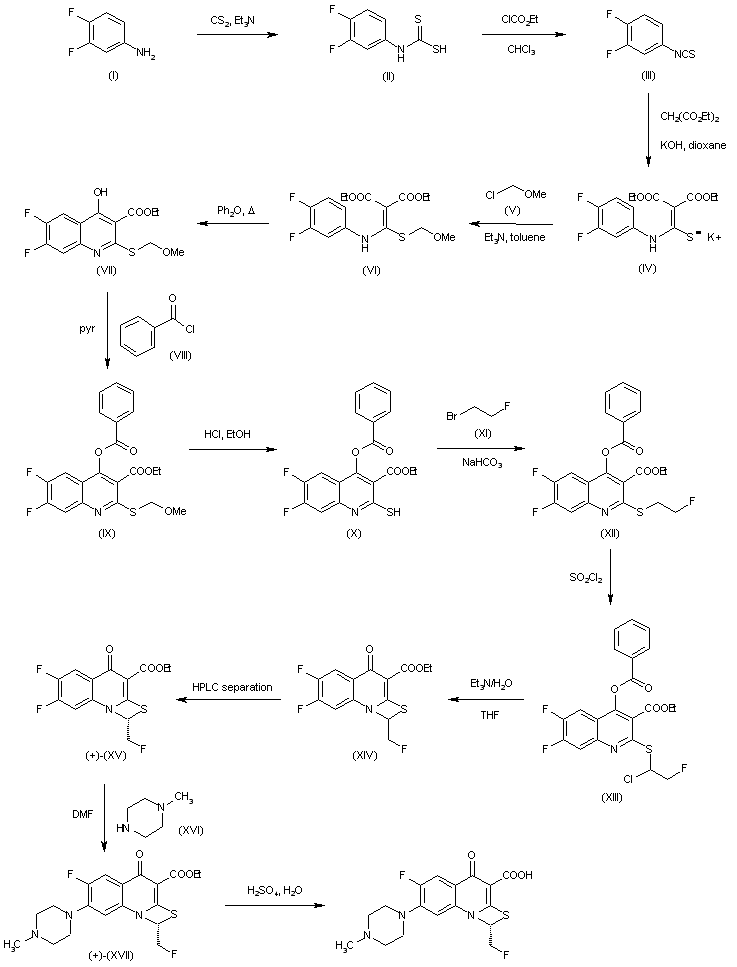






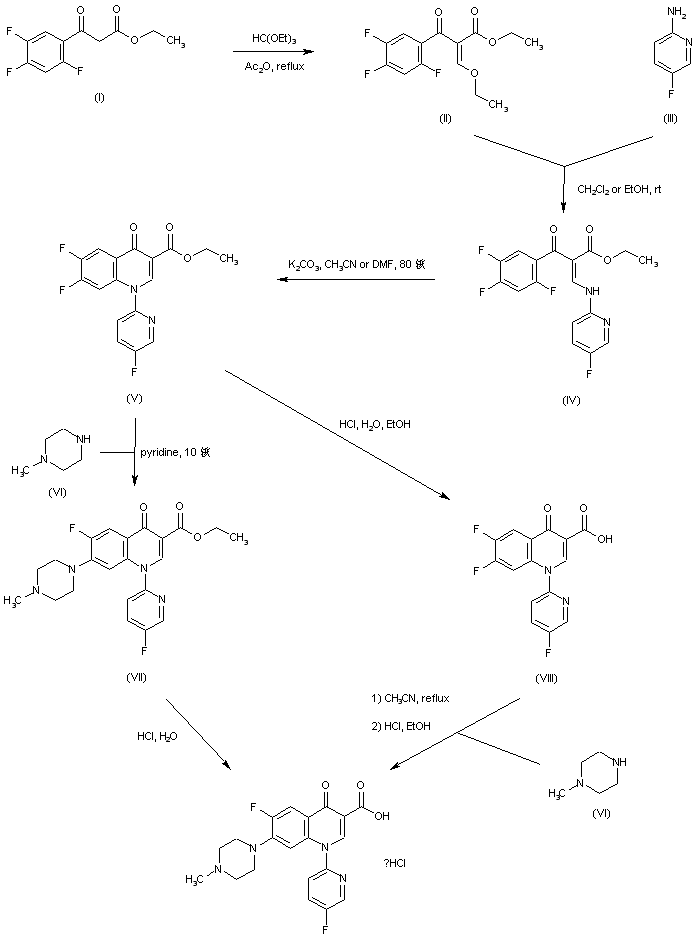
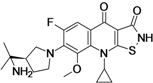

 ACH 702
ACH 702















 STEREOCENTERS SHOWN
STEREOCENTERS SHOWN


Chemical and Pharmaceutical Bulletin
![]()

17 WCK 2349
Votes
 . CH3SO3H
. CH3SO3H
 8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5
(S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic
acid of structural Formula I can be used to treat bacterial
Gram-positive, Gram-negative and anaerobic infections; especially
infections caused by resistant Gram-positive organism and Gram-negative
organism, mycobacterial infections and emerging nosocomial pathogen
infections.
8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5
(S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic
acid of structural Formula I can be used to treat bacterial
Gram-positive, Gram-negative and anaerobic infections; especially
infections caused by resistant Gram-positive organism and Gram-negative
organism, mycobacterial infections and emerging nosocomial pathogen
infections.
Formula I
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en
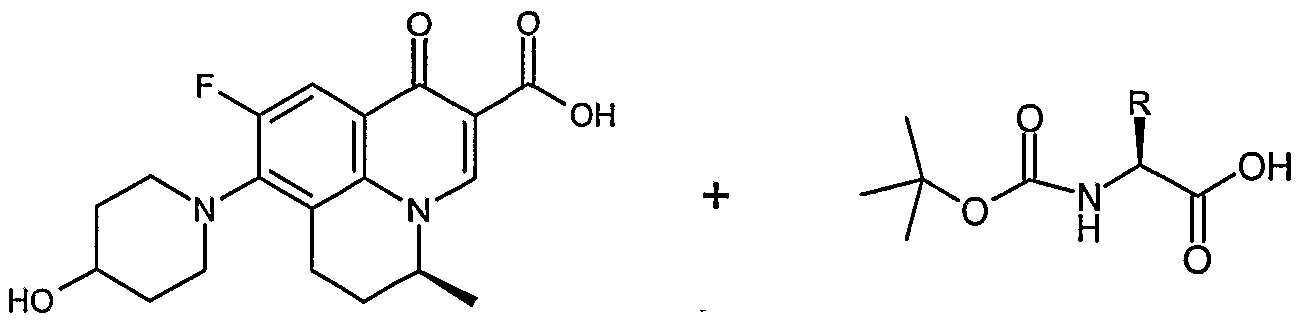
 Scheme 1
Scheme 1
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC



Clinafloxacin
7-(3-Aminopyrrolidin-1-yl)-8-chloro-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid
7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
(±)-7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid
105956-99-8 cas no
Clinafloxacin (INN) is a fluoroquinolone antibiotic. Its use is associated with phototoxicity and hypoglycaemia.[1]
Clinafloxacin is a novel quinolone with wide activity against the plethora of microorganisms encountered in intraabdominal infections.
Clinafloxacin is a chlorofluoroquinolone with excellent bioavailability and activity against gram-positive, gram-negative, and anaerobic pathogens . Typical MICs for α-streptococci are 0.06–0.12 µg/mL . MIC90 values for methicillin-resistant Staphylococcus aureus (MRSA) average 1.0 µg/mL. The MIC90 for enterococci is typically 0.5 µg/mL . Both intravenous and oral formulations have been developed . Several studies have demonstrated the efficacy of clinafloxacin monotherapy for serious infections Clinafloxacin was also active in animal models of endocarditis, including endocarditis due to ciprofloxacin-resistant S. aureus infection .
Clinafloxacin HCl, CI-960 HCl, 105956-99-8, Clinafloxacin hydrochloride (USAN), Clinafloxacin hydrochloride [USAN], AC1L1SJB,
Molecular Formula: C17H18Cl2FN3O3 Molecular Weight: 402.247523
............................................
EP 0195316
http://www.google.com/patents/EP0195316A1?cl=en
preparation process for the compound of the invention.

- Example 28 7-(3-Amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-l,4-dihydro-4-oxo-3-quinolinecarboxylic acid
- A mixture of 8-chloro-1-cyclopropyl-6,7-difluoro-1,4-di- hydro-4-oxo-3-quinolinecarboxylic acid (0.6 g), anhydrous acetonitrile (6 ml), 3-aminopyrrolidine (0.35 g) and DBU (0.31 g) was refluxed for an hour. Then, 3-aminopyrrolidine (0.2 g) was more added and further refluxed for 2 hours. After cooling, the resulting precipitate was collected by filtration, dissolved in water (9 ml) containing sodium hydroxide (0.12 g) and neutralized with acetic acid. The resulting precipitate was collected by filtration and washed with water and acetonitrile successively to give the title compound (0.52 g) as colorless powder, mp 237-238 °C (decompd.).
- Analysis (%) for C17H17ClFN3O3·H2O, Calcd. (Found): C, 53.20 (52.97); H, 4.99 (4.62); N, 10.95 (10.83).
- To a suspension of 7-(3-amino-1-pyrrolidinyl)-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid (100 mg) in ethanol (2 ml) was added 0.2 ml of ethanol solution of hydrogen chloride (7.0 mmol HC1/ml) and then the mixture was concentrated. The resulting residue was recrystallized from methanol to give the title compound (79 mg) as light yellow prisms, mp 263-265 °C (decompd.).
- Analysis (%) for C17H17ClFN3O3.HCl, Calcd. (Found): C, 50.76 (50.50); H, 4.51 (4.44); N, 10.45 (10.38).
J. Med. Chem., 23, 1358 (1980)
- structural formula D
may be readily prepared from the known starting material methyl 5-oxo-l-(phenylmethyl)-3-pyrrolidinecarboxylate, A, [J. Org. Chem., 26, 1519 (1961)] by the following reaction sequence.

- The compound wherein R3 is hydrogen, namely 3-pyrrolidinemethanamine, has been reported in J. Org. Chem., 26, 4955 (1961).
Journal of Medicinal Chemistry, 1988 , vol. 31, p. 983 - 991
References
- Rubinstein, E. (2001). "History of quinolones and their side effects.". Chemotherapy. 47 Suppl 3: 3–8; discussion 44–8.doi:10.1159/000057838. PMID 11549783.
| EP0106489A2 * | Sep 6, 1983 | Apr 25, 1984 | Warner-Lambert Company | Antibacterial agents |
| EP0153163A2 * | Feb 15, 1985 | Aug 28, 1985 | Warner-Lambert Company | 7-Substituted-1-cyclopropyl-6,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids; 7-substituted-1-cyclopropyl-1,4-dihydro-6-fluoro-4-oxo-1,8-naphthyridine-3-carboxylic acids; their derivatives; and a process for preparing the compounds |
| BE899399A1 * | Title not available | |||
| GB2057440A * | Title not available |
11
PRULIFLOXACIN
PRULIFLOXACIN
(RS)-6-Fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
6-Fluoro-1-methyl-7-(4-(5-methyl-2-oxo-1,3-dioxelen-4-yl)methyl-1-piperazinyl)-4-oxo-4H-(1,3)thiazeto(3,2-a)quinoline-3-carboxylic acid
123447-62-1 CAS NO
NM 441, Quisnon, Pruvel, Sword, Prulifloxacin [INN], 123447-62-1, NM-441, CCRIS 7686, NCGC00164615-01NAD-441A
OPT-99
OPT-99
Molecular Formula: C21H20FN3O6S
Molecular Weight: 461.463403
Launched - 2002 BY NIPPON SHINYAKU
SYNTHESIS.......http://www.drugfuture.com/synth/syndata.aspx?ID=151640
Prulifloxacin is an older synthetic chemotherapeutic antibiotic of the fluoroquinolone drug class[1][2] undergoing clinical trials prior to a possible NDA (New Drug Application) submission to the U.S. Food and Drug Administration (FDA). It is a prodrug which is metabolized in the body to the active compound ulifloxacin.[3][4] It was developed over two decades ago by Nippon Shinyaku Co. and was patented in Japan in 1987 and in the United States in 1989.[5][6]
It has been approved for the treatment of uncomplicated and complicated urinary tract infections, community-acquired respiratory tract infections in Italy and gastroenteritis, including infectious diarrheas, in Japan.[3][7] Prulifloxacin has not been approved for use in the United States.
Prulifloxacin is a novel fluoroquinolone antibiotic that was launched pursuant to a collaboration between Meiji Seika and Nippon Shinyaku in 2002 for the oral treatment of systemic bacterial infections, including acute upper respiratory tract infection, bacterial pneumonia, prostatitis, cholecystitis, bacterial enteritis, internal genital infections, otitis media, sinusitis and others. It is currently marketed in a tablet formulation. A once-daily formulation to be taken over a three-day period is in phase III clinical trials at Optimer Pharmaceuticals to be used in the treatment of bacterial gastroenteritis, including traveler's diarrhea. The formulation had been in phase II trials at the company for the treatment of urinary tract infections, however, no recent development for this indication have been reported. The drug has also been studied at Optimer for the treatment of community-acquired respiratory tract infections, but recent progress reports for this indication have not been made available.
Prulifloxacin has in vitro activity against a wide range of gram-negative and gram-positive microorganisms. Its antibacterial action results from inhibition of DNA gyrase and topoisomerase IV, both Type II isomerases. DNA gyrase is an essential enzyme that is involved in the replication, transcription, and repair of bacterial DNA. Topoisomerase IV is an enzyme known to play a key role in the partitioning of the chromosomal DNA during bacterial cell division. Together, the Type II topoisomerases remove the positive supercoils that accumulate ahead of a translocating DNA polymerase, allowing DNA replication to continue unhindered by topological strain. Fluoroquinolones may be active against pathogens that are resistant to penicillins, cephalosporins, aminoglycosides, macrolides and tetracyclines, as they possess a distinct mechanism of action from these antibiotics.
Prulifloxacin was discovered by Nippon Shinyaku and codeveloped with Meiji Seika in Japan. Nippon Shinyaku granted Angelini a manufacturing and marketing license for Italy in 1993. Exclusive Korean manufacturing and commercialization rights were acquired by Yuhan from Nippon Shinyaku in March 2003. In June 2004, Optimer was granted exclusive development and commercialization rights to prulifloxacin in the U.S. from Nippon Shinyaku. Finally, Recordati signed a nonexclusive licensing agreement with Angelini for the marketing and sale of prulifloxacin in Spain in October 2004. In March 2009, the product was licensed to Lee's Pharmaceuticals by Nippon Shinyaku for marketing in China as an oral treatment of bacterial infection. In 2010, prulifloxacin was licensed to Algorithm by Nippon Shinyaku in North Africa and the Middle East for the development and marketing for the treatment of bacterial infections.
History
In 1987 a European Patent (EP 315828) for prulifloxacin (Quisnon ) was issued to the Japanese based pharmaceutical company, Nippon Shinyaku Co., Ltd (Nippon). Ten years after the issuance of the European patent, marketing approval was applied for and granted in Japan (March 1997). Subsequent to being approved by the Japanese authorities in 1997 prulifloxacin (Quisnon) was co-marketed and jointly developed in Japan with Meiji Seika as licensee (Sword).[6]
In more recent times, Angelini ACRAF SpA, under license from Nippon Shinyaku, has fully developed prulifloxacin, for the European market.[8] Angelini is the licensee for the product in Italy. Following its launch in Italy, Angelini launched prulifloxacin in Portugal (January 2007) and it has been stated that further approvals will be sought in other European countries.[9][10]
Prulifloxacin is marketed in Japan and Italy as Quisnon (Nippon Shinyaku); Sword (Meiji); Unidrox (Angelini) and generic as Pruquin.
In 1989 and 1992 United States patents (US 5086049) were issued to Nippon Shinyaku for prulifloxacin. It was not until June 2004, when Optimer Pharmaceuticals acquired exclusive rights to discover, develop and commercialize prulifloxacin (Pruvel) in the U.S. from Nippon Shinyaku Co., Ltd., that there were any attempts to seek FDA approval to market the drug in the United States. Optimer Pharmaceuticals expects to file an NDA (new drug application) for prulifloxacin some time in 2010. As the patent for prulifloxacin has already expired, Optimer Pharmaceuticals has stated that this may have an effect on the commercial prospects of prulifloxacin within the United States market.[11]
Licensed uses
Prulifloxacin has been approved in Italy ,Japan,China,India and Greece (as indicated), for treatment of infections caused by susceptible bacteria, in the following conditions:
- Italy
- Acute uncomplicated lower urinary tract infections (simple cystitis)
- Complicated lower urinary tract infections
- Acute exacerbation of chronic bronchitis
- Japan
- Gastroenteritis, including infectious diarrheas
- Other countries
- Prulifloxacin has not been approved for use in the United States, but may have been approved in other Countries, other than that which is indicated above.
Availability
Prulifloxacin is available as:
- Tablets (250 mg, 450 mg or 600 mg)
In most countries, all formulations require a prescription.
Prulifloxacin is chemically known as 6-fluoro-1-methyl-7-{4-[(5-methyl-2-oxo-1 ,3-dioxol- 4-yl)methyl]piperazin-1-yl}-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid, and it has the structure as shown below as formula I:
FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent.
Prulifloxacin was first disclosed in US 5,086,049. The patent discloses a process for the preparation of prulifloxacin by the condensation of ulifloxacin with a 4-halomethyl-5- methyl-1 ,3-dioxolen-2-one of formula III
wherein X is halo selected form chloro, bromo or iodo, in the presence or absence of an aprotic solvent and a base to obtain prulifloxacin free base which is recrytallised with chloroform-methanol. In an exemplified process, ethyl 6,7-difluoro-1-methyl-4-oxo-4H- (1 ,3)-thiazeto-(3,2-a)-quinoline-3-carboxylate is condensed with piperazine in the presence of dimethyl formamide and purified by column chromatography followed by basic hydrolysis to give ulifloxacin, which is then converted to prulifloxacin.
The above process involves column chromatography. Prulifloxacin prepared by this method has a purity of 60-65% containing impurities in unacceptable levels. Removal of these impurities by usual purification procedures, such as recrystallisation, distillation and washing, is difficult and requires extensive and expensive multiple purification processes. This further decreases the overall yield. A method involving column chromatographic purifications and multiple purifications cannot be used for large-scale operations, thereby making the process commercially non-viable.
European Patent No. 315828 disclosed a variety of quinoline carboxylic acid derivatives and pharmaceutically acceptable salts thereof. These compounds are exhibiting antibacterial activity and useful as remedies for various infectious diseases. Among them prulifloxacin, chemically (+)-6-Fluoro- 1 -methyl-7-[4-(5-methyl-2-oxo-1 ,3-dioxolen-4-ylmethyl)-1 -piperazinyl]-4-oxo-4H- [1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid is a fluoroquinolone antibacterial prodrug which shows potent and broad-spectrum antibacterial activity both in vitro and in vivo. Prulifloxacin also showed superior activity against strains of Enterobacteriaceae and Pseudomonas aeruginosa. Prulifloxacin is represented by the following structure:
Processes for the preparation of prulifloxacin and related compounds were disclosed in European Patent No. 315828 and UK Patent Application No. GB 2190376.
In - the preparation of prulifloxacin, 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid of formula I:
is a key intermediate. According to the UK Patent Application No. GB 2190376, the compound of the formula I was prepared by the reaction of 3,4-difluroaniline with carbon disulfide and triethylamine to give triethylammonium N-(3,4- difluorophenyl)dithio carbamate, which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate, followed by reaction with diethyl malonate and KOH in dioxane affords the potassium salt, which is then treated with methoxymethyl chloride in dimethylformamide to give diethyl 1-(3,4-difluorophenylamino)-1- (methoxymethylthio)-rnethylene-rnalonate. The cyclization of the thio compound at 2400C in diphenyl ether affords ethyl 6,7-difluoro-4-hydroxy-2- methoxymethylthioquinoline-3-carboxylate, which by treatment with HCI in ethanol gives ethyl δy-difluoro^-hydroxy^-mercaptoquinoline-S-carboxylate. The cyclization of the mercapto compound with 1,1-dibromoethane by means of potassium carbonate and potassium iodide in hot dimethylformamide yields ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate, which is condensed with piperazine in dimethylformamide to afford ethyl 6- fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate, which is then subjected to hydrolysis with potassium hydroxide in hot tert-butanol to give the compound of formula I.
The compound of formula I obtained by the process described in the UK Patent Application No. GB 2190376 is not satisfactory from purity point of view, the reaction between ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2- a]quinoline-3-carboxylate and piperazine requires longer time about 48 hours to complete, the yield obtained is not satisfactory, and the process also involves column chromatographic purifications. Methods involving column chromatographic purifications cannot be used for large-scale operations, thereby making the process commercially not viable. According to the European Patent No. 315828, prulifloxacin is prepared by reacting 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a] quinoline-3-carboxylic acid with 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one in presence of potassium bicarbonate in dimethylformamide. However, a need still remains for an improved and commercially viable process of preparing pure prulifloxacin that will solve the aforesaid problems associated with process described in the prior art and will be suitable for large- scale preparation, in terms of simplicity, purity and yield of the product.
Prulifloxacin is chemically 6-fluoro-l-methyl-7-{4-[(5-methyl-2-oxo-l,3-dioxol-4- yl)methyl]piperazin-l-yl}-4-oxo-4H-[l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula I having the structure as depicted below:
FORMULA I
Prulifloxacin has significant antibacterial activity and has been marketed as a synthetic antibacterial agent. U.S. Patent No. 5,086,049 provides a process for the preparation of prulifloxacin by reacting 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H- [l,3]thiazeto[3,2-α]quinoline-3-carboxylic acid of Formula II,
FORMULA II and 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III,
FORMULA III using N,N-dimethylformamide as a solvent. 4-(Bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III is used in excess to one mole of the compound of Formula II. The process provided in U.S. Patent No. 5,086,049 further involves concentrating the reaction mixture, pouring the residue into water and isolating prulifloxacin by filtration. The resulting prulifloxacin is recrystallized from chloroform-methanol.
However, U.S. Patent No. 5,086,049 does not provide any method to remove the unreacted or the excess of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one of Formula III used as a starting material. The present inventors have observed that it is difficult to obtain prulifloxacin with pharmaceutically acceptable purity by following the process provided in U.S. Patent No. 5,086,049, which is typically contaminated by process related impurities including 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one
A need still remains for an improved and commercially-viable process for preparing pure prulifloxacin that will solve the aforesaid problems associated with the process described in the prior art and that will be suitable for large-scale preparation, in terms of simplicity, purity and yield of the product.
EP1626051 A1 mentions that Type I, Type II and Type III crystals of prulifloxacin are obtained by crystallization from acetonitrile as reported in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. However, the conditions of crystallization from acetonitrile for preparing Type I, Type II and Type III crystals are not disclosed in lyakuhin Kenkyu, Vol. 28 (1), (1997), 1-11. EP1626051A1 further mentions that Type III crystals have been marketed by considering the solubility, absorbability, therapeutic effect and the like of the respective crystal forms.
US 2007/0149540 discloses a crystal of prulifloxacin acetonitrile solvate (Compound B) which is an intermediate for producing preferentially the type III crystal of prulifloxacin. A crystal of Compound B can be preferentially precipitated by controlling the supersaturation concentration in crystallization using acetonitrile as a solvent, subsequently; the type III crystal of Compound A can be produced by performing desolvation of the crystal.
WO 2008/111018 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin. There is disclosed a process for preparing Type I crystals by controlled cooling over a period of 7 to 9 hours and prolonged drying over 24 hours. The inventors of the present invention have found that Type I and Type III crystals prepared according to the WO 2008/111018 process are unstable and the process is non-reproducible.
WO 2010/0084508 discloses processes for the preparation of Type I, Type II and Type III crystals of prulifloxacin.
WO 2008/059512 discloses a process for the preparation of prulifloxacin using novel intermediates.
WO 2008/111016 discloses a process for the preparation of prulifloxacin having purity of about 99% or above. It would be a significant contribution to the art to provide a crystalline form of prulifloxacin, which is consistent and to provide industrially viable methods of preparation, pharmaceutical formulations, and methods of use thereof.
.....................
SYNTHESIS
http://www.google.com/patents/WO2012001357A1?cl=en
Scheme 1.
Formula I
[PRULIFLOXACIN]
Example 1
Preparation of ethyl-6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]- quinoline-3-carboxylate (formula III)
5,6-difluoro-1-methyl-4-oxo-4H-[1 ,3]-thiazeto-[3,2-a]-quinoline-3-carboxylic acid ethyl ester of formula (II) (100 gms, 0.321 moles) was stirred in 500 ml of DMF at room temperature. Piperazine (76 gms, 0.882 moles) was added at room temperature and stirred for 10 minutes. The temperature was slowly raised to 50-55°C and the reaction mass was stirred at 50-55°C for 5 hours. After completion of the reaction, the reaction mass was cooled to 25-30°C and stirred for 2 hours. The reaction mass was further chilled to 10-15°C and stirred for 2 hours. The precipitated solid was filtered, washed of chilled DMF (2 x 50 ml). The solid was slurry washed with water (300 ml), filtered, washed with water ( 2 x 100 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 74 % yield, 95% HPLC purity].
Example 2
Preparation of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid (formula IV)
Ethyl-6-fluoro-1 -methyl-4-oxo-7-(1 -piperazinyl)-4H-(1 ,3)-thiazeto-[3,2-a]-quinoline-3- carboxylate (100 gms, 0.265 moles) was stirred in water (600 ml) at 25-30°C. To this potassium hydroxide solution (50 gms of potassium hydroxide flakes is dissolved in 200 ml of water) was added and the reaction mass was heated to 80-85°C. The contents were stirred for 1 hour and after completion of reaction, the reaction mass was cooled to 25-30°C. The pH of the reaction mass was adjusted to 6.5-7.0 using 1:1 aqueous acetic acid solution. The contents were stirred at room temperature for 1 hour. The precipitated solid was filtered, washed with water (2 x 100 ml). The solid was slurried in methanol (300 ml) for 1 hour at 25-30°C, filtered, washed with methanol (2 x 50 ml) and dried under vacuum at 70-75°C to yield the title compound [90 gms, 97% yield, 96% HPLC purity]. Example 3
Preparation of prulifloxacin
To a solution of 4-(chloromethyl)-5-methyl-1,3-dioxol-2-one (55 gms, 0.371 moles) in 50 ml of DMF at 25-30°C, sodium bromide (77 gms, 0.748 moles) was added and the reaction mass was slowly heated to 40-45°C. The contents were stirred at 40-45°C for 2 hours, acetone ( 500 ml) was added at 40-45°C and stirred for 3 hours. The reaction mass was filtered over hyflo, and the bed washed with acetone (100 ml). The solvent was completely distilled off under vacuum below 45°C to yield 4-(bromomethyl)-5- methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 4.0 It of acetonitrile, DIPEA (70 ml , 0.402 moles)) was added at room temperature, stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl- 1 ,3-dioxol-2-one (formula V) in 500 ml of acetonitrile was slowly added at 10-15°C over a period of 1 hour. The contents were stirred at 25-30°C for 20 hour, filtered over hyflo, and the bed washed with 200 ml of acetonitrile. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was slowly chilled to 0-5°C and the precipitated solid was filtered, washed with acetonitrile (25 ml) and dried to yield 65 gms of prulifloxacin. Example 4
Preparation of Type I crystals of prulifloxacin
Prulifloxacin (65 gms) was added to 200 ml of DMF at 25-30°C and heated to 80-85°C for 1 hour. The mixture was then slowly cooled to 25-30°C, stirred for 2 hours, chilled to 0-5°C for 2 hours. The precipitated solid was filtered and dried under vacuum at 70- 75°C to yield Type I crystals of prulifloxacin (55 gms, 99.5 % HPLC purity).
Example 5
Preparation of prulifloxacin
(55 gms, 0.371 moles) of 4-(chloromethyl)-5-methyl-1 ,3-dioxol-2-one is taken in 5.0 ml of DMF at 25-30°C. (77 gms, 0.748 moles) of sodium bromide is added and slowly heated the reaction mass to 40-45°C. The contents are stirred at 40-45°C for 2 hours, 500 ml of acetone is added at 40-45°C and stirred for 3 hours. The reaction mass is clarified over hyflo, and the bed washed with 100 ml of acetone to yield a solution of 4- (bromomethyl)-5-methyl-1 ,3-dioxol-2-one (formula V).
To a solution of 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]-thiazeto-[3,2-a]- quinoline-3-carboxylic acid of formula IV (100 gms, 0.286 moles) in 3.5 Its of acetone was at room temperature DIPEA (70 ml, 0.402 moles) and stirred for 10 minutes. The reaction mass was cooled to 10-15°C and a solution of 4-(bromomethyl)-5-methyl-1 ,3- dioxol-2-one (formula V) in acetone was slowly added to the reaction mass at 10-15°C over a period of 1 hour. The contents were further stirred at 25-30°C for 20 hour, filtered over hyflo and the bed washed with 200 ml of acetone. The solvent was distilled off completely under vacuum below 50°C. Acetonitrile (100 ml) was added at 50°C and the contents were stirred for 30-60 minutes. The reaction mass was further chilled to 0- 5°C and stirred for 2 hours. The precipitated solid was filteredand dried to yield prulifloxacin.
......................
http://www.google.com/patents/WO2008059512A1?cl=en
novel process for preparing 6-fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid of formula I:
which comprises: a) reacting the difluoro-quinoline compound of formula
wherein R represents hydrogen atom or alkyl containing 1 to 4 carbon atoms; with boric acid of formula III:
in presence of acetic anhydride and acetic acid to give borane compound of formula IV:
b) reacting the borane compound of formula IV with piperazine of formula V:
HN NH V
to give piperazine compound of formula Vl:
c) treating the compound of formula Vl with an alkaline metal hydroxide, carbonate or bicarbonate to obtain the compound of formula I.
Prulifloxacin and pharmaceutically acceptable acid addition salts of prulifloxacin can be prepared by using the compound of formula I by known methods for example as described in the European Patent No. 315828. Borane compound of the formula IV and Vl are novel and forms part of the invention. Preferably the reaction in step (a) is carried out at about 300C to reflux temperature more preferably at about 800C to reflux temperature and still more preferably at reflux temperature.
Example 1 Step-I:
Acetic anhydride (24 ml) and acetic acid (11 ml) are added to boric acid (3.5 gm) under stirring at 25 - 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, ethyl 6,7- difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate (20 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 2 hours. The reaction mass is cooled to 25 - 350C, toluene (200 ml) is added under stirring, the reaction mass is cooled to 50C and then stirred for 1 hour at 5 - 100C. Filtered the solid, washed with 20 ml of toluene and then dried to give 25.5 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3,04/bis/acetato-0/-borone. Step-I I: Acetonitrile (125 ml), dimethylsulfoxide (125 ml) and piperazine (13.8 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (25.5 gm, obtained in step-l) under stirring at 25 - 350C, the contents are heated to 850C and then stirred for 3 hours at 80 - 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 - 150C. Filtered the solid, washed with 25 ml of acetonitrile and then dried to give 26 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill: Water (155 ml), potassium hydroxide (17 gm) are added to 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate- O3,O4/bis/ acetato-0/-borone (26 gm, obtained in step-ll) under stirring at 25 - 350C, the contents are heated to 650C and then stirred for 4 hours at 60 - 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 - 7.5 with 50% HCI solution at 25 - 300C. The separated solid is stirred for 1 hour at 25 - 300C, filtered the solid, washed with 35 ml of water and then dried to give 17 gm of 6-fluoro-1- methyl-4-oxo-7-(1 -piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.5%). Example 2 Step-I:
Acetic anhydride (12 ml) and acetic acid (5.5 ml) are added to boric acid (1.25 gm) under stirring at 25 - 300C, the contents are heated to reflux and then stirred for 3 hours at reflux. The reaction mass is cooled to 1000C, 6,7-difluoro-1- methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylic acid (10 gm) is added at 1000C, the contents are heated to reflux and then refluxed for 3 hours. The reaction mass is cooled to 500C, toluene (100 ml) is added under stirring at 500C, the resulting mass is cooled to 100C and then stirred for 1 hour at 10 - 150C. Filtered the solid, washed with 20 ml of toluene and then dried to give 10 gm of 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate -O3 , 04/bis/acetato-0/-borone . Step-I I:
Acetonitrile (50 ml), dimethylsulfoxide (50 ml) and piperazine (5.5 gm) are added to 6,7-difluoro-1-methyl-4-oxo-4H-[1 ,3]thiazeto[3,2-a]quinoline-3- carboxylate-03,04/bis/acetato-0/-borone (10 gm, obtained in step-l) under stirring at 25 - 350C, the contents are heated to 850C and then stirred for 3 hours at 80 - 850C to form a clear solution. The solution is cooled to 100C and then stirred for 1 hour at 10 - 150C. Filtered the solid, washed with 10 ml of acetonitrile and then dried to give 10.4 gm of 6-fluoro-1-methyl-4-oxo-7-(1- piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/acetato-0/- borone. Step-Ill :
Water (62 ml), potassium hydroxide (7 gm) are added to 6-fluoro-1-methyl-4- oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto[3,2-a]quinoline-3-carboxylate-03,04/bis/ acetato-OAborone (10.4 gm, obtained in step-ll) under stirring at 25 - 350C, the contents are heated to 650C and then stirred for 4 hours at 60 - 650C. The reaction mass is cooled to 250C, filtered the undesired solid on hi-flow bed and then pH of the resulting filtrate is adjusted to 7 - 7.5 with 50% HCI solution at 25 - 300C. The separated solid is stirred for 30 minutes at 25 - 300C, filtered the solid, washed with 20 ml of water and then dried to give 68 gm of 6-fluoro-1- methyl-4-oxo-7-(1-piperazinyl)-4H-[1 ,3]thiazeto [3,2-a]quinoline-3-carboxylic acid (HPLC Purity: 98.6%). Example 3
Acetonitrile (560 ml) and potassium bicarbonate (8 gm) are added to 6- fluoro-i-methyM-oxo-y-CI-piperazinyO^H-CI .SKhiazetofS^-alquinoline-S- carboxylic acid (14 gm, obtained as per the processes described in examples 1 and 2) under stirring at 25 - 300C, the contents are cooled to 150C and then the solution of 4-bromomethyl-5-methyl-1 ,3-dioxolen-2-one (10 gm) in acetonitrile (140 ml) is added at 15 - 200C for 30 to 45 minutes. The contents are stirred for 25 hours at 25 to 300C, filtered and the resulting filtrate is distilled under vacuum. To the residue added acetonitrile (70 ml), cooled the mass to 200C and then stirred for 1 hour to 1 hour 30 minutes at 20 - 250C. Filtered the solid, washed the solid with 15 ml of chilled acetonitrile and then dried to give 16 gm of prulifloxacin crude (HPLC Purity: 98.8%).
To the prulifloxacin crude (obtained above) added acetonitrile (200 ml) at 25 - 300C, the contents are heated to reflux and then refluxed for 30 minutes. To the reaction mass added activated carbon (5 gm) and refluxed for 15 minutes. The reaction mass is filtered on hi-flo bed, the resulting filtrate is cooled to 200C and then stirred for 3 - 4 hours at 20 - 250C. Filtered the solid, washed with 20 ml of acetonitrile and then dried to give 14 gm of prulifloxacin (HPLC Purity: 99.9%).
.....................
http://www.google.com/patents/WO2008111016A1?cl=en
In a first aspect, a process for the preparation of prulifloxacin is provided, the process comprising: a) reacting a compound of Formula II with a compound of Formula III to obtain prulifloxacin;
FORMULA III
FORMULA II
b) contacting the prulifloxacin obtained in step a) with an acid in a biphasic solvent system, wherein the biphasic solvent system comprises water and a water- immiscible organic solvent; c) separating the aqueous layer from the reaction mixture obtained in step b); d) treating the aqueous layer with a base; and e) isolating prulifloxacin.
The process described in steps b - e above may be carried out with prulifloxacin made from any process however.
The compounds of Formula II and Formula III may be prepared according to the methods provided in U.S. Patent No. 5,086,049.
Example 1: Process for the Preparation of Prulifloxacin:
Step A): A solution of 4-(bromomethyl)-5-methyl-l,3-dioxol-2-one (35.5 g, 0.184 mole) in N,N-dimethylformamide (200 ml) was added dropwise at 0 to 5° C to a stirred solution of 6-fluoro-l-methyl-4-oxo-7-piperazin-l-yl-4H-[l,3]thiazeto[3,2-α]quinoline-3- carboxylic acid (50 g, 0.143 mole and potassium bicarbonate (15.8 g, 0.1578 mole) in N,N-dimethylformamide (200 ml). The resulting mixture was stirred at 25° to 28°C for 3 to 4 hours. After the completion of the reaction, the reaction mixture was poured into water (1250 ml). The solid obtained was filtered, washed with water (100 ml), and subsequently dissolved in a mixture of chloroform: methanol (7:3; 1250 ml). The lower organic layer was separated and water (500 ml) was added to the organic layer. A dilute aqueous solution of hydrochloric acid was added to the biphasic reaction mixture to adjust pΗ to 0.8 to 1.0. The reaction mixture was stirred for 15 minutes, allowed to settle and the upper aqueous layer was separated. The process was repeated twice and the aqueous layers were combined. Activated charcoal (10%) was added to the combined aqueous layer and stirred for 30 minutes, filtered and cooled to 20° to 25° C. The pΗ of the reaction mixture was adjusted to 6.5 to 7.0 by adding an aqueous solution of sodium bicarbonate. The solid obtained was extracted with chloroform (375 ml), stirred for 15 minutes and the organic layer was separated. The aqueous layer was further extracted with a mixture of chloroform: methanol (7:3 ratio; 50 ml). The combined organic layer was distilled under vacuum at 35° to 40° C to recover the solvent up to 125 ml. The reaction mass so obtained was stirred for 3 to 4 hours at 28° to 30° C, filtered and washed with chilled chloroform (50 ml). The wet cake obtained was dried at 45° C for 12 hours to obtain the title compound. Step B): The prulifloxacin (30 g) obtained in Step A) was suspended in a mixture of chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml) and heated to reflux temperature. Activated carbon (3.9 gm) was added to the partially cleared solution and refluxed for 30 minutes, followed by filtration through Celite bed. The bed was further washed with chloroform: ethanol (10:1, v/v, 585 ml: 58.5 ml). The filtrate so obtained was distilled at atmospheric pressure till to partially remove the solvent. The concentrate so obtained was stirred at about 25° C for 1 hour, and filtered. The solid obtained was washed with chloroform: ethanol (39 ml X 2), dried under vacuum at 45° C for 12 hours to obtain the title compound. Yield: 22 g
HPLC Purity: 99%
...............................
SEE
Studies on pyridonecarboxylic acids. 1. Synthesis and antibacterial evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto[3, 2-a]quinoline-3-carboxylic acids
J Med Chem 1992, 35(25): 4727
J Med Chem 1992, 35(25): 4727
http://pubs.acs.org/doi/pdf/10.1021/jm00103a011
The reaction of 3,4-difluoroaniline (I) with carbon disulfide and triethylamine gives triethylammonium N-(3,4-difluorophenyl)dithiocarbamate (II), which by reaction with ethyl chloroformate and triethylamine in chloroform is converted into 3,4-difluorophenyl isothiocyanate (III). The reaction of (III) with diethyl malonate and KOH in dioxane affords the potassium salt (IV), which is treated with chloromethyl methyl ether in DMF to give the corresponding methoxymethylsulfanyl compound (V). The cyclization of (V) at 240 C in diphenyl ether affords 6,7-difluoro-4-hydroxy-2-(methoxymethylsulfanyl)quinoline-3-carboxylic acid ethyl ester (VI), which by treatment with HCl in ethanol gives the corresponding mercapto compound (VII). The cyclization of (VII) with 1,1-dibromoethane by means of K2CO3 and KI in hot DMF yields 5,6-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid ethyl ester (VIII), which is condensed with piperazine (IX) in DMF to afford the corresponding piperazino-derivative (X). The hydrolysis of (X) with KOH in hot tert-butanol gives the corresponding free acid (XI) , which is finally condensed with 4-(bromomethyl)-5-methyl-1,3-dioxol-2-one (XII) by means of KHCO3 in DMF.
......................
Treatment of 3,4-difluoroaniline (I) with CS2 and Et3N gives triethylammonium dithiocarbamate (II), which reacts with ethyl chloroformate in chloroform to yield (III). Isothiocyanate (III) is converted into the potassium salt (IV) by reaction with diethyl malonate and KOH in dioxane and then transformed into methoxymethyl thioether (VI) by means of reagent (V) and Et3N in toluene. Cyclization of (VI) by heating in diphenyl ether affords quinoline (VII), which then reacts with benzoyl chloride (VIII) in pyridine to furnish (IX). Benzoyloxy derivative (IX) is converted into (X) by means of HCl in EtOH, and its reaction with 1-bromo-2-fluoroethane (XI) and NaHCO3 yields compound (XII). Chlorination of (XII) with SO2Cl2 in hexane provides (XIII), which by simultaneous hydrolysis and intramolecular cyclization by means of Et3N /H2O in THF provides the mixture of isomers (XIV). (+)-(XV) is obtained by HPLC chromatography of (XIV) on a chiral stationary phase. Treatment of (+)-(XV) with 1-methylpiperazine (XVI) in DMF provides ethyl ester (+)-(XVII), which is finally hydrolyzed by means of H2SO4 in H2O.
INTERMEDIATES
154330-67-3
Ethyl 6,7-difluoro-2-ethylmercapto-4-hydroxyquinoline-3-carboxylate
154330-68-4
Ethyl 4-acetoxy-6,7-difluoro-2-(ethylthio)quinoline-3-carboxylate
113046-72-3
Ethyl 6,7-difluoro-1-methyl-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate
113028-17-4
Ethyl 6-fluoro-1-methyl-4-oxo-7-(1-piprazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylate
112984-60-8
6-Fluoro-1-methyl-4-oxo-7-(1-piperazinyl)-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid
REFERENCES
- Nelson, Jennifer M.; Chiller, Tom M.; Powers, John H.; Angulo, Frederick J. (2007). "Food Safety: Fluoroquinolone‐Resistant Campylobacter Species and the Withdrawal of Fluoroquinolones from Use in Poultry: A Public Health Success Story". Clinical Infectious Diseases 44 (7): 977–80. doi:10.1086/512369. PMID 17342653.
- Kawahara S (1998). "[Chemotherapeutic agents under study]". Nippon Rinsho (in Japanese) 56 (12): 3096–9. PMID 9883617.
- Fritsche, T. R.; Biedenbach, D. J.; Jones, R. N. (2008). "Antimicrobial Activity of Prulifloxacin Tested against a Worldwide Collection of Gastroenteritis-Producing Pathogens, Including Those Causing Traveler's Diarrhea". Antimicrobial Agents and Chemotherapy 53 (3): 1221–4. doi:10.1128/AAC.01260-08. PMC 2650572.PMID 19114678.
- Giannarini, Gianluca; Tascini, Carlo; Selli, Cesare (2009). "Prulifloxacin: clinical studies of a broad-spectrum quinolone agent". Future Microbiology 4 (1): 13–24.doi:10.2217/17460913.4.1.13. PMID 19207096.
- JP patent 1294680, Kise Masahiro; Kitano Masahiko; Ozaki Masakuni; Kazuno Kenji; Matsuda Masato; Shirahase Ichiro; Segawa Jun, "Quinolinecarboxylic Acid Derivative", issued November 28, 1989
- Prulifloxacin. Drugfuture.com. Retrieved on 2010-11-03.
- Anonymous (2002). "Prulifloxacin ['Quisnon'; Nippon Shinyaku] has been approved in Japan". Inpharma 1 (1362): 22.
- Research and Development Department of Angelini. Angelinipharma.com. Retrieved on 2010-11-03.
- Nippon Shinyaku, Annual Report 2007
- "Prulifloxacin. NAD-441A, NM 441, Quisnon". Drugs in R&D 3 (6): 426–30. 2002.PMID 12516950.
- Annual Report 2008, p. 34
Segawa,J,Mashiko kitano, Kenji Kazuno et al, Studies on Pyridonecarboxylic acids,1.Sythesis and antibacterial Evaluation of 7-substituted-6-halo-4-oxo-4H-[1,3]thiazeto [3,2-]quionoline- 3-caroboxylic acids[J].J Med Chem. 1992,35(25):4727-4738.
Masato Matsuoka, Jun Segawa, Yoshihiko.et al, Studies on Pyridone Carb oxylic acids. V.A Practial synthesis of Ethyl 6,7–Difuoro-1-methyl-4-oxo-[1,3] Thiazeto [3,2-a]quinoline-3- Caroboxylate a key intermediate for the new tricyclic quinolone, prulifloxacin (NM441) and Versatile new syntheses of the 2-thioquinoline Skeleton[J].J Heterocyclic Chem.1997,34,1773-1779.
3-13-1996
|
Sustained release capsule
| |
10-11-1995
|
Method of manufacturing solid dispersion
| |
2-5-1992
|
7(4-(5 METHYL-2-OXO-1,3-DIOXALEN-4-YL)METHYL 1-PIPERZINYL)-4-OXO-4H-(1,3)THIAZETO(3,2-A)QUINOLINE-3-CARBOXYLIC ACIDS
|
6-31-2011
|
PHARMACEUTICAL COMPOSITION
| |
2-11-2011
|
PROCESS FOR THE PREPARATION OF PURE PRULIFLOXACIN
| |
8-6-2010
|
PROCESS FOR PREPARATION OF PRULIFLOXACIN USING NOVEL INTERMEDIATES
| |
5-7-2010
|
PROCESS FOR THE PREPARATION OF CRYSTALS OF PRULIFLOXACIN
| |
4-9-2010
|
COMPOSITION COMPRISING AN ANTIBIOTIC AND A CORTICOSTEROID
| |
12-11-2009
|
Compounds and Methods for modulating the Silencing of a Polynucleotide of Interest
| |
8-24-2007
|
PHARMACEUTICAL COMPOSITION
| |
6-29-2007
|
PHARMACEUTICAL COMPOSITION
| |
7-15-2005
|
Pharmaceutical composition
| |
2-6-2004
|
Medicinal composition
|
| WO2008059512A1 | Nov 17, 2006 | May 22, 2008 | Hetero Drugs Ltd | Process for preparation of prulifloxacin using novel intermediates |
| WO2008111016A1 | Mar 14, 2008 | Sep 18, 2008 | Ranbaxy Lab Ltd | Process for the preparation of pure prulifloxacin |
| WO2008111018A2 | Mar 14, 2008 | Sep 18, 2008 | Ranbaxy Lab Ltd | Process for the preparation of crystals of prulifloxacin |
| WO2010084508A2 | Dec 10, 2009 | Jul 29, 2010 | Elder Pharmaceuticals Ltd. | Process for the preparation of type i, type ii and type iii crystalline prulifloxacin |
| EP0315828A1 * | Oct 26, 1988 | May 17, 1989 | Nippon Shinyaku Company, Limited | Quinolinecarboxylic acid derivatives |
| EP1626051A1 | Apr 28, 2004 | Feb 15, 2006 | Nippon Shinyaku Co., Ltd. | Crystals of quinolinecarboxylic acid derivative solvate |
| US5086049 | Apr 8, 1991 | Feb 4, 1992 | Nipponshinyaku Co., Ltd. | 7[4-(5 methyl-2-oxo-1,3-dioxalen-4-yl)methyl 1-piperzinyl]-4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acids |
| US20070149540 | Apr 28, 2004 | Jun 28, 2007 | Nippon Shinyaky Co., Ltd. | Crystals of quinolinecarboxylic acid derivative solvate |
EXTRA INFO
http://www.google.com/patents/EP2524922A1?cl=en
- formula 1 is S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl]-4-oxo-4H-[1,3]thiazet o[3,2-α]quinoline-3-carboxylic acid (levo-prulifloxacin for short); its stereo configuration is S configuration; it has optical property of levorotatory polarized light:
 S-(-) ulifloxacin (as shown in formula 2 below) as raw material and the compound as shown in the following formula 3 are reacted in organic solvent in the presence of alkaline material. The reaction formula is shown below:
S-(-) ulifloxacin (as shown in formula 2 below) as raw material and the compound as shown in the following formula 3 are reacted in organic solvent in the presence of alkaline material. The reaction formula is shown below:

- S-(-)-ulifloxacin and R-(+)-ulifloxacin are prepared according to the method disclosed in CN101550142A .
- Japanese scholars Masato Matsuoka et al. have proved the absolute configuration of optically pure prulifloxacin. The study (see the publication Chem. Pharm. Bull. 43(7) 1238-1240 (1995)) verifies that (-)-ulifloxacin is S configuration while (+)-ulifloxacin is the enantiomer of R configuration by applying chemical methods together with single-crystal X-ray diffraction.
- Accordingly, R-prulifloxacin can be prepared from R-(+)-ulifloxacin and the compound of formula 3 by the method described hereinbefore.
- [0022]The reaction formula is depicted below:

- S-prulifloxacin prepared in accordance with the present invention is determined to be laevorotatory by optical rotation measurement, so it is S-(-)-prulifloxacin. R-prulifloxacin prepared in accordance with the present invention is determined to be dextrorotatory by optical rotation measurement, so it is R-(+)-prulifloxacin.
- The present invention studied the absorption features of S-(-)-prulifloxacin and R-(+)-prulifloxacin on circular polarized light by circular dichroism spectroscopy. The two spectrograms are mirror images of each other, which proves that S-(-)-prulifloxacin and R-(+)-prulifloxacin are enantiomer of each other.
- Comparing the circular dichroism spectrogram as depicted in figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it is found that (-)-prulifloxacin has similar Cotton effect to the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
- The compound of the present invention and physiologically acceptable acid can be prepared to salts: dissolving or suspending S-(-)-prulifloxacin in solvent such as chloroform, DMF and the like; adding into acid or acid solution (for example, hydrochloric acid or hydrogen chloride-methanol solution and the like) while stirring; precipitating and filtering to obtain solid salt from the solvent solution, or alternatively removing solvent from the salt solution directly by concentration, spray drying and the like to obtain the salt of S-(-)-prulifloxacin. The obtained solid may be further recrystallized.
- 105 g of racemic uliflourxacin was dissolved in 1,500 mL of dimethyl sulfoxide. 27 g of D-tartaric acid was dissolved in 405 mL of dimethyl sulfoxide dropwise while stirring. After stirring at room temperature for 20 hours, the precipitate was filtrated. The collected solid was dried under vacuum to obtain 86 g solid, which was recrystallized in dimethyl sulfoxide to obtain 37 g of levoulifloxacin-D-tartrate, with C49.08%, H5.06%, N9.50%, S7.44% shown by elemental analysis (molecular formula: C16H16FN3O3S·1/2C4H6O6·H2O, calculated values: C48.86%, H4.78, N9.50%, S7.25%). Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After precipitation, filtration, and drying, 24.5 g of (S)-uliflourxacin was obtained, having a chemical name (S)-(-)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto [3,2-α]quinoline-3-carboxylic acid.
- Specific rotation [α]20 D= -133° (c=0.5, 0.1 mol/L methanesulfonic acid); 1H-NMR (DMSO-d6) δ2.11 (3H, d, j=6.2 Hz), 2.87 (4H, m), 3.19 (4H, m), 6.40 (1H, q, j=6.2 Hz), 6.89 (1H, d, j=7.4Hz), 7.79 (1H, d, j=13.9Hz), optical purity e.e. 96%.
- 105 g of racemic uliflourxacin was dissolved in 1,500 mL of DMSO. 27 g of L-tartaric acid was dissolved in 405 mL dimethyl sulfoxide dropwise while stirring to allow that the solution became turbid and the precipitation occurred. The solution was stirred at room temperature for 20 hours and then filtered. The collected solid was dried under vacuum to obtain 82 g solid which was recrystallized in dimethyl sulfoxide to obtain 34 g of dextrouliflourxacin-L-tartarte. Said salt was added into water to obtain a suspension, and the pH value was adjusted to 7-8 with 2% NaOH aqueous solution while stirring. After filtration and drying, 22 g of (R)-uliflourxacin was obtained, having a chemical name (R)-(+)-6-fluoro-1-methyl-4-oxo-(1-piperazinyl)-1H,4H-[1,3]thiazeto[3,2-a]quinoline -3-carboxylic acid.
- Specific rotation [α]20 D= +132.4° (c=0.5, 0.1 mol/L methanesulfonic acid), optical purity e.e. 96%.
- 3.49 g (0.01 mol) of S-(-)-uliflourxacin prepared in Example 1, 2.02 g (0.02 mol) of triethylamine and 20 ml of dimethylformamide (hereinafter referred to as DMF) were mixed and stirred. After the solution was cooled to -5∼5 °C, 0.012 mol of 4-bromomethyl-5-methyl-1,3-dioxolen-2-one (hereinafter referred to as DMDO-Br) in DMF (5 ml) solution was added thereinto, followed by stirring at -5∼5 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, stirred for 30 minutes, and then filtered. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.9 g of S-(-)-prulifloxacin was obtained, having a chemical name: S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl ]-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylic acid, with a purity of 98% and a yield rate of 63%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
- R-(+)-prulifloxacin prepared in Example 2 was used as raw material to prepare 2.7 g of target product R-(+)-prulifloxacin in accordance with the method as described in Example 3, with a yield rate of 60.7% and a purity of 98%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
- Comparing the circular dichroism spectrogram as depicted in Figure 4 with the circular dichroism spectrogram of analogue of the similar structure with known absolute configuration as disclosed in the publication Chem. Pharm. Bull. 47(12) 1765-1773 (1999), it was found that (-)-prulifloxacin has similar Cotton effect with the two analogues reported in the publication, ethyl S-(-)-6,7-difluoro-1-methyl-4-oxo-4H-[1,3] thiazeto[3,2-α]quinoline-3-carboxylate and ethyl S-(-)-6, 7-difluoro-1-fluoromethyl-4-oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylate; so does (+)-prulifloxacin. The results also verify on the other hand that the absolute configuration of levo-prulifloxacin of the present invention is S type while the absolute configuration of dextro-prulifloxacin is R type.
- Conclusion: The absolute configuration of the sample prepared in Example 3 is S configuration, as shown in the formula below:

- 3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.2 g (0.012 mol) of anhydrous potassium bicarbonate and 20 ml of dimethylsulfoxide were mixed and stirred. 0.012 mol of DMDO-Br in DMSO (5 mL) solution was added dropwise at -20 °C. Stirring proceeded at -20 °C for 3 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
- 3.49 g (0.01 mol) of S-(-)-uliflourxacin, 1.04 g (0.008 mol) of N,N-diisopropylethylamine and 20 mL of N,N-dimethylformamide (DMF) was mixed and stirred, 0.008 mol of DMDO-Br in DMF (5 mL) solution was added thereinto. The solution was heating to 60 °C and reacted for 15 minutes. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.0 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 43%.
Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid)
- 10 g (0.029 mol) of S-(-)-uliflourxacin, 30 ml of N,N-dimethylacetylamide and 14.7 g (0.145mol) of triethylamine was mixed and cooled to 5~10 °C. 8.5 g (0.03 mol) 4-(p-toluenesulfonic acid-1-methyl ester)-5-methyl-1,3-dioxolen-2-one in 25 ml of N,N-dimethylacetylamide solution was added dropwise while stirring. After addition, the solution was reacted at room temperature for 10 hours. The reaction solution was poured into 200 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 7.46 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 57%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
- 3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.05 mol) of potassium carbonate and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.012 mol of DMDO-Br in DMF (5ml) solution was added at -10 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.2 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 48%. Specific rotation [α]20 D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
- 3.49 g (0.01 mol) of S-(-)-uliflourxacin, 0.79 g (0.02 mol) of diisopropylamine and 20 ml of dimethylformamide (DMF) was mixed and stirred. 0.02 mol of DMDO-Br in DMF (5ml) solution was added at 0 °C. At the same temperature, the solution was reacted for 2 hours. The reaction solution was poured into 100 ml of ice water, and the pH value was adjusted to 7 with 20% acetic acid. The solution was filtered after stirring for 30 minutes. The filter residue was washed with water. The solid was collected and dried under vacuum. After recrystallization from acetonitrile, 2.5 g of the target product levo-prulifloxacin was obtained with a purity of 98% and a yield rate of 54%. Specific rotation [α]20D= -108° (c=0.5, 0.1 mol/L methanesulfonic acid).
- In accordance with the method as described in Example 5, the raw material R-(+)-prulifloxacin was prepared to 2.5 g of the target product R-(+)-prulifloxacin with a purity of 98% and a yield rate of 54%. Specific rotation [α]20 D= +108° (c=0.5, 0.1 mol/L methanesulfonic acid).
- S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl] -4-oxo-4H-[1,3]thiazeto[3,2-a]quinoline-3-carboxylic acid hydrochloride
- 0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 33% (v/v) hydrochloric acid- methanol solution was added while stirring. The solution was filtered and the filtration residue was washed with methanol. The collected solid was dried to obtain 450 mg said compound with a yield rate of 83%. The melting point of the product is higher than 220 °C (the sample became darker during the test).
- S-(-)-6-fluoro-1-methyl-7-[4-(5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl-1-piperazinyl] -4- oxo-4H-[1,3]thiazeto[3,2-α]quinoline-3-carboxylic acid mesylate
- 0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 50% methanesulfonic acid- methanol solution was added while stirring. The solution was filtered and the filtration residue was washed with methanol. The collected yellow solid was dried with calcium chloride under vacuum for 24 hours and further dried with calcium chloride at 80 °C under vacuum for 5 hours to obtain 470 mg said compound with a yield rate of 78%. The melting point of the product is higher than 220 °C (the sample became darker during the test).
- 0.5 g of S-(-)-prulifloxacin was dissolved in 15 mL of chloroform and then 0.5 mL of 33% (v/v) hydrochloric acid- methanol solution was added while stirring. The solution was dried by evaporation. Methanol was added to the residue and stirred for 10 minutes. The solution was filtered and the filtration residue was washed with methanol. The collected solid was dried to obtain 460 mg said compound with a yield rate of 85%.
12
FANDOFLOXACIN
DW-116; fandofloxacin
164150-99-6 FREE BASE ,
164151-00-2., 164150-85-0
6-fluoro-1-(5-fluoropyridin-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxoquinoline-3-carboxylic acid
6-fluoro-1-(5-fluoropyridin-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxoquinoline-3-carboxylic acid
6-Fluoro-1-(5-fluoropyridin-2-yl)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Dong Wha Pharmaceutical Co Ltd
Molecular Formula: C20H18F2N4O3
Molecular Weight: 400.378726
synthesis.............http://www.drugfuture.com/synth/syndata.aspx?ID=226498
http://www.google.com.mx/patents/WO1995005373A1
synthesis 1
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
synthesis 2
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
Synthesis, pharmacokinetics, and biological activity of a series of new pyridonecarboxylic acid antibacterial agents bearing a 5-fluoro-2-pyridyl group or a 3-fluoro-4-pyridyl group at N-1
J Heterocycl Chem 1997, 34(3): 1021
J Heterocycl Chem 1997, 34(3): 1021
6-31-2011
|
PHARMACEUTICAL COMPOSITION
| |
8-24-2007
|
PHARMACEUTICAL COMPOSITION
| |
6-29-2007
|
PHARMACEUTICAL COMPOSITION
| |
10-28-2005
|
Identification and use of effectors and allosteric molecules for the alteration of gene expression
| |
7-15-2005
|
Pharmaceutical composition
| |
2-6-2004
|
Medicinal composition
| |
4-20-2000
|
NOVEL QUINOLONE CARBOXYLIC ACID DERIVATIVES
| |
3-6-1996
|
Quinolone carboxylic acid derivatives
| |
2-24-1995
|
NOVEL QUINOLONE CARBOXYLIC ACID DERIVATIVES
|
13 ACH 702
ACH-702
7-[3(R)-(2-Aminopropan-2-yl)pyrrolidin-1-yl]-9-cyclopropyl-6-fluoro-8-methoxy-2,3,4,9-tetrahydroisothiazolo[5,4-b]quinoline-3,4-dione
(7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE
(R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione
922491-46-1 free base
922491-09-6 (hydrochloride)468.973, C21 H25 F N4 O3 S . Cl H
ACH-0139586
ACH-702
ACH-702
Achillion Pharmaceuticals (USA)
pre clinical
Achillion Pharmaceuticals is working on the discovery of compounds in a new subclass of quinolones, the isothiazoloquinolones. The most advanced compound is ACH-702, which is at the pre-clinical stage of development [1-3].
ACH 702
The utility of isothiazoloquinolines as pharmaceutical agents has been discussed in the literature. For example, Pinol, et al discussed the use of isothiazoloquinolines as medical bactericides in US Patent 5,087,621, including
The Proctor & Gamble Company discussed antimicrobial quinolones including the following compound:
in published application no. US 2003008894.
The use of isothiazoloquinoline compounds as TNF production inhibitors has also been discussed, for example by Sankyo Co., Ltd. in JPl 010149, which includes the following compound
Bayer Aktiengesellschaft has discussed bicycle[3.3.0]oct-7-yl containing compounds useful for treating H. pylori infections in WO 98/26768, including isothiazoloquinolines, having the general structure shown below in which Y may be sulfur joined to the carboxamide group to form a 5-membered ring
Otsuka Pharmaceutical Co., Ltd. has discussed the use of isothiazoloquinolines as antibacterial agents in JP 01193275, including the following carbamate-containing compound
Abbott Laboratories has discussed the use of isothiazoloquinolines as antineoplastic agents in US Patent No. 5,071,848 and has discussed the use of tricyclic quinolones as antibacterial agents in US 4,767,762. The Abbott compounds have hydrogen, halogen, or lower alkyl as substituents at the 6- and 8- positions of the isothiazoloquinoline core.
..................
Synthesis
WO2008021491A2
http://www.google.com/patents/WO2008021491A2?cl=en
EXAMPLE 1. SYNTHESIS OF (7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-
CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE (5). Step 1. Ethyl l-cyclopropyl-6, 7-difluoro-2-methanesulfonyl-8-methoxy-4-oxo-l,4-dihydro- quinoline-3-carboxylate (6)
Oxonβ®
MeOHZH2O
1
6
Water (180 mL), followed by Oxone® (Dupont Specialty Chemicals) (170 g, 277 mmol), is added to a suspension of 1 in MeOH (510 mL). The reaction mixture is heated with stirring at 55-60 0C for 3 h. The reaction mixture is cooled to room temperature, diluted with water (40 mL), and stirred at 5 0C (ice bath) for 30 min. The resulting crystals are collected by filtration, washed with water (2 x 100 mL), and dried to afford 6 (13.8 g). This material was used in the next step without further purification, mp 177-178 0C. 1H NMR (DMF-^7): J0.62 (m, IH), 1.11 (m, 2H), 1.29 (m, IH), 1.32 (t, JH-H = 7.0 Hz, 3H), 3.76 (s, 3H), 4.18 (m, IH), 4.21 (d, JH-F = 2.0 Hz, 3H), 4.33 (q, JH-H = 7.0 Hz, 2H), 7.64 (dd, JH-F = 10.0 Hz, 8.5 Hz, IH). Step 2 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2- methanesulfonyl-8-methoxy-4-oxo-l , 4-dihydro-quinoline-3-carboxylic acid ethyl ester (7)
10 6 7
A mixture containing compound (6) (3.88 g, 9.67 mmol), compound 10 (1.64 g, 12.8 mmol), anhydrous DIEA (5.05 g, 39.1 mmol, dried over 4A sieves), and anhydrous DMF (40 mL) is heated at 70 0C under an atmosphere of argon gas. After heating for 4.5 h (LC-MS analysis shows ~7% compound (6) remained), the reaction mixture is cooled to room temperature, diluted with EtOAc (200 mL), and washed with water (100 mL). The aqueous layer is extracted with EtOAc (100 mL), and the combined organic layers are washed with a saturated aqueous solution of sodium bicarbonate (100 mL). The organic layer is diluted with water (100 mL) and treated with an aqueous solution of HCl (4 N) until the aqueous layer is acidic (pH 2—3 after shaking the mixture vigorously). The organic layer is separated, and this process is repeated. The combined aqueous layers are diluted with EtOAc (100 mL) and treated with an aqueous solution of sodium hydroxide (6 N) until the aqueous layer is basic (pH ~8 after shaking the mixture vigorously). The aqueous layer is separated, and this process is repeated. The combined organic layers are dried over magnesium sulfate, filtered, and concentrated under reduced pressure giving an orange solid (3.27 g of an~80:20 mixture of compound (7) and impurity B). This solid is recrystallized from hot EtOAc (~60 mL) furnishing 2.18 g (44% yield) of pure compound 7 as a bright yellow solid. LC-MS mlz calcd for C24H32FN3O6S 509 ([M+]); found 510 ([M + H]+).
This reaction should not be allowed to proceed for more than a few hours (not overnight) as prolonged reaction time can lead to the formation of more side products. The product should be —95% pure (based on HPLC), with only a trace amount of impurity B. Step 3. (R)-7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2-mercapto-8- methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid ethyl ester (8)
7 8
Compound 7 (1.04 g, 2.04 mmol) is partially dissolved in DME (40 mL) under an atmosphere of argon. Sodium hydrosulfide hydrate (Aldrich, 72.6% by titration, 465 mg, 6.02 mmol) in water (3.0 mL) is added to this solution. The resulting mixture is sparged slowly with argon for 30 min.
The progress of the reaction is monitored by HPLC-MS, and judged to be complete (<2% of 7 remains) after 11.5 h. Excess sodium hydrosulfide is quenched upon addition of aq HCl (4.5 mL, 4 N).
The resulting orange solution (pH ~2) is sparged with argon (30 min) to remove the generated hydrogen sulfide. Step. 4 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione (5)
5= ACH 702
A solution of potassium carbonate (4.26 g, 30.8 mmol) in water (25 mL) is next added to this solution to give a clear yellow solution (pH 9-10). The clear yellow solution is then sparged with argon for ~5 min. Finally, hydroxylamine-0-sulfonic acid (0.93 g, 8.2 mmol) is added portionwise as a solid, with immediate evolution of gas and formation of the product as a yellow precipitate. After stirring for 16 h, the reaction mixture (pH 10.2) is acidified with aq HCl to pH 8.3 (the approximate isoelectric point of 5) causing additional product to precipitate from solution. The reaction mixture is concentrated under reduced pressure (final volume -40 mL). The yellow precipitate is collected by centrifugation, washed with water (3 x 40 mL, with sonication), and lyophilized to give 0.80 g of 5.
..........................
WO2007014308A1
http://www.google.com/patents/WO2007014308A1?cl=en
EXAMPLE 5. SYNTHESIS OF I-METHYL-I-PYRROLIDIN-3-YL ETHYLAMINE (5)
1 -Methyl- l-pyrrolidin-3-yl-ethylamine is prepared in accordance with the synthetic scheme below.
N O
5 P
Step 1. Synthesis of (S)-l-benzylpyrrolidin-3-yl methanesulfonate (N).
Methanesulfonyl chloride (15 mL, 0.19 mol) is added to a cooled (0 0C) solution of toluene (300 mL) containing (5)-l-benzylpyrrolidin-3-ol (24.5 g, 0.14 mol) and triethylamine (80 mL, 0.57 mol). The resulting mixture is stirred at 0 °C for 15 min, and allowed to warm to room temperature with stirring for 2h. The mixture is quenched with a 5% aqueous solution of sodium bicarbonate (250 mL). The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 250 mL), washed with water (I x 250 mL), dried over magnesium sulfate, and concentrated under reduced pressure to give N (35.1 g, 99 %) as an orange oil. 1H NMR (300 MHz, CDCl3): £2.07 (m, IH), 2.30 (m, IH), 2.49 (m, IH), 2.75-2.90 (m, 3H), 2.98 (s, 3H), 3.61 (d, J= 13.0 Hz, IH), 3.68 (d, J= 13.0 Hz, IH), 5.18 (m, IH), 7.15-7.30 (m, 5H). LCMS mlz calcd for C12H17NO3S 255 ([M+]); found 256 ([M + H]+, 100%), 160 (40%). Steps 2 and 3. Syntheses of(R)-l-benzylpyrrolidine-3~carbonitrile (O) and 2-((R)-I- benzylpyrrolidin-3-yl)propan-2-amine (P).
The syntheses of O and P are described previously by Fedij et al. (Fedij, V.; Lenoir, E. A., Ill; Suto, M. J.; Zeller, J. R.; Wemple, J. Tetrahedron: Asymmetry 1994, J, 1131- 1134). Step 4. Synthesis ofl-((R)-Methyl~l-pyrrolidin-3-yl)-ethylamine (5).
A mixture containing P (7.4 g), 20% palladium hydroxide on carbon (7.5 g), and ethanol (75 niL) is stirred under an atmosphere of hydrogen gas (50 psi) at 45 °C for 24 h. The mixture is filtered and the filtrate is concentrated under reduced pressure to give 5 (4.1 g, 95 %) as a yellow oil. This material is stored under an atmosphere of argon gas. 1H NMR (300 MHz, CDCl3): J1.09 (s, 6H), 1.51 (m, IH), 1.64 (br s, 3H), 1.81 (m, IH), 2.06 (apparent pentet, J= 8.5 Hz, IH), 2.69 (dd, J= 11.0 Hz, J= 8.5 Hz, IH), 2.94 (m, 2H), 3.00 (dd, J= 11.0 Hz, J= 8.5 Hz, IH). LCMS mlz calcd for C7H16N2 128 ([M+]); found 129 ([M + H]+, 60%), 112 (100%).
EXAMPLE 6. GENERAL METHOD FOR THE FINAL AMINE-COUPLING STEP: SYNTHESIS OF 7-((R)-3-
(2-AMINOPROPAN-2-YL)PYRROLIDIN- 1 -YL)-9-CYCLOPROPYL-6-FLUORO-8- METHOXYISOTHIAZOLO[5,4-B]QUINOLINE-3 ,4(2H,9H)-DIONE HYDROCHLORIDE
[0261 ] 7-((R)-3-(2-Aminopropan-2-yl)pyrrolidin- 1 -yl)-9-cyclopropyl-6-fluoro-8- methoxyisothiazolo[5,4-b]quinoline-3,4(2H,9H)-dione hydrochloride is prepared in accordance with the synthetic scheme below.
Synthesis ofJ-ffRJS-^-aminopropan^-ylJpyrrolidin-l-ylJ-P-cyclopropyl-o-fluoro-S- methoxyisothiazolofS, 4-bJguinoline-3,4(2H, 9H)-dione hydrochloride (6).
Under an atmosphere of argon, a reaction vessel is charged with 5 (206.0 mg, 1.6 mmol), 3 (328.6 mg, 1.0 mmol), dimethyl sulfoxide (4.5 mL), and ΛζN-diisopropylethylamine (750 μL, 4.3 mmol). The resulting mixture is irradiated with microwaves (CEM Discover) at 125 0C for 1 h (conventional heating may also be used — 115 °C in an oil bath for 14 h), allowed to cool, and evaporated to dryness under reduced pressure (-70 °C/2-3 mm Hg). The oily residue is triturated with ethyl acetate (15 mL) and the resulting powder is collected by centrifugation. This solid is purified using preparative HPLC to give the desired product. Preparative HPLC is performed using a YMC Pack Pro C18 150 x 30.0 mm 5//m column coupled to a YMC Pack Pro 50 x 20 mm 5/an column with an isocratic elution of 0.37 min at 95:5 H2OiCHsCN containing 0.1% TFA followed by a 15.94 min linear gradient elution from 95:5 to 25:75, followed by a 0.69 min linear gradient from 25:75 to 5:95 at a flow rate of 30.0 mL/min with UV detection at 254. The crude material is loaded as a solution containing acetic acid (~2 mL), methanol (~1 mL), and water (~1 mL). The purified product is isolated as the TFA salt and is converted to the corresponding hydrochloride salt by addition of a solution of hydrogen chloride (~1.25 M in methanol) followed by evaporation; this process is repeated twice to give a yellow solid. Purity by HPLC: >99%; tR = 10.08 min. 1H NMR (300 MHz, TFA-d): δ 1.28 (m, 2H), 1.53 (m, 2H), 1.66 (s, 6H), 2.43 (m, IH), 2.57 (m, IH), 3.35 (m, IH), 3.97 (s, 3H), 4.01-4.38 (m, 5H), 8.17 (d, J= 12.0 Hz, IH, aromatic). 19F(1H) (282 MHz, TFA-J): δ-\ 18.0 (s). 13C(1H) (75 MHz, TFA-d): £13.5, 13.9, 25.0, 25.1, 29.1, 39.7, 49.6, 59.4 (br, W1/2 « 14 Hz), 59.8 (br, W1/2 « 14 Hz), 60.0, 66.8, 106.0, 112.1 (dJc_F = 23.0 Hz), 137.5 (br m, W1/2 « 24 Hz), 138.4, 144.8 (br, W1/2 » 10 Hz), 155.3 (dJc_F = 255.0 Hz), 169.8, 170.1, 171.5 (br, W1/2 « 9 Hz). LCMS mlz calcd for C21H25FN4O3S 432 ([M+]); found 433 ([M + H]+). Anal. Calcd for C21H25FN4O3S-l.5HCM.5H2O: C, 49.05; H, 5.78; N, 10.90; Cl, 10.34. Found: C, 49.30; H, 5.60; N, 10.83; Cl, 10.00.
EXAMPLE 3. SYNTHESIS OF 9-CYCLθPRθPYL-6,7-DiFLUθRθ-8-METHθχγ-9H-isoτHiAzθLθ[5,4- 5]QUlNOLlNE-3,4-DIONE (Compound 3).
9-Cyclopropyl-6,7-difluoro-8-methoxy-9H-isothiazolo[5,4-&]quinoline-3,4-dione (3) is prepared in accordance with the synthetic scheme below.
Step 1. Synthesis of 2,4, 5-trifluoro-3-methoxybenzoyl chloride (A)
A mixture of 2,4,5-trifluoro-3-methoxybenzoic acid (154 mg, 0.75 mmol) and thionyl chloride (8 mL) is refluxed for 4 h. Excess thionyl chloride is removed in vacuo, and the remaining residue is used directly in the next synthetic step. Step 2. Synthesis of (Z)-ethyl 3-hydroxy-3-(2,4,5-trifluoro-3-methoxyphenyl)aaγlate (B).
Compound B is prepared using the general method of Wierenga and Skulnick (Wierenga, W.; Skulnick, H. I. J. Org. Chein. (1979) 44: 310-311). H-Butyllithium (1.6 M in hexanes) is added to a cooled (-78 °C) solution of tetrahydrofliran (10 mL) containing ethyl hydrogen malonate (180 juL, 1.50 mmol) and 2,2'-bipyridyl (~1 mg as indicator). The temperature of the reaction mixture is allowed to rise to ca. -5 0C during the addition of n- butyllithium. Sufficient n-butyllithium (2.8 mL, 4.48 mmol) is added until a pink color persists at -5 0C for 5-10 min. A solution of 2,4,5-trifluoro-3-methoxybenzoyl chloride (0.75 mmol, vide supra) in tetrahydrofuran (~3 mL) is added in one portion to the reaction mixture that had been recooled to -78 0C. The resulting mixture is allowed to warm to room temperature, diluted with ethyl acetate (50 mL), and quenched with a 1 M aqueous solution of hydrochloric acid. The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 30 mL), followed by brine (2 x 50 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 20% v/v ethyl acetate in hexanes) to give pure B as a white solid. 1H NMR (300 MHz, CDCl3): (enol, predominant tautomer, >90%) δ 1.32 (t, JH-H = 7.0 Hz, 3H, CO2CH2CH3), 4.02 (apparent t, JH-F = 1.0 Hz, 3H, OCH3), 4.25 (q, JH-H = 7.0 Hz, 2H, CO2CH2CH3), 5.79 (s, IH, CH3C(OH)=CH- CO2CH2CH3), 7.39 (ddd, JH_F= 11.0 Hz, 8.5 Hz, 6.5 Hz, IH, aromatic), 12.68 (s, IH, OH). 19F(1H) NMR (282 MHz, CDCl3): <5-146.8 (dd, JF_F = 21.5 Hz, 10.5 Hz, IF), -140.2 (dd, JF_F = 21.5 Hz, 13.5 Hz, IF), -131.3 (dd, JF_F = 13.5 Hz, 10.5 Hz, IF).
Step 3. Synthesis ofζEyethy^-^ZJ-N-cyclopropy^methylthioJcarbonoimidoylJS-hydroxyS- (2, 4, 5-trifluoro-3-methoxyphenyl)acrylate (C)
Sodium hydride (60% in mineral oil, 31 mg, 0.78 mmol) is added portionwise to a cooled (0 °C) solution containing B (200 mg, 0.73 mmol), cyclopropyl isothiocyanate (120 /JL, 1.2 mmol), and dimethylformamide (2 mL). The resulting mixture is allowed to warm to room temperature with stirring overnight (18 h). Methyl iodide (80 juL, 1.2 mmol) is added to the resulting solution and stirred for an additional 4 h (until TLC indicated the complete consumption of B). The reaction mixture is diluted with ethyl acetate (100 mL) and quenched by addition of a saturated aqueous solution of ammonium chloride (30 mL). The organic layer is washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 40% v/v ethyl acetate in hexanes) to give C as a yellow oil. 1H NMR (300 MHz, CDCl3): (50.86 (m, 2H, cyclopropyl CH2), 0.97 (m, 5H), 2.52 (s, 3H, SCH3), 3.00 (m, IH, cyclopropyl CH), 3.96 (q, JH-H = 7.0 Hz, 2H, CO2CH2CH3), 4.02 (apparent t, JH-F = 1.0 Hz, 3H, OCH3), 6.96 (m, IH, aromatic), 11.71 (s, IH). 19F(1H) NMR (282 MHz, CDCl3): £-149.9 (br, IF), -141.4 (br, IF), -135.7 (br, IF).
Step 4. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylthio)-4-oxo-l,4- dihydroquinoline-3-carboxylate (D)
Sodium hydride (60% in mineral oil, 82 mg, 2.1 mmol) is added portionwise to a solution of C (760 mg, 1.95 mmol) in dimethylformamide (15 mL) at room temperature. The reaction mixture is heated at 80 0C for 3 d (until TLC indicates the complete consumption of B), cooled to room temperature, and quenched by addition of a saturated aqueous solution of ammonium chloride (10 mL). The mixture is extracted with ethyl acetate (3 x 50 mL). The combined organic extracts are washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give crude D. This material is purified by flash column chromatography (eluting with 30% v/v ethyl acetate in hexanes) to D as a pale yellow oil.1H NMR (300 MHz, CDCl3): £0.73 (m, 2H, cyclopropyl CH2), 1.19 (m, 2H, cyclopropyl CH2), 1.38 (t, JH-H = 7.0 Hz, 3H, CO2CH2CH3), 2.66 (s, 3Η, SCH3), 3.74 (m, IH, cyclopropyl CH), 4.08 (d, JH-F = 2.5 Hz 3H, OCH3), 4.40 (q, JH_H = 7.0 Hz, 2H, CO2CH2CH3), 7.76 (dd, JH_F = 10.5 Hz, 8.5 Hz IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-146.8 (d, JF_F = 21.0 Hz, IF), - 137.7 (d, JF-F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO4S 369 ([M+]); found 370 ([M + H]+).
Step 5. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylsulfinyl)-4-oxo-l,4- dihydroquinoline-3-carboxylate (E)
m-Chloroperoxybenzoic acid (<77%, 34 mg, 0.15 mmol) is added in one portion to a solution of D (50 mg, 0.14 mmol) in methylene chloride (3 mL) at room temperature. The reaction mixture is stirred for 1 h, diluted with ethyl acetate (20 mL), and washed with a 5% aqueous solution of sodium bicarbonate (2 x 10 mL). The organic layer is dried over sodium sulfate and evaporated under reduced pressure to give the crude product. This material is purified by preparative thin-layer chromatography (eluting with 10% v/v hexanes in ethyl acetate) to give pure E as a white solid. 1H NMR (300 MHz, CDCl3): £0.62 (m, IH, cyclopropyl CH2), 1.00 (m, IH, cyclopropyl CH2), 1.13 (m, IH, cyclopropyl CH2), 1.29 (m, IH, cyclopropyl CH2), 1.36 (t, JH_H = 7.5 Hz, 3H, CO2CH2CH3), 3.22 (s, 3Η, S(O)CH3), 3.85 (m, IH, cyclopropyl CH), 4.09 (d, JH-F = 2.5 Hz, 3H, OCH3), 4.37 (q, JH-H = 7.5 Hz, 2H, CO2CH2CH3), 7.75 (dd, JH-F = 10.0, 8.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-145.2 (d, JF_F = 21.0 Hz, IF), -136.2 (d, JF_F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO5S 385 ([M+]); found 386 ([M + H]+).
Step 6. Synthesis of ethyl l-cyclopropyl-βJ-difluoro-l-mercaptoS-methoxy-^oxo-lA- dihydroquinoline-3-carboxylate (F).
Anhydrous sodium hydrogen sulfide (Alfa Aesar, 20 mg, 0.36 mmol) is added in one portion to a solution of DMF (6 mL) containing E (93 mg, 0.24 mmol) at room temperature. The resulting solution is heated at 40 0C for 2-3 h (until TLC indicated complete consumption of E) and allowed to cool to room temperature. The reaction mixture is quenched by addition of a 5% aqueous solution of hydrochloric acid (20 mL) and extracted with ethyl acetate (2 x 25 mL). The combined organic extracts are washed with brine (4 x 25 mL), dried over sodium sulfate, and evaporated to dryness under reduced pressure to give crude F in quantitative yield. This material is used directly in the next synthetic step to prevent its oxidative degradation. LCMS mlz calcd for C16H15F2NO4S 355 ([M+]); found 356 ([M + H]+) Step 7. Synthesis of9-cyclopropyl-6,7-difluoro-8-methoxyisothiazolo[5,4-b]quinoline- 3,4(2H,9H)-dione (3).
A solution of sodium bicarbonate (820 mg, 9.8 mmol) in water (14 mL) is added to a solution of F (348 mg, 0.98 mmol) in tetrahydrofuran (10 mL) at room temperature. Hydroxylamine-O-sulfonic acid (465 mg, 4.1 mmol) is added in one portion to this mixture. The reaction mixture is stirred at room temperature for ~3 h and quenched by addition of an aqueous solution of 5% hydrochloric acid (100 mL). The precipitate that formed is collected by filtration, washed with water (3 x 5 mL), and dried in vacuo to give 3 as a white solid. This product is of sufficient purity (>95% by 1H NMR spectroscopy) to use directly in the final amine-coupling step. 1HNMR (300 MHz, DMSO-J6): Jl.12 (m, 4H, cyclopropyl CH2), 3.85 (m, IH, cyclopropyl CH), 4.01 (d, JH-F= 1.5 Hz, 3H, OCH3), 7.85 (dd, JH_F = 11.0 Hz, 9.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, DMSO-J6): £-146.4 (d, JF_F = 23.0 Hz, IF), -140.2 (d, JF_ F = 23.0 Hz, IF). LCMS mlz calcd for C14H10F2N2O3S 324 ([M*]); found 325 ([M + H]+).
REFERENCES
- Achillion Pharmaceuticals. About ACH-702. Available online: http://www.achillion.com/PL/pdf/04_ach_702_bg.pdf (accessed on 2 May 2013).
- Pucci, M.J.; Podos, S.D.; Thanassi, J.A.; Leggio, M.J.; Bradbury, B.J.; Deshpande, M. In vitro and in vivoprofiles of ACH-702, an isothiazoloquinolone, against bacterial pathogens. Antimicrob. Agents Chemother. 2011, 55, 2860–2871, doi:10.1128/AAC.01666-10.
- Achillion Pharmaceuticals, Inc. SEC filling form 10-Q quarterly report filed August 7, 2013. Available online: http://ir.achillion.com/secfiling.cfm?filingID=1193125–13–324297 (accessed on 28 September 2013).
- An efficient method for the synthesis of (R)-3-(1-amino-1-methylethyl)pyrrolidines for the antiinfective agent, PD 138312
Tetrahedron Asymmetry 1994, 5(7): 1131 - WO 2007014308
- WO 2008021491
WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs HASHIMOTO, A. ET AL.: "Practical synthesis and molecular structure of a potent broad-spectrum antibacterial isothiazoloquinolone" ORG. PROCESS RESEARCH & DEVELOPMENT, vol. 11, 16 March 2007 (2007-03-16), pages 389-398, XP002465315 2 * WANG, Q. ET AL.: "Isothiazoloquinolones with Enhanced Antistaphylococcal Activities against Multidrug-Resistant Strains: Effects of Structural Modifications at the 6-, 7-, and 8-Positions" J. MED. CHEM., vol. 50, 2007, pages 199-210, XP002465316 WO2005019228A1 * Aug 4, 2004 Mar 3, 2005 Achillion Pharmaceuticals Inc Isothiazoloquinolones and related compounds as anti-infective agents WO2006118605A2 * Nov 10, 2005 Nov 9, 2006 Achillion Pharmaceuticals Inc 8a, 9-dihydro-4a-h-isothiazolo[5,4-b] quinoline-3, 4-diones and related compounds as anti-infective agents WO2007014308A1 * Jul 27, 2006 Feb 1, 2007 Achillion Pharmaceuticals Inc 8-methoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones and related compounds as anti-infective agents Citing Patent Filing date Publication date Applicant Title WO2008021491A2 * Aug 16, 2007 Feb 21, 2008 Achillion Pharmaceuticals Inc Method for synthesis of 8-alkoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs EP2488532A2 * Oct 15, 2010 Aug 22, 2012 Rib-X Pharmaceuticals, Inc. Antimicrobial compounds and methods of making and using the same US7902365 Aug 16, 2007 Mar 8, 2011 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones US8138346 Mar 4, 2011 Mar 20, 2012 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones
14 levonadifloxacin , wck 771
STEREOCENTERS SHOWN
Levonadifloxacin arginine salt, WCK 771
S-(−)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]quinolizine-2-carboxylic Acid l-Arginine Salt Tetrahydrate
RN: 306748-89-0
- C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
- L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)
WCK 771………..S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt tetrahydrate
(-)-9-Fluoro-8-(4-hydroxypiperidin-1-yl)-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic acid L-arginine salt hydrate
L-arginine salt of (S)-nadifloxacin
S-(-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[ij]qumorizine-2-carboxylic acid L-arginine salt is a broad-spectrum antibiotic, medically grouped together with the fluoroquinolone class of antibiotics, which is disclosed and claimed in U.S. patent 6,514,986 B2 as being isolated in a less crystalline anhydrate form and a more crystalline hydrate form.
U.S. patent 6,664,267 describes a crystalline monohydrate form of S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j] quinolizine-2-carboxylic acid L-arginine salt that is disclosed as having advantages over the anhydrate and hydrate forms described in US 6,514,986 B2.
SYNTHESIS
A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus
J Med Chem 2005, 48(16): 5232………..http://pubs.acs.org/doi/abs/10.1021/jm050035f
J Med Chem 2005, 48(16): 5232………..http://pubs.acs.org/doi/abs/10.1021/jm050035f
There is an urgent medical need for novel antibacterial agents to treat hospital infections, specially those caused by multidrug-resistant Gram-positive pathogens. The need may also be fulfilled by either exploring antibacterial agents having new mechanism of action or expanding known classes of antibacterial drugs. The paper describes a new chemical entity, compound 21, derived from hitherto little known “floxacin”. The choice of the entity was made from a series of synthesized prodrugs and salts of the active chiral benzoquinolizine carboxylic acid, S-(−)-nadifloxacin. The chemistry, physicochemical characteristics, and essential bioprofile of 21 qualifies it for serious consideration as a novel drug entity against hospital infections of multi-drug-resistant Staphylococcus aureus, and its progress up to clinical phase I trials in humans is described.
S-(−)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]quinolizine-2-carboxylic Acid l-Arginine Salt Tetrahydrate (Crystalline Form) (21). To a three-necked round-bottom flask fitted on an oil bath and equipped with a mechanical stirrer, a thermometer pocket, and a reflux condenser was charged 1 (100 g, 0.278 mol) followed by acetone (300 mL). Stirring was started and to the stirred suspension was charged powderedl-arginine (48.4 g, 0.278 mol) followed by distilled water (250 mL). The reaction mixture was stirred at a temperature between 50 and 60 °C for 1 h to obtain a clear solution. Activated charcoal (3 g) was added to the solution and the solution was filtered hot. To the filtrate was then added acetone (700 mL) and the reaction mixture was allowed to cool to 30−35 °C. The reaction mixture was stirred for an additional 2 h at this temperature. The crystalline solid was filtered under reduced pressure and the wet cake was washed with acetone (100 mL). The resulting solid was dried under vacuum at 65−70 °C to furnish 21 (137 g, 92% yield):
mp 236−240 °C;
1H NMR (DMSO-d6) δ 1.4 (d, 3H, J = 7.0 Hz), 1.5−2.2 (m, 8H), 2.8−4.2 (m, 16H), 4.8 (m, 1H), 7.8 (d, 1H, J = 13.0 Hz), 8.8 (s, 1H). MS (ES+) m/z 535 (M + H).
Anal. (C25H35FN6O6·4H2O) C, H, N. HPLC assay of free base (theoretical free base content) 67.41%, found 67.16%. Estimated l-arginine by HPLC (theoretical l-arginine content) 32.59%, found 32.14%.
S-(−)-Nadifloxacin is S-(−)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid (1). Prodrugs and aqueous soluble salts of 1were synthesized and explored for possible use in parenteral or oral formulations………….De Souza, N. J.; Agarwal, S. K.; Patel, M. V.; Bhawsar, S. B.; Beri, R. K.; Yeole, R. D.; Shetty, N.; Khorakiwala, H. F. Chiral Fluoroquinolone Arginine Salt Form. US patent 6,514,986, 2003.
(b) De Souza, N. J.; Deshpande, P. K.; Shukla, M. C.; Mukarram S. M. J.; Kulkarni, D. G.; Rahman, A.; Yeole, R. D.; Patel, M. V.; Gupte, S. V. Crystalline Fluoroquinolone Arginine Salt Form. US patent 6,664,267, 2003.
………………………………………………………….
CN 102532131,
quinolones has now grown to four generations, the first generation to nalidixic acid is represented as the representative of the second generation to PPA, only the Gram-negative bacteria effectively, the third generation is the development of these drugs the peak period, there has been a lot of drugs, and is a broad-spectrum antibiotic, which to norfloxacin, ciprofloxacin and other representatives. The fourth-generation quinolone antibiotics is in the third generation on the basis of a broad spectrum of antibacterial spectrum further expanded to make it available against mycoplasma and chlamydia infections.
[0003] R & D has been relatively popular domestic antibiotics, the most widely used on the market today is the third generation fluoroquinolones. Nadifloxacin developed by the Japanese company Otsuka, belongs to the third-generation quinolone antibacterial drugs, topical treatment of acne and folliculitis. 1993 for the first time in Japan (trade name: Acuatim), 2004 in the German market (trade name: Nadixa), 2005 in China listed (trade name: By Union, ointment).
[0004] nadifloxacin irritation due to its absorption and vascular problems, only made of topical formulations for in vitro Propionibacterium acnes (propionibacterium acnes) caused by acne. Wherein the S-(-) – that is the main role difloxacin isomer, the antibacterial activity of the R-isomer of 64 to 256 times that of racemic 2 times.
[0005] fine that gatifloxacin is S-(-) _ nadifloxacin salt on the basis of the system.Significantly improved solubility nadifloxacin well absorbed by the body, so it retains nadifloxacin broad spectrum antimicrobial, antibacterial activity, especially methicillin-sensitive Staphylococcus aureus and methicillin-resistant Staphylococcus aureus Effective characteristics (Antimicrobial Agents and Chemotherapy, 2004,3188 ~ 31920; J. Med. Chem. 2005 (48), 5232 ~ 5242). Pre-clinical tests prove that the product on the market anti-methicillin-resistant Staphylococcus aureus Antibiotic better compare the efficacy, including vancomycin, trovafloxacin, quinupristin + dalfopristin, linezolid amine.
[0006] fine molecular structure that gatifloxacin following formula:
[0007]
[0008] S-(-) _ nadifloxacin (C19H21FN2O4) with L-arginine salt, the further improve the play a major role in antibacterial s-(-) – nadifloxacin isomer content, and improved oral bioavailability, so that it can develop an oral or injectable preparations.
[0009] the literature (J. Med. Chem. 2005 (48), 5232 ~ 5242) discloses the synthesis of S_ (_) _ Nadifloxacin-L-arginine salt, S-(-) _ that fluoride gatifloxacin and L-arginine salt in the reaction solvent system, which solvent system is mainly methanol – water system, according to the paper reported in S-(-) – Nadifloxacin-L-arginine salt, yields were and related substances are not high enough.
Example 1
[0026] In equipped with oil bath, magnetic stirrer, thermometer, reflux condenser flask at 25 ° C was added (S) – (-) – nadifloxacin (100. 0g, 278mmol), dioxane ring (300ml), and the reaction solution was added dropwise to the L-arginine 4g, 278mmol) in distilled water (250ml) was added. Then heated to 50_60 ° C stirred 1.5 hours, and then adding activated carbon (3. Og) for 5 minutes, filtered hot, and then added dropwise at 55-60 ° C dioxane (700ml), and the natural cooling to 30 -35 ° C for 2 hours crystallization. The solid was collected by filtration and acetone (IOOml) wash. Dried at room temperature M hours. To give a white solid 137g, yield: 92%.
……………………………………
WO 2005023805,
Example 1
Preparation of the single crystal of S-(- -9-fluoro-6,7-dihvdro-8-(4-hvdroxypiperidin-l-ylV5- methyl-l-oxo-lH,5H-benzo[“i,ι‘lquinolizine-2-carboxylic acid L-arginine salt terahvdrate.
S-(-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid L-arginine salt (1.0 g) was dissolved in a mixture of acetone (40 ml) and water (10 ml) by heating the suspension at 70 °C for 15 minutes. The clear solution thus obtained was left for slow evaporation at room temperature in a beaker covered with a perforated aluminum foil. The crystal formation started after 2 days. Finally the single crystal was selected for X-ray crystal analysis from a cluster left after complete evaporation of the solvent. The ORTEP diagrams are described in Figures 1 and 2.
………………………………………………………………
WO 2002009758,
…………………………………………………
WO 2001085095,
EXAMPLE 1
S-(-)-9-Fluoro-6,7-dihvdro-8-(4-hvdroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo Ti l quinolizine-2-carboxylic acid arginine salt Synthesis of SubstantiaUy CrystaUine product A solution of L-(+)-arginine (48.372 g, 0.278 mole) in distilled water (600 ml) was added dropwise over a period of 30 min to the stirred solution/suspension of finely powdered S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxylic acid (100 g, 0.278 mole) in acetone (1250 ml). The obtained clear solution was stirred for 30 min and concentrated on a water bath in vacuum (175 mbar) at 80°C. When product started solidifying, the concentration was carried out in vacuum (50 mbar) at 80°C up to dryness. Hexane (1 liter) was added, the reaction mixture was stirred for 4 hr, the solid thus separated was filtered and dried in vacuum (0.7 mbar) for 12 hrs at 70 °C. Yield 145 g (96.9%), m.p. 238-242 °C, and solubility 6 mg/ml (pH 9.5 buffer solution).
The substantially crystalline S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5- methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid arginine salt prepared according to Example 1 possesses the following properties: a) Crystalline form, with a degree of crystallinity as determined by X-ray powder diffraction and as shown in Fig. 1. , b) A thermogram as determined by Differential scanning calorimetry and as shown in Fig. 3. c) Particle size measured as mean mass diameter (MMD) of 83.92 μm, as determined by laser diffraction technique. d) Density of 0.51 g/cm3 (untapped) and 0.7 g/cm3 (tapped). e) Hygroscopicity of 0% increase of weight upon storage for 14 days up to 22% relative atmospheric humidity as determined gravimetricaUy. f) A content of moisture water of 0.1 % by weight as determined by titration according to Karl Fischer. g) A content of acetone of 0.014 % by weight as determined by gas chromatography
……………………………………………………..
WO 2000068229
Example 1
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-car boxy lie acid anhydrate
Method A
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yI)-5-methyl-l-oxo-lH,5H-benzo [ij] quinoIizine-2-carboxylic acid (3.0 g) obtained according to the process described in literature [K Hashimoto et al., Chem.Pharm.Bull.44, 642-5(1996)] was dissolved in acetonitrile (250 ml) at 85 °C. The resulting clear solution was filtered (to remove if any fibrous material is in suspension). The filtrate was concentrated to 125 ml and left at room temperature for crystallization. The crystals thus separated were filtered and dried in a drying cabinet at 40 °C for 2 hr in vacuum at 50 mm of Hg to obtain constant weight. Yield 2.6 g (86%).
Method B:
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxyIic acid (2.0 g) obtained according to the process described in literature [K.Hashimoto etal., Chem.Pharm.Bull.44, 642-5(1996)] was dissolved in ethyl alcohol (95 %; 200 ml) at 80 °C. The obtained clear solution was filtered (to remove if any fibrous material is in suspension), concentrated to 100 ml and left for crystallization. The separated solid was Altered and dried in a drying cabinet at 40 °C for 3 hr in vacuum at 50 mm of Hg to obtain constant weight. Yield 1.7 g (85 %).
M.p.258-62 °C, moisture content 0 % (by Karl Fisher method) [CXJD 26 -299°, HPLC purity 99.8%
Example 8
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxylic acid, L-arginine salt 0.75 hydrate
L-(+)-Arginine (0.958 g., 5.5 mmoles) was added in portions to a suspension solution of S- (-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo [ij] quinoIizine-2-carboxyIic acid 0.2 hydrate (2.0 g., 5.5 mmole) in methanol (400 ml). The obtained solution was concentrated in vacuum to give the desired product as a yellow solid, which was dried at 50 °C at 50 mm/Hg for 5 hours. Yield 3.0 g. (100%), m.p. 220- 223 °C (dec), m/z 535 (M+H), moisture content 2.3% (by Karl Fisher, required 2.46%), [CIJD 25 -144 ° (1% methanol c=l), solubility 93 mg/ml.
……………………………..
Chemical and Pharmaceutical Bulletin
Vol. 44 (1996) No. 4 P 642-645
A Practical Synthesis of (S)-(-)-Nadifloxacin : Novel Acid-Catalyzed Racemization of Tetrahydroquinaldine Derivative
(S)-(-)-Nadifloxacin [(S)-(-)-9-fluoro-6, 7-dihydro-8-(4-hydroxy-1-piperidyl)-5-methyl-1-oxo-1H, 5H-benzo[i, j]quinolizine-2-carboxylic acid, (S)-(-)-OPC-7251], an antibacterial agent, was synthesized from (S)-(-)-5, 6-difluoro-2-methyl-1, 2, 3, 4-tetrahydroquinoline (DFTQ), which was prepared by the optical resolution of recemic DFTQ with 2, 3-di-O-benzoxyl-L-tartaric acid. Racemization of the undesired enantiomer [(R)-(+)-DFTQ] was studied in the presence of various acids and the best result was obtained in the case of methanesulfonic acid. The absolute configuration of (-)-nadifloxacin was determined as S by X-ray crystallographic analysis.
31 Aug, 2014,
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
Read more at:
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
- Ishikawa, H.; Tabusa, F.; Miyamoto, H.; Kano, M.; Ueda, H.; Tamaoka, H.; Nakagawa, K. Studies on antibacterial agents. I. Synthesis of substituted 6,7-dihydro-1-oxo-1H,5H-benzo[i,j]-quinolizine-2-carboxylic acids. Chem. Pharm. Bull. 1989, 37, 2103-2108.
(b) Kurokawa, I.; Akamatsu, H.; Nishigima, S.; Asada, Y.; Kawabata, S. Clinical and Bacteriologic Evaluation of OPC-7251 in Patients with Acne: A Double Blind Group Comparison Study vs Cream Base. J. M. Acad. Dermatol. 1991, 25, 674−81.
(c) Morita, S.; Otsubo, K.; Matsubara, J.; Ohtnai, T.; Uchida, M. An Efficient Synthesis of a Key Intermediate towards (S)-(−)-Nadifloxacin. Tetrahedron: Asymmetry 1995, 6 (1), 245−254.
- (7) (a) Patel, M. V.; Gupte, S. V.; Sreenivas, K.; Chugh, Y.; Agarwal, S. K.; De Souza, N. J. S-(−)-Nadifloxacin: Oral Bioavailbility and Bioefficacy in Mouse Model of Staphylococcal Septicemia. Abstract of Papers, 40th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego, CA, September 2000; American Society for Microbiology: Washington, DC, 2000; Poster F-558.
- (8) A preliminary version of this work was described in a poster. Deshpande, P. K.; Desai, V. N.; Bhavsar, S. V.; Chaturvedi, N. C.; Ghalsasi, S. A.; Aher, S.; Yeole, R. D.; Pawar, D.; Shukla, M. C.; Patel, M. V.; Gupte, S. V.; De Souza, N. J.; Khorakiwala, H. F. WCK 771
 A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430
A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430
Some quinolones introduced for clinical use.
15
QUARFLOXACIN


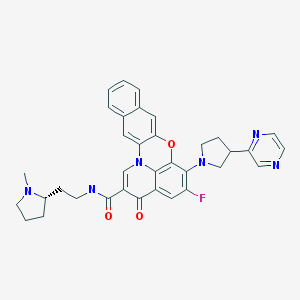
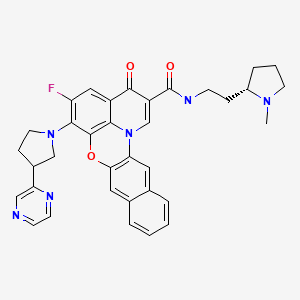












16
GATIFLOXACIN



WO 2005009970
http://www.google.com/patents/WO2005009970A1?cl=en
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
STAGE – I:
 Ethyl l-cyclopropyl-6,7-difluoro-8-met oxy l-Cycloproρyl-6,
7-difluoro-8-methoxy -4-oxo-l, -dihydro-3-quinoline -4-oxo-l,
4-dihydro-3-quinoline carboxylate carboxylic acid boron difluoride
chelate
Ethyl l-cyclopropyl-6,7-difluoro-8-met oxy l-Cycloproρyl-6,
7-difluoro-8-methoxy -4-oxo-l, -dihydro-3-quinoline -4-oxo-l,
4-dihydro-3-quinoline carboxylate carboxylic acid boron difluoride
chelate
STAGE – II :
 l-Cycloprop l-7- ( 3-methylpiperazin-l-yl.
l-Cycloprop l-7- ( 3-methylpiperazin-l-yl.
 6-fluoro~8-methoxy-4-oxo-l , 4-dihydro-3- carboxylicacid
borondifluoride chelate quinoline carboxylicacid borondifluoride chelate
6-fluoro~8-methoxy-4-oxo-l , 4-dihydro-3- carboxylicacid
borondifluoride chelate quinoline carboxylicacid borondifluoride chelate
STAGE -III :
 l-Cyclopropyl-7- (3- ethylpiperaz.in-l-yl . GATIFLOXACIN
l-Cyclopropyl-7- (3- ethylpiperaz.in-l-yl . GATIFLOXACIN
-6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3- quinoline carboxylicacid borondifluoride chelate
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
Stage-2: Preparation of 1- Cyclopropyl-7- (3-methylpiperazin-l-yl) – 6-fluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylic acid boron difluoride chelate
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate.
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
……………………….
WO 2005047260
http://www.google.co.in/patents/WO2005047260A1?cl=en
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:
 European patent application EP-A-230295 discloses a process for
obtaining gatifloxacin that consists on the reaction of compound (II)
with 2-
European patent application EP-A-230295 discloses a process for
obtaining gatifloxacin that consists on the reaction of compound (II)
with 2-
 In this process the gatifloxacin is isolated in the form of a
hemihydrate after a laborious process of column chromatography and
recrystallisation in methanol, which contributes towards making the
final yield lower than 20% by weight. Moreover, in said process an
undesired by-product is formed, resulting from demethylation at position
8 of the ring. European patent application EP-A-241206 discloses a
process for preparing gatifloxacin, whose final steps are as follows:
In this process the gatifloxacin is isolated in the form of a
hemihydrate after a laborious process of column chromatography and
recrystallisation in methanol, which contributes towards making the
final yield lower than 20% by weight. Moreover, in said process an
undesired by-product is formed, resulting from demethylation at position
8 of the ring. European patent application EP-A-241206 discloses a
process for preparing gatifloxacin, whose final steps are as follows:
 (III) H ft N Me H DMSO
(III) H ft N Me H DMSO
Gatifloxacin (I)
 (IV) This process uses the intermediate compound (III) , which has
been prepared and isolated in a separate operation, while the
intermediate compound (IV) is also isolated before proceeding to its
conversion into gatifloxacin by treatment with ethanol in the presence
of triethylamine. The overall yield from these three steps is lower than
40%. These disadvantages — a synthesis involving several steps, low
yields, and the need to isolate the intermediate products — hinder the
production of gatifloxacin on an industrial scale. There is therefore a
need to provide a process for preparing gatifloxacin with a good
chemical yield, without the need to isolate the intermediate compounds
and that substantially avoids demethylation in position 8 of the ring.
The processes termed in English “one pot” are characterised in that the
synthesis is carried out in the same reaction vessel, without isolating
the intermediate compounds, and by means of successive addition of the
reacting compounds. The authors of the present invention have discovered
a simplified process for preparing gatifloxacin which does not require
isolation of the intermediate compounds .
(IV) This process uses the intermediate compound (III) , which has
been prepared and isolated in a separate operation, while the
intermediate compound (IV) is also isolated before proceeding to its
conversion into gatifloxacin by treatment with ethanol in the presence
of triethylamine. The overall yield from these three steps is lower than
40%. These disadvantages — a synthesis involving several steps, low
yields, and the need to isolate the intermediate products — hinder the
production of gatifloxacin on an industrial scale. There is therefore a
need to provide a process for preparing gatifloxacin with a good
chemical yield, without the need to isolate the intermediate compounds
and that substantially avoids demethylation in position 8 of the ring.
The processes termed in English “one pot” are characterised in that the
synthesis is carried out in the same reaction vessel, without isolating
the intermediate compounds, and by means of successive addition of the
reacting compounds. The authors of the present invention have discovered
a simplified process for preparing gatifloxacin which does not require
isolation of the intermediate compounds .
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.

Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
PAPER
 An improved process to obtain gatifloxacin (1) through use of
boron chelate intermediates has been developed. The methodology involves
an initial activation step which accelerates the formation of the first
chelate under low-temperature conditions and prevents demethylation of
the starting material. To increase the overall yield and to avoid the
isolation and manipulation of the resulting intermediates, the process
has been designed to be carried out in one pot. As a result, we present
here an easy, scaleable and substantially impurity-free process to
obtain gatifloxacin (1) in high yield.
An improved process to obtain gatifloxacin (1) through use of
boron chelate intermediates has been developed. The methodology involves
an initial activation step which accelerates the formation of the first
chelate under low-temperature conditions and prevents demethylation of
the starting material. To increase the overall yield and to avoid the
isolation and manipulation of the resulting intermediates, the process
has been designed to be carried out in one pot. As a result, we present
here an easy, scaleable and substantially impurity-free process to
obtain gatifloxacin (1) in high yield.
Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.

QUARFLOXACIN
Quarfloxin, Itarnafloxin
CAS: 865311-47-3.
Chemical Formula: C35H33FN6O3
Exact Mass: 604.25982
Molecular Weight: 604.67
Elemental Analysis: C, 69.52; H, 5.50; F, 3.14; N, 13.90; O, 7.94
Synonym: CX 3543; CX3543; CX-3543; QuarfloxacinTA1-1B
- CX 3543
- CX-3543
- Itarnafloxin
- Quarfloxacin
- Quarfloxin
- UNII-8M31J5031Q
IUPAC/Chemical name:
5-fluoro-N-(2-((S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-((R)-3-(pyrazin-2-yl)pyrrolidin-1-yl)-3H-benzo[b]pyrido[3,2,1-kl]phenoxazine-2-carboxamide.
- 5-Fluoro-N-(2-((2S)-1-methylpyrrolidin-2-yl)ethyl)-3-oxo-6-(3-(pyrazin-2- yl)pyrrolidin-1-yl)-3H-benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide
- 3H-Benzo(b)pyrido(3,2,1-kl)phenoxazine-2-carboxamide, 5-fluoro-N-(2-((2S)- 1-methyl-2-pyrrolidinyl)ethyl)-3-oxo-6-(3-pyrazinyl-1-pyrrolidinyl)-
Quarfloxin, also known as Quarfloxacin and CX-3543, is a fluoroquinolone derivative with antineoplastic activity. Quarfloxin disrupts the interaction between the nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis that is overexpressed in cancer cells; disruption of this G-quadruplex DNA:protein interaction in aberrant rRNA biogenesis may result in the inhibition of ribosome synthesis and tumor cell apoptosis.
CX-3543, developed at Cylene Pharmaceuticals, is a multi-targeting oncogene inhibitor evaluated in phase II clinical studies for the treatment of low or intermediate grade neuroendocrine carcinoma, including carcinoid and islet cell cancer. In 2008, a trial for the treatment of chronic lymphocytic leukemia (CLL) was withdrawn prior to patient enrollment. In 2010, phase I clinical studies for the treatment of solid tumors and for the treatment of lymphoma were terminated upon observation that the modified dose schedule presented no advantage over previously studies schedule solid tumors.
CX-3543 was developed using the company’s Quadruplex Targeting technology which is based on quadruplex motifs in genomic DNA that regulate the expression of clusters of key oncogenes but not normal cellular genes. In 2013, the product was licensed to TetraGene by Cylene Pharmaceuticals on an exclusive, worldwide basis for development for the treatment of cancer. Cylene ceased operations in 2013.
Current developer: Cylene Pharmaceuticals Inc. phase 2
Clinical trial news: Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function. According to news released on June 19, 2011, Cylene Pharmaceuticals announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
Cylene Pharmaceuticals today announced the initiation of a Phase II clinical trial of quarfloxin (CX-3543) in patients with carcinoid/neuroendocrine tumors (C/NET), which are malignant cancers arising from neural crest cells.
“Quarfloxin (CX-3543) is a small molecule that disrupts a protein:rDNA complex that forms in the abnormal nucleoli of cancer cells, thereby selectively inducing apoptotic cell death in cancers,” said Dr. William Rice, President and Chief Executive Officer of Cylene Pharmaceuticals. “Many commercialized cancer therapeutics act on or through the nucleolus, but quarfloxin is the first agent designed to directly target a key function within the nucleolus. Quarfloxin has been well tolerated in humans and has demonstrated signs of biological benefit for patients with C/NET in Phase I clinical trials. Moreover, biodistribution studies revealed that quarfloxin accumulates in the tissues in which C/NET arise.”
In this open-label Phase II trial, quarfloxin will be administered to patients with low or intermediate grade C/NET, including those receiving concomitant treatment with a stable dose of octreotide. This multi-centered study will include an assessment of improvements in patients’ symptoms and biochemical markers, in addition to RECIST tumor response measurements. The first patient was enrolled and treated at Front Range Cancer Specialists in Fort Collins, CO under the care of Robert Marschke Jr., M.D. This study is expected to enroll up to 25 patients at several leading cancer centers.
“The initiation of this Phase II trial with quarfloxin is a major milestone for Cylene, but more importantly, we hope that quarfloxin will be an effective treatment for cancer patients with limited therapeutic alternatives,” added Dr. Daniel Von Hoff, Cylene’s Co-Founder and Vice President, Medical Affairs. “Quarfloxin has demonstrated potent in vivo efficacy against a broad range of tumors and a considerable therapeutic window in preclinical antitumor models, and has a unique profile of concentrating in neural crest tissues. For these reasons, we are enthusiastic about offering a Phase II clinical trial for patients with carcinoid/neuroendocrine tumors.”
About Quarfloxin (CX-3543), a Nucleolus Targeting Agent (NTA)
Quarfloxin is a ground-breaking small-molecule targeted cancer therapeutic derived from the validated fluoroquinolone class of drugs. Rationally designed to selectively inhibit ribosomal RNA (rRNA) biogenesis in cancer cells, quarfloxin disrupts the interaction between the Nucleolin protein and a G-quadruplex DNA structure in the ribosomal DNA (rDNA) template, a critical interaction for rRNA biogenesis and one that is amplified in cancer cells. As a result, quarfloxin selectively induces apoptotic cell death in cancers. Many commercialized cancer therapeutics act indirectly on rRNA Biogenesis through upstream modulators, but quarfloxin is the first agent to directly target this cancer-specific aberrant cell function.
About Cylene Pharmaceuticals, Inc.
Cylene Pharmaceuticals is a biotech pharmaceutical company dedicated to the discovery, development and commercialization of targeted small-molecule drugs to treat life-threatening cancers. Cylene has created a diverse portfolio of product candidates, including novel inhibitors of cancer-linked serine/threonine kinases, as well as innovative Nucleolus Targeting Agents (NTAs) that target the abnormal nucleolus functions of cancer cells and selectively kill cancer cells. More information can be found athttp://www.cylenepharma.com.
………………………………………………………..
To a series of solutions of the fluoroacid (0.5 mmol) in NMP (3.6 mL) was added the amines NHR1R2 (0.5-2.0 mmol) at room temperature. The vessels were sealed and heated on a 90° C. hotplate with constant stirring for 1-2 hours until the reactions were determined to be complete by HPLC/MS analysis. The reaction mixtures were allowed to cool to room temperature and water was added (20 mL). The resulting precipitates were collected by vacuum filtration and dried under vacuum. In cases where 1.0 equivalent of amine was used, the resulting reaction mixtures were used in the next step “as is.” The resulting solids or solutions were treated with HBTU (2.5 eq.) and DIEA in 3.6 mL NMP and allowed to stir for 30 minutes at room temperature under an inert atmosphere. These solutions were added to a series of amines NHR3R4 (2.5 equivalents) in a 96 well format (Whatman Uniplate, 2 mL) and allowed to react for 2 hours. Methanol was then added (50-100 μL) and the plate was filtered (Whatman Unifilter Polypropylene). The resulting liquids were directly chromatographed on reverse HPLC (Waters Xterra 19×50 mm) with mass directed collection (Micromass ZQ, Waters FCII). The fractions were analyzed for purity (MS TIC, UV) and dried by vacuum evaporation (Savant) with an average yield of 5-10 mg). Examples of substituted quinobenzoxazines analogs are described in Table 1.
Example 48Synthesis of CX-3092 and CX-3543
One method for synthesizing CX-3543 is shown below. As shown in Scheme 2, CX-3543 is synthesized in a convergent manner, assembling the substructures 1, 1A and 2A in the final two synthetic steps (Scheme 2), to form CX-3543 having a 50:50 ratio of RS and SS isomers. CX-3092 may be synthesized in a similar manner using a non-chiral form of 1A.
In more detail, pyrazinopyrrolidine 1A is synthesized via a [3+2] cycloaddition chemistry. Conversion of L-proline 7 to cyano-1-aminopyrrolidine 8 without loss of stereochemistry, followed by reduction provides the chiral 2-aminoethyl-1-methylpyrrolidine 2A in high yield. CX-3543 was found to have a formulated solubility of approximately 20 mg/mL.
Example 70This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding ester with an amine, and aluminum chloride.
To a solution of 2,3,4,5-tetrafluorobenzoic acid (100 g, 510 mmol), in methylene chloride (0.5 L) was added oxalyl chloride (68 g, 540 mmol) and DMF (ca 3 drops) and the reaction mixture was allowed to stir at room temperature overnight allowing for the produced gasses to escape. The solvent was removed in vacuo and the vessel was placed on high vacuum (ca 0.5 mm Hg) for 2 hours to afford the acid chloride as a viscous oil (105 g) and was used in the subsequent reaction without further purification.
To a suspension of potassium ethyl malonate (97 g, 570 mmol) and magnesium chloride (55 g, 570 mmol) in acetonitrile and the suspension was chilled to 0° C. To this suspension was added the crude 2,3,4,5-benzoyl chloride (105 g, 520 mmol) over 5 minutes. Triethylamine was slowly added at a rate sufficient to keep the reaction temperature below 10° C. and the mixture was allowed to warm to room temperature and was stirred overnight. The solvent was removed in vacuo and replaced with toluene (300 mL) and 1N HCl (500 mL) was added and the mixture was allowed to stir for 1 hour. The organic layer was separated and washed with 1N HCl (100 mL) and brine (100 mL) and dried over sodium sulfate, filtering over a pad of silica gel (50×100 mm), eluting with ethyl acetate. The solvent was removed in vacuo and the resulting oil was dissolved in ethanol/water (9:1) and was allowed to crystallize overnight. The resulting crystals were Isolated by filtration, washing with ethanol/water (8:2) to afford the ketoester (43.75 g, 166 mmol) as a white crystalline solid.
To a 250 mL round bottom flask was added the tetrafluoroketoester (10.0 g, 37.9 mmol), triethylorthoformate (8.6 mL, 56.8 mmol) and acetic anhydride (7.15 mL, 75.8 mmol) and the reaction mixture was heated to 145° C. for 2 hours. The reaction was allowed to cool to room temperature and placed on high vacuum (ca 0.5 mm Hg) for 1 hour. The resulting oil was dissolved in ethanol (100 mL) and 2-amino-1-naphthol (6.02 g, 37.9 mmol) was added at room temperature and the solution became briefly clear and then product began to precipitate. The reaction was allowed to stir for 2 hours and was then filtered and washed with ethanol (100 mL) to afford the enamine as a yellow solid (12.5 g, 28.9 mmol).
To a solution of the enamine (12.13 g, 27.95 mmol) in dry DMF (50 mL) was added potassium carbonate (4.24 g, 1.1 eq.) and the mixture was heated to 90° C., with constant stirring, for 2 hours. The mixture was allowed to cool to room temperature without stirring and was allowed to remain at room temperature for an additional hour. The crystalline solid was collected by filtration, washing with water. Recrystallization from THF afforded the difluoroester as a white crystalline solid (9.3 g, 23.6 mmol).
To a solution of the difluoroester (1.0 g, 2.5 mmol) in NMP (10 mL) was added N-Boc-3-(2-pyrazino)pyrrolidine (870 mg, 3.5 mmol) and the mixture was heated to reflux for 3 hours. The reaction mixture was then allowed to cool to room temperature and the product was collected by filtration. Crystallization from THF afforded the pyrazine ester as a yellow solid (910 mg, 1.74 mmol).
To a solution of the pyrazine ester (250 mg, 0.48 mmol) and 2-(2-aminoethyl)-1-methylpyrrolidine (80 mg, 0.63 mmol) in methylene chloride at room temperature was added aluminum chloride (83 mg, 0.63 mmol) and the reaction mixture was allowed to stir for 2 hours. The solvent was removed in vacuo and saturated L-tartaric acid was added (5 mL) and the mixture was allowed to stir for 1 hour. Methylene chloride (10 mL) was then added and the mixture was basified with 1N NaOH. The organic layer was separated and washed with a saturated solution of Rochelle’s salt, brine and dried over sodium sulfate. The solvent was removed in vacuo and the resulting solid was dissolved in THF and filtered and the solvent was removed again. The crude solid was recrystallized in ethyl acetate to afford the amide as a yellow solid (225 mg, 0.37 mmol, 98.5% pure).
Example 71
This example describes a method for preparing a substituted benzoxazine analog from reaction of the corresponding carboxylic acid with an amine, and aluminum chloride.
The pyrazinoester (2.0 g, 3.8 mmol) was dissolved in ethanol (100 mL) and conc HCl was added (20 mL) and the mixture was refluxed overnight. The mixture was allowed to cool to room temperature and the solid was collected by vacuum filtration, washing with ethanol to afford the pyrazinoacid as a light tan powder (1.6 g, 3.2 mmol).
To a mixture of the fluoroaminoacid (1.6 g, 3.2 mmol) and HBTU (2.0 g, 5.3 mmol) in NMP (20 mL) was added N,N-diisopropyl-N-ethylamine (1.0 mL, 6 mmol) and the mixture was allowed to stir at room temperature, under argon, for 1 hour (the solution became clear). (S)-2-(2-aminoethyl)-1-methylpyrrolidine (Mizuno, A.; Hamada, Y.; Shioiri, T., Synthesis, 1980, 12 1007)(1.0 mL, 6.9 mmol) was added and the mixture was allowed to stir for 30 minutes. Water (200 mL) was added and the resulting solid was collected by vacuum filtration, washing with water, and dried to afford the pyrazine as a yellow solid. The yellow solid was purified on silica gel (10% MeOH/CH2Cl2 first eluting off impurities followed by eluting with 5% NH4OH/15% MeOH/CH2Cl2. The combined fractions were evaporated to afford the compound as a yellow solid. (1.2 g, 2.0 mmol, 85% pure).
……………………………..
The present disclosure provides an improved method of treating cancer using a combination of a G-quadruplex-interactive compound that binds to G- quadruplexes in rDNA to release the nucleolin already bound to these G- quadruplexes together with a PARP inhibitor. This results in an increase in apoptosis in cancer cells. The PARP inhibitor can be administered to a patient (human or animal) in need of cancer treatment simultaneously or from 0.1 to 24 hours prior to or 0.1 to 24 after the administration of the G-quadruplex-interactive agent that releases the nucleolin bound to the G-quadruplex and triggers enhanced apoptosis of cancer cells, or the PARP inhibitor and the enhancer of nucleolin binding can be administered simultaneously, with each agent being administered in an amount sufficient to inhibit the growth and/or cell division of cancer (neoplastic) cells, and preferably to cause cancer cell death. In the methods provided herein, the PARP inhibitor can be benzamide (as specifically exemplified) or it can be 3- benzamide, 3-methoxybenzamide, carba-NAD+, nicotinamide, a dihydroisoquinolinone, an isoquinolinone such as 5-methyl-dihydroisoquinolinone, a benzimidazole-4-carboxamide, a 2-aryl-benzimidazole-4-carboxamide, a benzoxazole-4-carboxamide, an N,N-dimethylaminomethyl, pyrrolidinomethyl or bis- benzamide derivative, for example 1 ,5-di(3- carbamoylphenyl)aminocarbonyloxy)pentane, a phthalazinone, a quinazolinone, an isoindolinone, a phenanthhdinone, among others. The G-quadruplex-interactive agent that releases nucleolin from the rDNA bound to the G-quadruplexes and triggers apoptosis of cancer cells is desirably a substituted quinobenzoxazine analog; in an embodiment of the invention, it is CX-3543
(see also US Patent Publication 2006-0029950, which is incorporated by reference herein). This combination chemotherapy can be administered in a single dose, or it can be administered at intervals chosen by a medical or veterinary practitioner.
[0003] The present disclosure further provides compositions comprising a PARP inhibitor and a G-quadruplex-interactive compound that triggers the release of nucleolin from the G-quadruplexes in the rDNA and triggers apoptosis of cancer cells. The compositions desirably further comprises a pharmaceutically acceptable excipient, especially one which is compatible with intravenous administration in human patients. In an embodiment of the invention, the composition comprises benzamide and CX-3543.
……………………………
J. Med. Chem., 2013, 56 (3), pp 843–855
DOI: 10.1021/jm3013486
Nowadays, it has been demonstrated that DNA G-quadruplex arrangements are involved in cellular aging and cancer, thus boosting the discovery of selective binders for these DNA secondary structures. By taking advantage of available structural and biological information on these structures, we performed a high throughput in silico screening of commercially available molecules databases by merging ligand- and structure-based approaches by means of docking experiments. Compounds selected by the virtual screening procedure were then tested for their ability to interact with the human telomeric G-quadruplex folding by circular dichroism, fluorescence spectroscopy, and photodynamic techniques. Interestingly, our screening succeeded in retrieving a new promising scaffold for G-quadruplex binders characterized by a psoralen moiety.
……………………………..
US 20070293485
…………………………..
see
US 20130005720
…………………………….
see
WO 2004091504 or http://www.google.com/patents/EP1610759A2?cl=en
References:
|
1. Bayes, M.; Rabasseda, X.; Prous, J. R., Gateways to clinical trials. Methods Find Exp Clin Pharmacol 2007, 29, (1), 53-71.
2. Brennan, A. B.; Long, C. J.; Bagan, J. W.; Schumacher, J. F.; Spiecker, M. M. Surface topographies for non-toxic bioadhesion control. US20100226943A1.
3. Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C. B.; Proffitt, C.; Trent, K.; Whitten, J. P.; Lim, J. K. C.; Von, H. D.; Anderes, K.; Rice, W. G., Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, (19), 7653-7661.
4. Hurley, L. H.; Guzman, M. Combination cancer chemotherapy. WO2007137000A2, 2007.
5. Lim, J.; Whitten, J. P. Drug administration methods. WO2007143587A1, 2007.
6. Neidle, S., Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 277, (5), 1118-1125.
7. O’Brien, S.; Siddiqui-Jain, A. Targeting quadruplex sequences in human nucleic acids by identifying interacting quinoline and porphyrin derivatives. WO2007056113A2, 2007.
8. Ryckman, D. M.; Drygin, D.; Whitten, J. P.; Anderes, K.; Trent, K.; Darjania, L.; Haddach, M.; O’Brien, S.; Rice, W. G. Methods for treating aberrant cell proliferation disorders. US20080318938A1, 2008.
9. Tian, M.; Zhang, X.; Pan, R.; Zhao, C.; Tang, Y., Structure of G-quadruplex in the oncogene c-myc promoter and small ligands targeting the G-quadruplex. Huaxue Jinzhan 22, (5), 983-992.
10. Whitten, J. P.; O’Brien, S. Methods for treating ophthalmic disorders. US20080318939A1, 2008.
11. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. US20060074089A1, 2006.
12. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Preparation of fused quinolone analogs which inhibit cell proliferation and/or induce cell apoptosis. WO2006034113A2, 2006.
13. Whitten, J. P.; Pierre, F.; Regan, C.; Schwaebe, M.; Yiannikouros, G. P.; Jung, M. Process for the preparation of benzothiazole and phenoxazine compounds. US20060063761A1, 2006.
14. Whitten, J. P.; Schwaebe, M.; Siddiqui-Jain, A.; Moran, T. Preparation of substituted quinobenzoxazine analogs as antitumor agents. US20060029950A1, 2006.
| “CX-3543, 386705” ANNUAL DRUG DATA REPORT, PROUS, BARCELONA, ES, vol. 27, no. 4, 2005, page 379, XP009092663 ISSN: 0379-4121 | ||
| 2 | * | CEPEDA, V. ET AL.: “Poly(ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors in Cancer Chemotherapy” RECENT PATENTS ON ANTI-CANCER DRUG DISCOVERY, vol. 1, no. 1, January 2006 (2006-01), pages 39-53, XP007903584 ISSN: 1574-8928 |
| 3 | * | DATABASE INTEGRITY [Online] Prous science; DailyDrugNews.com (Daily Essentials) 22 July 2005 (2005-07-22), “CX-3543 begins phase I cancer trial” XP007903594 retrieved from INTEGRITY.PROUS.COM Database accession no. 386705 |
| 4 | * | JIN ET AL: “In vivo efficacy of CX-3543, a novel c-Myc oncogene inhibitor” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 45, 2004, page ABS. LB-243, XP001537665 ISSN: 0197-016X |
| 5 | * | RICE WILLIAM G ET AL: “Design of CX-3543, a novel multi-targeting antitumor agent” PROCEEDINGS OF THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR CANCER RESEARCH, NEW YORK, NY, US, vol. 46, April 2005 (2005-04), pages 594-ABS. 2530, XP001536592 ISSN: 0197-016X |
| 6 | * | SHIOKAWA D ET AL: “Inhibitors of poly(ADP-ribose) polymerase suppress nuclear fragmentation and apoptotic-body formation during apoptosis in HL-60 cells” FEBS LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 413, no. 1, 11 August 1997 (1997-08-11), pages 99-103, XP004261237 ISSN: 0014-5793 |
| 7 | * | VALERIOTE F ET AL: “SYNERGISTIC INTERACTION OF ANTICANCER AGENTS: A CELLULAR PERSPECTIVE” CANCER CHEMOTHERAPY REPORTS, vol. 59, no. 5, September 1975 (1975-09), pages 895-900, XP009019750 |
| S7910600 | Aug 29, 2008 | Mar 22, 2011 | Cylene Pharmaceuticals, Inc. | Therapeutic kinase modulators |
| US7956064 | Aug 31, 2007 | Jun 7, 2011 | Cylene Pharmaceuticals, Inc. | Fused tricyclic compounds as serine-threonine protein kinase and PARP modulators |
| US8481529 | May 9, 2007 | Jul 9, 2013 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | Combination cancer chemotherapy |
| EP2023935A1 * | Jun 1, 2007 | Feb 18, 2009 | Cylene Pharmaceuticals, Inc. | Drug administration methods |
| WO2007137000A2* | May 9, 2007 | Nov 29, 2007 | Univ Arizona | Combination cancer chemotherapy |
| WO2007143587A1* | Jun 1, 2007 | Dec 13, 2007 | Cylene Pharmaceuticals Inc | Drug administration methods |
16
GATIFLOXACIN
GATIFLOXACIN
BMS-206584, CG-5501, AM-1155, Zymar, Bonoq, Gatiflo, AM-1155
(±)-1-Cyclopropyl-6-fluoro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Gatifloxacin sold under the brand names Gatiflo, Tequin and Zymar, is an antibiotic of the fourth-generation fluoroquinolonefamily,[1] that like other members of that family, inhibits the bacterial enzymes DNA gyrase and topoisomerase IV. Bristol-Myers Squibb introduced Gatifloxacin in 1999 under the proprietary name Tequin for the treatment of respiratory tract infections, having licensed the medication from Kyorin Pharmaceutical Company of Japan. Allergan produces it in eye-drop formulation under the names Zymar and Zymaxid. In many countries, gatifloxacin is also available as tablets and in various aqueous solutions forintravenous therapy.
Originally developed at Kyorin, gatifloxacin was first licensed to
Gruenenthal in Europe, and that company still maintains rights to the
oral and injectable formulations of the product. In October 1996, Kyorin
licensed gatifloxacin to BMS, granting the company development and
marketing rights in the U.S., Canada, Australia, Mexico, Brazil and
certain other markets. In 2006, rights to the compound were returned by
BMS. Subsequently, Senju and Kyorin signed a licensing agreement
regarding the development of ethical eye drops containing the
fluoroquinolone. In April 2000, Sumitomo Dainippon Pharma agreed to
comarket the oral formulation in Japan. In August of that year, Allergan
in-licensed gatifloxacin from Kyorin, gaining development and
commercialization rights to the drug in all territories except Japan,
Korea, China and Taiwan. The India-based Lupin Pharmaceuticals signed an
agreement in June 2004 with Allergan to promote the ophthalmic solution
of gatifloxacin in the pediatric specialty area in the U.S. PediaMed
Pharmaceuticals also holds rights to the drug. In 2009, Kyorin licensed
the drug candidate to Senju in China.
Gatifloxacin is the common name for
(±)-1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic
acid (1), one of the most important broad-spectrum antibacterial
agents and a member of the fourth-generation fluoroquinolone
family.(1)Fluoroquinolones inhibit the enzyme DNA gyrase (topoisomerase
II), which is responsible for the supercoiling of the DNA double helix,
preventing the replication and repair of bacterial DNA and
RNA.(2) Gatifloxacin (1) reached the market in 1999 under the
brand name Tequin for the treatment of respiratory tract infections. The
drug is available as tablets and aqueous solutions for intravenous
therapy as well as eye drop formulation (Zymar).
To date, there are several processes described for the preparation
of gatifloxacin, which can be grouped into two main categories: direct
substitution of the 7-position fluorine atom of
1-cyclopropyl-6,7-difluoro-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic
acid (2) by 2-methylpiperazine (Scheme 1),(3-5) and through
boron chelate-type intermediates to overcome the diminished reactivity
induced by the 8-methoxy group, which uses as starting material the
ethyl ester derivative 3 (Scheme 2).(6-9)
SCHEME1
SCHEME2
- 1.
Mather, R.; Karenchak, L. M.; Romanowski, E. G.; Kowalski, R. P. Am. J. Ophthalmol.2002, 133 ( 4) 463
- 2.
Corey, E. J.; Czakó, B.; Kürti, L. Molecules and Medicine; Wiley: NJ, 2007; p 135.
- 3.
Masuzawa, K.; Suzue, S.; Hirai, K.; Ishizaki, T. 8-Alkoxyquinolonecarboxylic acid and salts thereof excellent in the selective toxicity and process of preparing the same EP 0 230 295 A3, 1987.
- 4.
Niddam-Hildesheim, V.; Dolitzky, B.-Z.; Pilarsky, G.; Steribaum, G. Synthesis of Gatifloxacin WO 2004/069825 A1, 2004.
- 5.
Ruzic, M; Relic, M; Tomsic, Z; Mirtek, M. Process for the preparation of Gatifloxacin and regeneration of degradation products WO 2006/004561 A1, 2006.
- 6.
Iwata, M.; Kimura, T.; Fujiwara, Y.; Katsube, T. Quinoline-3-carboxylic acid derivatives, their preparation and use EP 0 241 206 A2, 1987.
- 7.
Sanchez, J. P.; Gogliotti, R. D.; Domagala, J. M.; Garcheck, S. J.; Huband, M. D.; Sesnie,J. A.; Cohen, M. A.; Shapiro, M. A. J. Med. Chem. 1995, 38, 4478
- 8.
Satyanarayana, C.; Ramanjaneyulu, G. S.; Kumar, I. V. S. Novel crystalline forms of Gatifloxacin WO 2005/009970 A1 2005.
- 9.
Takagi, N.; Fubasami, H.; Matsukobo, H.; (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture EP 0 464 823 A1, 1991.
WO 2005009970
http://www.google.com/patents/WO2005009970A1?cl=en
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
STAGE – I:
STAGE – II :
STAGE -III :
-6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3- quinoline carboxylicacid borondifluoride chelate
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
Stage-2: Preparation of 1- Cyclopropyl-7- (3-methylpiperazin-l-yl) – 6-fluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylic acid boron difluoride chelate
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate.
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
……………………….
WO 2005047260
http://www.google.co.in/patents/WO2005047260A1?cl=en
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:
Gatifloxacin (I)
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.
Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-cyclopropyl-6-fluoro- 8-methoxy-7-(3-methylpiperazin-1-yl)- 4-oxo-quinoline-3-carboxylic acid | |
| Clinical data | |
| Trade names | Zymar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605012 |
|
|
| Oral (discontinued), Intravenous(discontinued) ophthalmic |
|
| Pharmacokinetic data | |
| Protein binding | 20% |
| Half-life | 7 to 14 hours |
| Identifiers | |
| 112811-59-3 |
|
| J01MA16 S01AE06 | |
| PubChem | CID: 5379 |
| DrugBank | DB01044 |
| ChemSpider | 5186 |
| UNII | 81485Y3A9A |
| KEGG | D08011 |
| ChEBI | CHEBI:5280 |
| ChEMBL | CHEMBL31 |
| NIAID ChemDB | 044913 |
| Chemical data | |
| Formula | C19H22FN3O4 |
| 375.394 g/mol | |
A High-Throughput Impurity-Free Process for Gatifloxacin
Department of Research & Development, Química Sintética S.A.,
c/ Dulcinea s/n, 28805 Alcalá de Henares, and Department of Organic
Chemistry, University of Alcalá, 28871 Madrid, Alcalá de Henares, Spain
Org. Process Res. Dev., 2008, 12 (5), pp 900–903
DOI: 10.1021/op800042a
gatifloxacin (1) as white crystals. Yield 32.3 kg, (93%); purity by HPLC 99.87%; Assay by HPLC 100.8%; mp 167−168 °C(18) (Lit. (J. Med. Chem. 1995, 38, 4478)159−162 °C).
18
DSC analysis showed two endothermic peaks at 166.2 °C (T onset = 164.3 °C) and 190.0 °C (T onset
= 188.2 °C) and an exothermic one at 168.1 °C. The shape of this DSC
curve is characteristic of a monotropic transition between crystalline
forms
Water content by Karl Fischer 3.0%(19) MS m/z 376 (M+ + H);
19
Although there are several hydrates described for gatifloxacin such
as, among others, the hemimydrate, sesquihydrate, and pentahydrate(Raghavan, K.
S.; Ranadive, S. A.;Gougoutas, J. Z.; Dimarco, J. D.; Parker, W.
L.; Dovich, M.; Neuman, A.Gatifloxacin pentahydrate. WO 2002/22126
A1, 2002) , the Gatifloxacin obtained by the present procedure does not
seem to form a stoichometric hydrate, but instead it retains moisture.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
1H NMR (DMSO-d6) δ 0.97 (d, J = 6.1 Hz, 3H), 1.04 (m, 2H), 1.15 (m, 2H), 2.75−2.94 (m, 4H) 3.14 (m, 1H), 3.30 (m, 2H), 3.74 (s, 3H), 4.15 (m, 1H), 7.70 (d, JH−F = 12.2 Hz, 1H), 8.67 (s, 1H).
13C NMR (DMSO-d6) δ 8.40, 8.42, 18.66, 40.28, 45.46, 50.17, 50.29 (d, JC−F = 3.44 Hz), 57.36 (d, JC−F = 3.74 Hz), 62.15, 106.0 (d, JC−F = 22.7 Hz), 106.04, 120.05 (d, JC−F = 8.6 Hz), 133.6 (d, JC−F = 1.1 Hz), 138.9 (d, JC−F = 11.9 Hz), 145.2 (d, JC−F = 5.87 Hz), 149.88, 155.06 (d, JC−F = 249.2 Hz), 165.56, 175.56 (d, JC−F = 3.3 Hz).
19F NMR (DMSO-d6) δ −120.4 (d, J = 12.2 Hz).
Anal. Calcd for C19H22N3O4F + 3.0% H2O; C, 58.95; H, 6.07; N, 10.85. Found: C, 58.90; H, 5.82; N, 10.90.
Boron content 170 ppm.(The respective boron contents found for batches 2 and 3 were 160 and 300 ppm)
Side-effects and removal from the market
A Canadian study published in the New England Journal of Medicine in March 2006 claims Tequin can have significant side effectsincluding dysglycemia.[2] An editorial by Dr. Jerry Gurwitz in the same issue called for the Food and Drug Administration (FDA) to consider giving Tequin a black box warning.[3] This editorial followed distribution of a letter dated February 15 by Bristol-Myers Squibb to health care providers indicating action taken with the FDA to strengthen warnings for the medication.[4] Subsequently it was reported on May 1, 2006 that Bristol-Myers Squibb would stop manufacture of Tequin, end sales of the drug after existing stockpiles were exhausted, and return all rights to Kyorin.[5]Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
Contraindications
Diabetes[7]Availability
Gatifloxacin is currently available only in the US and Canada as an ophthalmic solution.In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.
- The reaction of 1-bromo-2,4,5-trifluoro-3-methoxybenzene (I) with CuCN and N-methyl-2-pyrrolidone at 150 C gives 2,4,5-trifluoro-3-methoxybenzonitrile (II), which by treatment with concentrated H2SO4 yields the benzamide (III) The hydrolysis of (III) with H2SO4 -. water at 110 C affords 2,4,5-trifluoro-2-methoxybenzoic acid (IV), which by reaction with SOCl2 is converted into the acyl chloride (V). The condensation of (V) with diethyl malonate by means of magnesium ethoxide in toluene affords diethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) malonate (VI), which by treatment with p-toluenesulfonic acid in refluxing water gives ethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) acetate (VII). The condensation of (VII) with triethyl orthoformate in refluxing acetic anhydride yields 3-ethoxy -2- (2,4,5-trifluoro-3-methoxybenzoyl) acrylic acid ethyl ester (VIII), which is treated with cyclopropylamine (IX) to afford the corresponding cyclopropylamino derivative (X). The cyclization of (X) by means of NaF in refluxing DMF gives 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (XI), which is hydrolyzed with H2SO4 in acetic acid to yield the corresponding free acid (XII). Finally, this compound is condensed with 2-methylpiperazine (XIII) in hot DMSO.
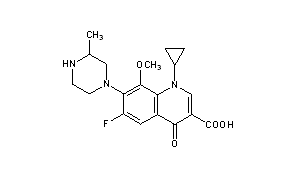 |
Title: Gatifloxacin
CAS Registry Number: 112811-59-3
CAS Name: 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid
Trademarks: Tequin (BMS); Zymar (Allergan)
Molecular Formula: C19H22FN3O4
Molecular Weight: 375.39
Percent Composition: C 60.79%, H 5.91%, F 5.06%, N 11.19%, O 17.05%
Literature References: Fluorinated quinolone antibacterial. Prepn: K. Masuzawa et al., EP 230295; eidem, US 4980470 (1987, 1990 both to Kyorin); J. P. Sanchez et al., J. Med. Chem. 38, 4478 (1995); of the sesquihydrate: T. Matsumoto et al., US5880283 (1999 to Kyorin). In vitro antibacterial activity: A. Bauernfeind, J. Antimicrob. Chemother. 40, 639 (1997); H. Fukuda et al., Antimicrob. Agents Chemother. 42, 1917 (1998). Clinical pharmacokinetics: M. Nakashima et al., ibid. 39, 2635 (1995). Clinical study in urinary tract infection: H. Nito, 10th Mediterranean Congr. Chemother. 1996, 327; in respiratory tract infection: S. Sethi, Expert Opin. Pharmacother. 4, 1847 (2003).
Properties: Pale yellow prisms from methanol as hemihydrate, mp 162°.
Melting point: mp 162°
Derivative Type: Sesquihydrate
CAS Registry Number: 180200-66-2
Manufacturers’ Codes: AM-1155
Molecular Formula: C19H22FN3O4.1½H2O
Molecular Weight: 384.40
Percent Composition: C 59.37%, H 6.03%, F 4.94%, N 10.93%, O 18.73%
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Quinolones and Analogs
|
References
- Burka JM, Bower KS, Vanroekel RC, Stutzman RD, Kuzmowych CP, Howard RS (July 2005). “The effect of fourth-generation fluoroquinolones gatifloxacin and moxifloxacin on epithelial healing following photorefractive keratectomy”. Am. J. Ophthalmol. 140 (1): 83–7. doi:10.1016/j.ajo.2005.02.037.PMID 15953577.
- Park-Wyllie, Laura Y.; David N. Juurlink; Alexander Kopp; Baiju R. Shah; Therese A. Stukel; Carmine Stumpo; Linda Dresser; Donald E. Low; Muhammad M. Mamdani (March 2006).“Outpatient Gatifloxacin Therapy and Dysglycemia in Older Adults”. The New England Journal of Medicine 354 (13): 1352–1361. doi:10.1056/NEJMoa055191. PMID 16510739. Retrieved 2006-05-01. Note: publication date 30 March; available on-line 1 March
- Gurwitz, Jerry H. (March 2006). “Serious Adverse Drug Effects — Seeing the Trees through the Forest”. The New England Journal of Medicine 354 (13): 1413–1415.doi:10.1056/NEJMe068051. PMID 16510740. Retrieved2006-05-01.
- Lewis-Hall, Freda (February 15, 2006). “Dear Healthcare Provider:” (PDF). Bristol-Myers Squibb. Retrieved May 1, 2006.
- Schmid, Randolph E. (May 1, 2006). “Drug Company Taking Tequin Off Market”. Associated Press. Archived from the original on November 25, 2007. Retrieved 2006-05-01.[dead link]
- “Two drugs banned”. The Hindu (Chennai, India). 19 March 2011.
- Peggy Peck (2 May 2006). “Bristol-Myers Squibb Hangs No Sale Sign on Tequin”. Med Page Today. Retrieved 24 February2009.
| EP0610958A2 * | 20 Jul 1989 | 17 Aug 1994 | Ube Industries, Ltd. | Intermediates in the preparation of 4-oxoquinoline-3-carboxylic acid derivatives |
| ES2077490A1 * | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008126384A1 | 31 Mar 2008 | 23 Oct 2008 | Daiichi Sankyo Co Ltd | Method for producing quinolone carboxylic acid derivative |
| CN101659654B | 28 Aug 2008 | 6 Nov 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | 18 Aug 2011 | 15 Feb 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
| EP1832587A1 * | 2 Mar 2007 | 12 Sep 2007 | Quimica Sintetica, S.A. | Method for preparing moxifloxacin and moxifloxacin hydrochloride |
| US7365201 | 2 Mar 2006 | 29 Apr 2008 | Apotex Pharmachem Inc. | Process for the preparation of the boron difluoride chelate of quinolone-3-carboxylic acid |
| US7875722 | 30 Sep 2009 | 25 Jan 2011 | Daiichi Sankyo Company, Limited | Method for producing quinolone carboxylic acid derivative |
| EP0464823A1 * | Jul 4, 1991 | Jan 8, 1992 | Kyorin Pharmaceutical Co., Ltd. | (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture |
| US4997943 * | Mar 31, 1987 | Mar 5, 1991 | Sankyo Company Limited | Quinoline-3-carboxylic acid derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101659654B | Aug 28, 2008 | Nov 6, 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | Aug 18, 2011 | Feb 15, 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
* Cited by examiner
17 WCK 2349
Votes
. CH3SO3H
WCK 2349
Cas 948895-94-1 methane sulfonate
Base..706809-20-3
527.563., C22 H26 F N3 O5 . C H4 O3 S
8-[4-(L-Alanyloxy)piperidin-1-yl]-9-fluoro-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic
acid methanesulfonate
S-(-)-9-fluoro-6,7-dihydro-8-(4-L-alaninyloxypiperidin-
1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]quinolizine-2-
carboxylic acid methanesulfonate
(2’S,
5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl-
l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid
salt
Wockhardt Research Centre INNOVATOR
Oral broad-spectrum antibiotic
WO 2000068229, WO 2002009758, WO 2007102061, WO 2008053295, Indian (2015), IN 267210 , IN 2008MU00864,
Shetty, N.M.; Nandanwar, M.B.; Kamalavenkatesh, P.; et al.
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
PATENT
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC

PATENT
WO 2000068229
A S-(-)-optically pure benzoquinolizine
carboxylic acid, its derivatives, its pharmaceutically acceptable salts,
derivatives, pseudopolymorphs, polymorphs or hydrates thereof of
formula I,
Formula I
Patent
WO 2011101710
PATENT
The tablets may optionally be coated with film forming agents and/or
pharmaceutically acceptable excipients. Particularly suitable for use
are commercially available coating compositions comprising film-forming
polymers marketed under various trade names, such as Opadry® and Eudragit®.
The coating layers over the tablet may be applied as
solution/dispersion of coating ingredients using conventional techniques
known in the art.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1
 Procedure: The compound of Formula I or pharmaceutically acceptable
salts, esters or products thereof, lactose and croscannellose sodium
were sifted and dry mixed in a rapid mixer granulator. The above mass
was granulated by spraying aqueous solution of povidone. The granules
were dried in a fluidized bed drier, sifted and oversize granules were
milled in a Quadra mill. The resultant granules were mixed with talc,
croscarmellose sodium, microcrystalline cellulose and sodium stearyl
fumarate in a double cone blender. The lubricated granules were
compressed into tablets using suitable tooling. Tablets were coated with
aqueous dispersion of opadry.
Procedure: The compound of Formula I or pharmaceutically acceptable
salts, esters or products thereof, lactose and croscannellose sodium
were sifted and dry mixed in a rapid mixer granulator. The above mass
was granulated by spraying aqueous solution of povidone. The granules
were dried in a fluidized bed drier, sifted and oversize granules were
milled in a Quadra mill. The resultant granules were mixed with talc,
croscarmellose sodium, microcrystalline cellulose and sodium stearyl
fumarate in a double cone blender. The lubricated granules were
compressed into tablets using suitable tooling. Tablets were coated with
aqueous dispersion of opadry.
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data

The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data
//////////////////////////////
aChemical name: S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]
quinolizine-2-carboxylic acid L-arginine salt tetrahydrate. bChemical name: S-(–)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3,
3-dimethylpiperidin-1-yl)-1,4 dihydro-4-oxo-quinoline-3-carboxylic acid
hydrochloride monohydrate. cChemical name: R-(+)-1-cyclopropyl-6-fluoro-8-methoxy-7-(4-amino-3,3-dimethylpiperidin-1-yl)-1,4
dihydro-4-oxo-quinoline-3-carboxylic acid hydrochloride monohydrate.
31 Aug, 2014,
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
Read more at:
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
Levonadifloxacin arginine salt, WCK 771
RN: 306748-89-0
RN: 306748-89-0
-
C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
-
L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)
WCK 771………..S-(–)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j]
quinolizine-2-carboxylic acid L-arginine salt tetrahydrate
(-)-9-Fluoro-8-(4-hydroxypiperidin-1-yl)-5(S)-methyl-1-oxo-1,5,6,7-tetrahydropyrido[3,2,1-ij]quinoline-2-carboxylic
acid L-arginine salt hydrate
L-arginine salt of (S)-nadifloxacin
A chiral benzoquinolizine-2-carboxylic acid arginine salt active against vancomycin-resistant Staphylococcus aureus
J Med Chem 2005, 48(16): 5232
J Med Chem 2005, 48(16): 5232
CN 102532131, WO 2005023805, WO 2002009758, WO 2001085095, WO 2000068229
| WO1991012815A1 * | Feb 25, 1991 | Sep 5, 1991 | Squibb Bristol Myers Co | COMPOSITIONS AND METHODS FOR TREATING INFECTIONS CAUSED BY ORGANISMS SENSITIVE TO β-LACTAM ANTIBIOTICS |
| WO2000068229A2 * | May 8, 2000 | Nov 16, 2000 | S K Agarwal | (s)-benzoquinolizine carboxylic acids and their use as antibacterial agents |
| WO2001085095A2 * | May 3, 2001 | Nov 15, 2001 | Shiv Kumar Agarwal | Chiral fluoroquinolizinone arginine salt forms |
| WO2002009758A2 * | Jul 31, 2001 | Feb 7, 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| EP2062582A1 * | Aug 14, 2007 | May 27, 2009 | Tianjin Hemey Bio-Tech Co., Ltd. | The antibiotics composition comprising beta-lactam antibiotics and buffers |
| US4524073 * | Jul 20, 1983 | Jun 18, 1985 | Beecham Group P.1.C. | β-Lactam compounds |
| US6465428 * | Aug 25, 2000 | Oct 15, 2002 | Aventis Pharma S.A. | Pharmaceutical combinations based on dalfopristine and quinupristine, and on cefepime |
| US20040254381 * | Aug 15, 2003 | Dec 16, 2004 | Day Richard A. | Antibiotic compositions and methods of using the same |
| US20050148571 * | Nov 29, 2002 | Jul 7, 2005 | Nancy Niconovich | Method of treating bacterial infections using gemifloxacin or a salt thereof and a betha-Lactam antibiotic |
| US20090148512 * | Apr 17, 2008 | Jun 11, 2009 | Lannett Co Inc | Novel uses of chloramphenicol and analogous thereof |
| US20090232744 * | Feb 26, 2009 | Sep 17, 2009 | Pari Pharma Gmbh | Macrolide compositions having improved taste and stability |
| WO2002009758A2 * | 31 Jul 2001 | 7 Feb 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| US6750224 | 17 Aug 2000 | 15 Jun 2004 | Wockhardt Limited | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |
| Examples of | ||||||||
| reported trade | ||||||||
| names for products | ||||||||
| containing the 6- | ||||||||
| 6-Fluoroquinolin- | fluoroquinolin- | |||||||
| 4(1H)-one | 4(1H)-one | Structure | ||||||
| amifloxacin |  | |||||||
| balofloxacin |  | |||||||
| ciprofloxacin | Cipro®, Ciprobay, & Ciproxin |  | ||||||
| clinafloxacin |  | |||||||
| danofloxacin | Advocin & Advocid |  | ||||||
| difloxacin | Dicural® & Vetequinon |  | ||||||
| enrofloxacin | Baytril® |  | ||||||
| fleroxacin | Megalone |  | ||||||
| flumequine | Flubactin |  | ||||||
| garenoxacin |  | |||||||
| gatifloxacin | Tequin® & Zymar® |  | ||||||
| grepafloxacin | Raxar |  | ||||||
| ibafloxacin |  | |||||||
| levofloxacin | Levaquin®, Gatigol, Tavanic, Lebact, Levox, & Cravit |  | ||||||
| lomefloxacin | Maxaquin® |  | ||||||
| marbofloxacin | Marbocyl® & Zenequin |  | ||||||
| moxifloxacin | Avelox® & Vigamox® |  | ||||||
| nadifloxacin | Acuatin, Nadoxia, & Nadixa |  | ||||||
| norfloxacin | Noroxin®, Lexinor, Quinabic, & Janacin |  | ||||||
| ofloxacin | Floxin®, Oxaldin, & Tarivid |  | ||||||
| orbifloxacin | Orbax® & Victas |  | ||||||
| pazufloxacin |  | |||||||
| pefloxacin |  | |||||||
| pradofloxacin |  | |||||||
| prulifloxacin |  | |||||||
| rufloxacin | Uroflox |  | ||||||
| sarafloxacin | Floxasol, Saraflox, Sarafin |  | ||||||
| sitafloxacin |  | |||||||
| sparfloxacin | Zagam |  | ||||||
| temalioxacin | Omniflox |  | ||||||
| enoxacin | Penetrex & Enroxil |  |
| gemifloxacin | Factive |  |
| tosufloxacin |  | |
| trovafloxacin | Trovan |  |
No comments:
Post a Comment