WILL BE UPDATED………..
………………….
……………………….
PAPER
RSC Adv., 2014,4, 14468-14470
DOI: 10.1039/C4RA00840E
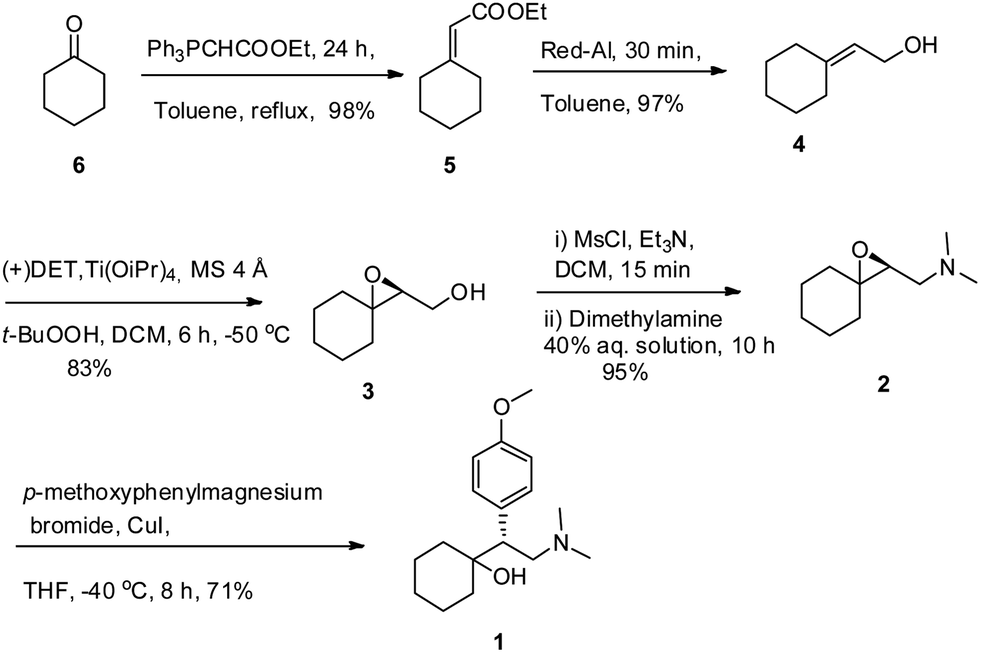
A protecting group free asymmetric total synthesis of (−)-venlafaxine is reported. The strategy employs Sharpless epoxidation and regio-selective epoxide ring opening by an in situgenerated Gilman reagent as key steps. This paper reports a 53% overall yield in 6 steps for total synthesis of (−)-venlafaxine.
…………………
Examples:Example 1 – Preparation of venlafaxine from N,N-didesmethyl venlafaxine hydrochloride
A 50 % aqueous NaOH solution (4 ml, 74 mmol) was added to a stirred solution of N,N-didesmethyl venlafaxine hydrochloride (5.72 g, 20 mmol) in water (16 ml) at room temperature. Formic acid (98 %, 11.5 ml, 305 mmol) and 37 % aqueous solution of formaldehyde (8.4 ml, 113 mmol) were added to this mixture. The mixture was stirred under reflux temperature and the conversion was completed in 5 h (HPLC: 98.67 area %). Then the solution was cooled to room temperature and adjusted with 50 % aequous NaOH to pH 12. The mixture was extracted twice with 66 ml of isopropyl acetate. The collected organic phases were washed three times with water (66 ml). The isolated solution of venlafaxine base was very pure (HPLC: 98.9 area%).
Example 2 – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
To the solution of venlafaxine base in isopropyl acetate from example 1 (66 ml, 10 mmol) 5 ml of 2 M aqueous HCl were added. The mixture was heated and water was removed by azeotropic distillation using a Dean-Starck trap. When all water was removed from the mixture, the product began slowly to crystallize. The obtained suspension was heated under reflux temperature for 1.5 h, then cooled and filtered. 2.75 g (88 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %) were obtained.
Example 3 (exemplary) – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
The solution of venlafaxine in isopropyl acetate from example 1 (66 ml, 10 mmol) was concentrated to ½ of the volume. Then 10 to 50 mg of venlafaxine hydrochloride form I was added to the solution. Subsequently, a 2.5 M solution of HCl in ethanol (4.0 ml) was slowly added within 30 min. After the whole amount of acid was added, the obtained suspension was stirred for another 2 h. Then the mixture was filtered and the product was washed with isopropyl acetate and dried. We obtained 2.69 g (86 % fromN,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %).
…………………..
PATENT
Venlafaxine is known by the chemical name 1-[2-dimethylamino-1-(4 methoxyphenyl ethyl Cyclohexanol hydrochloride and structure of formula (V).
(V)
Venlafaxine is a useful pharmaceutical agent as an antidepressant. Venlafaxine, the intermediates in the manufacture of Venlafaxine, the process of preparing said Venlafaxine and their intermediates are well known from US Patents 4,535,186, US Patent No. 6,350,912, and CN 1225356.
Further International Publication No. WO03/050074 discloses the manufacture of Venlafaxine Hydrochloride and crystalline polymorphs Form I, Form II, Form III and optically pure (R) and (S) enantiomers exhibiting different crystalline structures of Venlafaxine hydrochloride. The preparation of all the forms of Venlafaxine and their inter-conversion are also described in said WO03/0500074 publication. U.S. Patents 4535186, 4761501 disclose a process for manufacture of 1-[2- amino-i-(p-methoxyphenyl) ethyljcyclohexanol (free base of formula IV), an intermediate produced during the preparation of Venlafaxine in two stages by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of n- butyl lithium (Stage 1) to form 1-[cyano(p-methoxyphenyl) methyl] Cyclohexanol of formula III
(111)
This process is commonly used for the preparation of formula III. The US Patent 4,535,186 produces a yield of about 30% based on p-methoxyphenyl acetonitrile.
WO/03/050074 suggests an alternate way of preparing compound of formula III without using butyl lithium i.e. by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of alkali metal hydroxide in a mixture of toluene and hexane. The publication WO/03/050074 also suggests a material yield of 74% based upon p-methoxyphenyl acetonitrile and purity.
The drop wise addition of butyl lithium to p-methoxyphenyl acetonitrile is hazardous and hence it requires skill and safety measures to be taken by the person skilled in the art for handling butyl lithium over the addition period to avoid any accidents during the preparation process.
The second stage i.e. conversion of compound of formula III to formula IV described in US patent US 4,535,186 is by hydrogenating compound of formula III using Rhodium on alumina. The catalyst Rhodium is recycled by filtering and washing the catalyst with ethanol and the combined filtrate evaporated and dried under vacuum yielding free base as an oil. However, the cost of Rhodium catalyst is very high and hence the catalyst has to be recovered.
WO/02/500017 suggests the use of a Nickel or cobalt catalyst for the hydrogenation, which is highly economical when compared with the Rhodium catalyst as suggested by US Patent No. 4,535,186. The International Publication WO/02/500017 teaches that the hydrogenation reaction of Stage Il may be carried out in the presence of an organic solvent preferably an alcohol. The international publication also suggests the pretreatment of the catalyst with ethanol.
The US Patent 4,535,186 describes the third stage in the process of preparing Venlafaxine i.e. conversion of compound of formula IV (free base) to compound V i.e. Venlafaxine by methylating the compound of formula IV (free base) with a mixture of formaldehyde and formic acid in water.
(IV)
US Patent Publication No. 2005/0033088 describes a process for preparing phenylethylamine derivative, an intermediate of Venlafaxine hydrochloride; said process comprising steps of reduction of compound of formula III with palladium on charcoal in an organic acid selected from formic acid, acetic acid or propionic acid, preferably acetic acid in an autoclave at a pressure of 5 to 25 kg/cm2 preferably 10 to 15 kg/cm2 at a temperature in the range of 30 to 75°C, preferably at 50 to 55°C till the hydrogenation substantially complete, filtering the palladium catalyst and evaporating the filtrate. Extracting the filtrate with halogenated hydrocarbon solvent and purifying the same. The process also describes the preparation of Venlafaxine hydrochloride without isolation of freebase.
The route of synthesis for Venlafaxine (formula V) and intermediate of Venlafaxine (formula IV) is depicted in the following scheme:
Step -I
(I) (II) (III) Step-ll
…………..(III) ……………………(IV)
Step-ll I
(IV) (V)
Accordingly, the present invention relates to an improved process for the preparation of compound of formula IV
(IV)
comprising the step of hydrogenating a compound of formula
(Hi)
in the presence of toluene, water, and a catalyst wherein the said process yields 66% formula (IV) with 99% HPLC purity.
The compound of formula IV is further methylated using formaldehyde and formic acid mixture to form Venlafaxine (formula V) followed by the treatment with HCL gas dissolved in Isopropanol.
(IV) (V)
According to the preferred embodiment of the present invention, nickel catalyst, preferably Raney nickel catalyst is used. The catalyst is washed in water to remove the alkali. No pretreatment of the nickel catalyst is required.
(HI) (IV)
According to another embodiment of the present invention, compound of formula IV is prepared by hydrogenating compound of formula III in the presence of Raney Nickel and water. According to another embodiment of the present invention, compound of formula III is prepared by charging p-methoxyphenyl acetonitrile into butyl lithium at -70 to -75°C and tetrahydrofuran; cooling the reaction mixture to about -50°C to -750C; adding cyclohexanone at a temperature below -500C quenching with ice and saturated ammonium chloride solution below 0°C; and stirring and filtering the product of formula (III) wherein the said process yields 89% of compound of formula III with 99.8% purity. The reaction scheme is depicted as follows:
…………(I) …………(H)………………………………. (III)
Example 1 :
Preparation of 1-[cyano-(4-methoxyphenyl) methyl Icvclohexanol.
In a 2 ltr 4 necked round bottom flask equipped with a overhead stirrer, thermometer and dropping funnel, 100 ml dry THF followed by 210 ml Butylithium (1.6 M solution in Hexane) was charged. The reaction mixture was cooled to – 700C. Added gradually a solution of 50 gm p-methoxyphenyl acetonitrile dissolved in 50 ml dry THF at -70 to -75°C. After 30 min, added solution of 33.1 gm Cyclohexanone in 50 ml THF. After the addition, maintained at -65 to -700C and monitored by TLC. After 4 hrs, reaction mixture was gradually added over mixture of ice and 150 ml saturated ammonium chloride solution below 0°C and adjusted pH to 7 with dilute Hydrochloric acid. Stirred for 1 hr and filtered the product. Washed the product with 200 ml hexane and dried to obtain 74.3 gm. (The yield based on p-methoxyphenyl acetonitrile 89%, Melting range 123- 125°C, HPLC purity of 99.8%).
Example 2:
Preparation of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate
In an autoclave are charged 100 gm 1-[cyano(4- methoxyphenyl)ethyl]cyclohexanol, 100 ml toluene and 400 ml water at RT. Stirred and cooled to 10°C. Charged 20 gm Raney Nickel (which was prewashed with water to make it free of Alkali) and 100 ml liquor ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintained for 120 minutes below 120C. Then the reaction temperature slowly raised to bout 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 50°C for 8 hr. After the completion of the reaction, cooled the reaction to RT, released the hydrogen pressure and charged 400 ml toluene. Filtered the catalyst and washed bed with 100 ml toluene. Separated the organic layer from the filtrate. The organic layer was washed with 10% Sodium chloride solution. To the organic layer was added 40 ml methanol followed by 10 ml acetic acid. Stirred for 15 minutes and then again charged 10 ml acetic acid. Then heated to 75-8O0C and maintained for 15 minutes. Cooled to 0 – 50C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 83.5 gm (Yield 66%, Melting range 164-166°C, HPLC purity 99%).
Example 3:
Preparation of H2-dimethylamino-1-(4-methoxyphenyl) ethvH Cyclohexanol Hydrochloride
To a stirred solution of 100 gm of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate in 300 ml water was added 117 gm of formic acid (88%) and 91 gm of formaldehyde (40% solution). The solution was heated to 98°C and maintained for 20 hrs. Reaction mixture was cooled to about 100C and added 500 ml ethyl acetate. The pH was adjusted to about 7 with sodium hydroxide solution and further to 10 – 10.5 with ammonium hydroxide solution. Layers were separated. Aqueous layer was extracted with ethyl acetate. Combined organic layers were washed with water. Combined organic extract was stirred with activated carbon (5 gm) and filtered. Filtrate was concentrated in vacuum to completely remove ethyl acetate. Residue was dissolved in isopropanol (300 ml) and acidified at 300C (pH 1-1.5) with the solution of HCI in isopropanol. Temperature was then raised to 600C and maintained for 60 to 90 min. The reaction mass was cooled under agitation to 10°C and maintained under agitation at 10°C for 60 min. Product was isolated by filtration. Finally it was washed with isopropanol and dried at 60°C.
Dry wt. : 85 gm (84% yield, HPLC 99.9% purity with all individual impurities below 0.1% concentration). This material exhibited following characteristic x-ray powder diffraction pattern with characteristic peaks expressed in d-values (A) at.
(The abbreviations in brackets mean : (vs) = Relative intensity above 80%; (s) = 30% – 80%; (m) = 15% – 30%; (w) = 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)12.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
Example 4 :
Preparation of 1-f2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate (IV)
In an autoclave charged 150 gm 1-[cyano(4-methoxyphenyl)ethyl]cyclohexanol, and 675 ml water at RT. Stirred and cooled to 1O0C. Charged 30 gm Raney Nickel (prewashed with water to make it free of Alkali) and 150 ml liquor Ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintaind for 120 minutes below 12°C. After completion of 120 minutes slowly raised the temperature to about 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 5O0C for about 20 hrs. Monitored reaction by TLC to ensure disappearance of starting material. After the completion of the reaction cooled the reaction to RT, released the hydrogen pressure and filtered through celite bed. Washed bed with 300 ml toluene. To the filtrate added 300 ml toluene. Shaken well and separated the organic layer. The organic layer was washed with 5% Sodium chloride solution. To the organic layer was added 45 ml methanol and 15 ml acetic acid. Stirred for 15 minutes and then again charged 15 ml acetic acid. Then heated to 75-8O0C and maintained for 15 mins. Cooled to 0 – 5°C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 104 gm (Yield 53%, Melting range 152-153°C, HPLC purity 90%).
……………….
PATENT
Venlafaxine acts by inhibiting re-uptake of norepinephrine and serotonin. It has been reported that its (-) enantiomer is a more potent inhibitor of norepinephrine synaptosomal uptake while its (+) enantiomer is more selective in inhibiting serotonin uptake (J. Med. Chem. 1990, 33(10), 2899-2905) In humans, venlafaxine is transformed by a metabolic pathway into two minor metabolites, N-desmethylvenlafaxine of formula II, N,O-di- desmethylvenlafaxine of formula IV and one major metabolite, O-desmethylvenlafaxine of formula III.
formula I formula II
formula III formula IV
In the literature there are several processes reported for the synthesis of venlafaxine of formula I and venlafaxine hydrochloride of formula Ia.
formula Ia
The synthesis of venlafaxine from 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (hereinafter called as cyano-intermediate and represented by formula V) involving two step synthesis is known in the prior art.
formula V
US 4,535,186 discloses the preparation of venlafaxine of formula I by the reaction of p- methoxyphenylacetonitrile with cyclohexanone at -78 0C in the presence of n-butyllithium as a base which yields 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V. Reduction of the cyano-intermediate under hydrogen pressure with rhodium on alumina catalyst gives l-[2-amino-l-(4-methoxyphenyl)ethyl]cyclohexanol. N-Methylation of the amino compound is accomplished employing formaldehyde and formic acid (Eschweiler- Clarke reaction) to give venlafaxine of formula I.
The reaction is as shown in the Scheme- 1.
Reduction
Scheme-1
Another prior art reference, Zhou Jinpei et.al, J. China Pharm. University, 1999, 30(4), 249- 50) discloses the preparation of venlafaxine starting from anisole. Anisole is acylated to the chloroacetyl derivative, which is then animated using N,N-dirnethylamine. The carbonyl group of this compound is reduced to the alcohol using KBH4 and is converted to the bromo- derivative using PBr3 which in turn when reacted with Mg and cyclohexanone undergoes a Grignard reaction to provide venlafaxine of formula I.
The reaction is as shown in the Scheme-2.
Scheme-2
US 2005033088 discloses a two step process for venlafaxine starting from the cyano- intermediate. The cyano-intermediate is reduced in the presence of palladium on charcoal in acetic acid at a hydrogen pressure of 5-25 kg/cm2 at a temperature in the range of 30-75 0C. The product of step 1 is N-methylated using formic acid, formaldehyde solution at a temperature of 90-98 0C for 19 hrs to yield venlafaxine, which is then converted to its hydrochloride salt.
WO2006035457 also discloses a process of making venlafaxine and its intermediates. The process comprises the step of hydrogenating the cyano-intermediate in the presence of toluene, water, and Raney nickel where in the said process yields 66% of an intermediate with 99% HPLC purity. This reaction is carried out at 10-12 0C and at 4-5 kg/cm2 of hydrogen pressure for 2 hrs and further at 50 0C at 7-8 kg/cm2 for 7-8 hrs. This intermediate is N-methylated using formaldehyde and formic acid mixture to form venlafaxine, which is treated with IPA/HC1 to get venlafaxine hydrochloride.
US 6,350,912 discloses a one pot process for the preparation of venlafaxine in 15-28 % yield from the cyano-intermediate. In the said patent venlafaxine has been prepared by the reduction of cyano-intermediate in the presence of Raney nickel and without isolation of intermediate l-[2-amino-l-(4-methoxyphenyl) ethyl] cyclohexanol
CN 1850781 discloses a process for the preparation of venlafaxine by following steps: (1) carrying out condensation of 4-methoxyphenylacetonitrile and cyclohexanone in presence of base to obtain 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile, (2) reacting 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with cuprous chloride and dimethylamine to obtain 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)-N,N- dimethylacetimidamide, and (3) reacting 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)- N,N-dimethylacetimidamide with KBH4 to obtain 1 – [2-dimethylamino)- 1 -(4-methoxyphenyl) ethyl)cyclohexanol (venlafaxine).
The reaction is as shown in the Scheme-3.
Scheme-3
The processes disclosed in the prior art have many disadvantages. Most of the prior art processes employ formaldehyde as a reactant for N-methylation step which is known to be a carcinogen. Acute exposure of the same is highly irritating to the eyes, nose and throat. Ingestion of formaldehyde is fatal and long term exposure causes respiratory problems and skin irritation.
Another disadvantage is the formation of an impurity (represented by formula VI), which is formed during N-methylation step using formaldehyde as a reagent.
formula VI
As may be appreciated, all the above well-known processes share the same strategy of synthesis, consisting of two steps for the synthesis of venlafaxine from cyano-intermediate or are prepared in one pot with poor yield.
Yet another drawback of the processes disclosed in the prior art is the use of expensive catalysts like rhodium on alumina and use OfBF3 etherate which is highly corrosive.
The prior art references disclose the synthesis of alkoxyphenylethyldimethylamine from alkoxyphenylacetonitrile using excess dimethylamine and palladium catalyst in methanol solution which is firstly reported by Kindler and Hensse. (1. Kindler and Hesse; Arch. Pharm., 1933, 271, 439. 2. Johannes S. Buck, Richard Baltzly and Walter S. Ide; J. Am. Chem. Soc. 1938, 60(8), 1789-1792; 3. Albert J. Schuster and Eugene R. Wagner; J. Labelled Compounds and Radiopharmaceuticals 1992, XXXIII(3), 213-217).
The reaction is as shown in the Scheme-5.
Scheme-5
According to an aspect of the invention there is provided a novel single step process for the synthesis of venlafaxine of formula I and N-desmethylvenlafaxine of formula II from 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V comprising reaction of 2- (l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with an alkylamine and/or its salt in a solvent in the presence of a transition metal catalyst, under hydrogen atmosphere.
formula I formula II formula V DETAILED DESCRIPTION OF THE INVENTION
The present invention describes a single step process for venlafaxine and its analog starting from the cyano-intermediate. The present invention circumvents the difficulties encountered in the prior art and is an economically viable process.
The present invention particularly relates to a single step synthesis of venlafaxine of formula I, and N-desmethylvenlafaxine of formula II from the cyano-intermediate of formula V.
The reaction of the present invention is as shown in Scheme-4:
formula V formula L R = CH3 (Venlafaxine) formula II. R = H (N-desmethylvenlafaxine)
formula Ia. R = CH3, X = Cl formula Ha. R = H, X = Cl
Example-1:
General procedure for synthesis of venlafaxine and N-desmethylvenlafaxine To the, stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (1.0 equiv.) in methanol (10-20 volumes), alkylamine (3-5 equiv.) was added and the mixture was stirred for 5-10 minutes to get a clear solution. Palladium catalyst (10-50 wt %) was added under nitrogen atmosphere to the above reaction mixture. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 5 to 40 hrs. The progress of the reaction was monitored by TLC and HPLC. After completion of the reaction, the catalyst was filtered through celite and washed with methanol. The combined filtrate was concentrated to dryness under reduced pressure and the residue was poured in water. The aqueous layer was basifϊed with 10% aq. NaOH to pH 8-10 and extracted with ethyl acetate (3 times). The combined ethyl acetate layers were washed with brine and dried over sodium sulphate. Ethyl acetate was evaporated under vacuum to obtain the title compound.
Example-2: Venlafaxine
To the stirred solution of 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (460.0 ml), dimethylamine hydrochloride (26.6 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 20 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.Og) which was directly converted to its hydrochloride salt using IPA/HC1. HPLC purity of the crude reaction mixture (18.Og) = 81.64%.
Venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.05 (d, 2H), 6.81 (d, 2H), 3.79 (s, 3H), 3.27 (t, IH), 2.94 , (dd, IH), 2.31 (s, 6H), 2.28 (dd, IH), 1.71-0.94 (m, 10H). 13C NMR in CDCl3: δ 158.16,’ 132.65, 130.01, 113.20, 74.14, 61.14, 55.05, 51.52, 45.37, 37.99, 31.07, 25.91, 21.52, 21.23.
IR (KBr): 3152, 2980, 2941, 2895, 1728, 1607, 1512, 1462, 1439, 1358, 1279, 1204, 1186, 1177, 1146, 1103, 1040, 1011, 968, 851 cm“1. HPLC Purity: 99.37 % (area %). GC-MS: 178 (M+H+).
Venlafaxine hydrochloride salt:
1H NMR in D2O (300 MHz): δ 7.23 (d, 2H), 6.91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 6H), 1.41-1.0 (m, 10H).
13C NMR in D2O: δ 158.52, 130.86, 128.16, 114.25, 73.23, 58.26, 55.25, 50.38, 44.87, 41.41, 35.07, 33.34, 24.76, 21.08, 20.86.
IR (KBr): 3321, 2941, 2928, 2675, 2644, 2623, 2611, 2587, 2521, 2482, 2359, 2330, 1512, 1441, 1242, 1179, 1038, 829 cm“1. HPLC Purity: 99.64 % (area %). GC-MS: 178 (M+H+).
Example-3: N-Desmethyl venlafaxine To the stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (350.0 ml), monomethylamine hydrochloride (27.7 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 24 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.5g) which was directly converted to its hydrochloride salt using IPA/HCl.
HPLC conversion to N-desmethylvenlafaxine (in the crude reaction mixture = 18.5g): 35.46%. N-Desmethyl venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.23 (d, 2H), £91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 3H), 1.41-1.0 (m, 10H)
| CITED PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2003050074A1* | Mar 19, 2002 | Jun 19, 2003 | Cadila Healthcare Ltd | Manufacture of venlafaxine hydrochloride and crystalline polymorphs thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2008059525A2* | Oct 1, 2007 | May 22, 2008 | Calyx Chemicals And Pharmaceut | An improved process for the preparation of venlafaxine and its analogs |
| CITED PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2006035457A1* | Sep 16, 2005 | Apr 6, 2006 | Amoli Organics Ltd | A process for the manufacture of venlafaxine and intermediates thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| US6350912 * | Feb 28, 2001 | Feb 26, 2002 | Council Of Scientific And Industrial Research | One pot process for the preparation of 1-[2-dimethylamino-(4-methoxyphenyl)-ethyl]cyclohexanol |
| US20050033088 * | Jun 7, 2004 | Feb 10, 2005 | Dr. Reddy’s Laboratories Limited | Catalytic hydrogenation of phenylacetonitrile using palladium on carbon supports |
| REFERENCE |
|---|
| 1 | * | CHAVAN S P ET AL: “An efficient and green protocol for the preparation of cycloalkanols: a practical synthesis of venlafaxine” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 45, no. 39, 20 September 2004 (2004-09-20), pages 7291-7295, XP004558985 ISSN: 0040-4039 |
| 2 | * | J.S. BUCK ET AL: “beta-Phenylethylamine Derivatives. Tertiary and quaternary salts” J.AM.CHEM.SOC., 1938, pages 1789-1792, XP002478651 cited in the application |
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|
| WO2010100520A1* | Mar 4, 2009 | Sep 10, 2010 | Hikal Limited | A process for preparation of phenethylamine derivative |
| WO2011124190A2 | Apr 6, 2011 | Oct 13, 2011 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |
P.S.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
P.S.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
P.S.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.

COCK WILL TEACH YOU NMR

COCK SAYS MOM CAN TEACH YOU NMR

Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com

SEE

 2014, Vol. 35
2014, Vol. 35 



 .
. Kawran Bazar
Kawran Bazar .
. Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.
Dry fish sellers at the Karwan Dry Fish Market (Bazar), Dhaka, Bangladesh.































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
