1 AVOSENTAN
2 TEZOSENTAN
3 CLAZOSENTAN
4. ATRASENTAN
5. AMBRISENTAN
6. ZIBOTENTAN
7
8
WILL BE ADDED..................

1 AVOSENTAN

 AVOSENTAN
AVOSENTAN








2 TEZOSENTAN

 TEZOSENTAN
TEZOSENTAN

3 CLAZOSENTAN

 CLAZOSENTAN
CLAZOSENTAN
 CLAZOSENTAN
CLAZOSENTAN
 CLAZOSENTAN DI-NA SALT is discontinued
CLAZOSENTAN DI-NA SALT is discontinued





CLAZOSENTAN
CLAZOSENTAN
CLAZOSENTAN DI-NA SALT is discontinued
4

2 TEZOSENTAN
3 CLAZOSENTAN
4. ATRASENTAN
5. AMBRISENTAN
6. ZIBOTENTAN
7
8
WILL BE ADDED..................
1 AVOSENTAN
AVOSENTAN
N-[6-Methoxy-5-(2-methoxyphenoxy)-2-(4-pyridyl)pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidin-4-yl]-amide,
5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide,
Endothelin ETA Receptor Antagonists
M.Wt: 479.51
Formula: C23H21N5O5S
Formula: C23H21N5O5S
Roche (Originator)
CAS No.: 290815-26-8
- RO 67-0565
- SPP 301
- UNII-L94KSX715K
PHASE 3
CLINICAL TRIALS
SPP-301 is an oral, once-daily, second-generation endothelin ETA receptor antagonist which had been in phase III clinical development at Speedel for the treatment of diabetic nephropathy. In December 2006, the company reported that the phase III trial had been stopped based on the recommendation from the trial’s Data Safety Monitoring Board (DSMB) to stop the trial following incidence of a significant imbalance in fluid retention in patients in the study arms. Speedel reported that the compound will be evaluated for potential new clinical development for the treatment of diabetic kidney disease and other indications.
Originally developed by Roche and specifically optimized for improved liver safety, SPP-301 was licensed to Speedel in October 2000. In 2003, Speedel exercised its option to license from Roche all rights to SPP-301, including exclusive worldwide rights for the full development and commercialization of the ETA antagonist. SPP-301 has fast track designation and has undergone a special protocol assessment (SPA) by the FDA. Speedel had been studying the drug for the treatment of hypertension.
290815-26-8 CAS
PATENTS
2. WO 2004078104
3. WO 2005113543
4. WO 2007031501
5. WO 2008077916
Dutzler R, Ernstb B, Hediger MA, Keppler D, Mohr P, Neidhart W, Märki HP.Chimia (Aarau). 2010;64(9):662-6.
………………………
INTRODUCTION
- 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide corresponding to the formula
 is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2.
is an inhibitor of endothelin receptors. WO00/52007 describes the preparation of said compound which is crystallized from Me2Cl2. - Own investigations have shown that there exist two distinct crystalline forms, hereinafter referred to as form A and form B, as well as a number of further solvates, in particular the methanol, ethanol, isopropanol, dichloromethane, acetone, methyl ethyl ketone and tetrahydrofuran solvates.
- It was further surprisingly found that the thermodynamically stable crystalline form – form B – can be prepared under controlled conditions and that said form B can be prepared with a reliable method in an industrial scale, which is easy to handle and to process in the manufacture and preparation of formulations.
………………..
4,6-Dichloro-5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine (described in EP 0 799 209) can be transformed to the intermediate of formula (III)—according to scheme 1—on reaction with an appropriate sulfonamide of formula (II), wherein R1 is as defined in claim 1, in a suited solvent such as DMSO or DMF at room temperature or at elevated temperature and in the presence of a suited base such as potassium carbonate.
EXAMPLE 1
[0064] a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
[0065] C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
[0066] Preparation of the starting material:
[0067] b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide ( CHLORO STARTING MATERIAL) as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
……………………………….
EXAMPLE 1
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg2SO4, concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120° C. to give the desired 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as white crystals. Melting point 225-226° C. ISN mass spectrum, m/e 478.2 (M-1 calculated for C23H21N5O5S1: 478).
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
Preparation of the starting material:
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 19.66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40° C. until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60° C. for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl]-amide as yellow crystals. Melting point 177-179° C. ISN mass spectrum, m/e 482.2 (M-1 calculated for C22H18ClN5O5S1: 482).
…………………….
SYNTHESIS OF
4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine
A BASIC STARTING MATERIAL FOR AVOSENTAN
- Preparation of the starting material
- b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hours 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- c) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
- d) A suspension of 154.6 g of 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCl3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2Cl2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2Cl2. The combined CH2Cl2 extracts are dried with MgSO4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2Cl2remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine, melting point 178-180°C.
…………………………
Preparation of the starting material:
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide IE THE 6 CHLORO COMPD
b) 11.3 g of 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)-pyrimidine and 1 .66 g of 5-methylpyridyl-2-sulfonamide potassium salt (preparations described in EP 0 799 209) were dissolved in DMF (255 ml) under argon. The solution was stirred for 2 h at 40°C until completion of the reaction according to TLC analysis. The reaction mixture was cooled to RT and the solvent removed in a high vacuum. The residue was suspended in water (850 ml), acetic acid (85 ml) was added and the mixture was stirred for 30 minutes at RT. The solid that precipitated was collected by filtration and dried in a high vacuum at 60 °C for 16 h to give 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2- methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as yellow crystals. Melting point 177-179 °C. ISN mass spectrum, m/e 482.2 (M-l calculated for C22Hi8ClN5O5Sι: 482).
………………………………………………………………………………………….
NEXT
Example 1AVOSENTAN
a) To a solution of 6.9 g sodium in MeOH (300 ml) were added 14.52 g of
5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl- pyrimidin-4-yl] -amide at RT and the mixture was refluxed for 5 days until completion of the reaction according to TLC analysis. The reaction mixture was concentrated in vacuo to half its volume upon which the crude reaction product precipitated as a sodium salt. It was filtered off by suction and dried in a high vacuum. The solid was dissolved in water, which was then made acidic by addition of acetic acid. The precipitating free sulfonamide was extracted into Me2Cl2. The organic layer was dried over Mg SO , concentrated on a rotary evaporator, and the crystalline solid that had formed was filtered off. It was then dried in a high vacuum for 12 h at 120 °C to give the desired 5-methyl-pyridine-2-sulfonic acid [6- methoxy-5-(2-methoxy-phenoxy)-2-pyridin-4-yl-pyrimidin-4-yl] -amide as white crystals. Melting point 225-226 °C. ISN mass spectrum, m/e 478.2 (M-l calculated for
C23H21N5O5S1: Calc: C 57.61; H 4.41; N 14.61; S 6.69. Found: C 57.56; H 4.38; N 14.61; S 6.83
…………………………………………….
IS DESCRIBED IN
ALSO
- Diabetic nephropathy is the principle cause of end stage renal disease in the western world. It is a major cause of morbidity and mortality in Type-I Diabetes, but is an increasing problem in Type-II Diabetes and because the incidence of this is five times that of Type-I Diabetes, it contributes at least 50% of diabetics with end stage renal disease.
- The initial stage of subtle morphologic changes in the renal glomeruli is followed by microalbuminuria. This is associated with a modestly rising blood pressure and an increased incidence of cardiovascular disease. There follows a continued increase in urinary protein excretion and declining glomerular filtration rate. Diabetic nephropathy has many possible underlying pathophysiological causes including metabolic, glycosylation of proteins, haemodynamics, altered flow/pressure in glomeruli, the development of hypertension and cytokine production; all of these are associated with the development of extracellular matrix and increased vascular permeability leading to glomerular damage and proteinuria.
| WO2005113543A1 * | May 12, 2005 | Dec 1, 2005 | Alexander Bilz | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO2007031501A2 * | Sep 11, 2006 | Mar 22, 2007 | Speedel Pharma Ag | Pyridylsulfonamidyl-pyrimidines for the prevention of blood vessel graft failure |
| WO2008077916A1 * | Dec 21, 2007 | Jul 3, 2008 | Ovidiu Baltatu | Pharmaceutical composition using aliskiren and avosentan |
| EP1454625A1 * | Mar 6, 2003 | Sep 8, 2004 | Speedel Development AG | Pyridylsulfonamidyl-pyrimidines for the treatment of diabetic nephropathies |
| EP1595880A1 * | May 13, 2004 | Nov 16, 2005 | Speedel Pharma AG | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| EP1938812A1 * | Dec 22, 2006 | Jul 2, 2008 | Speedel Pharma AG | Pharmaceutical composition using aliskiren and avosentan |
| US6951856 | Jul 10, 2001 | Oct 4, 2005 | Actelion Pharmaceuticals Ltd. | Arylethene-sulfonamides |
| US7402587 | May 12, 2005 | Jul 22, 2008 | Speedel Pharma Ag | Crystalline forms of a pyridinyl-sulfonamide and their use as endothelin receptor antagonists |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
| EP0713875A1 * | Nov 13, 1995 | May 29, 1996 | F. Hoffmann-La Roche AG | Sulfonamides |
| EP0897914A1 * | Aug 10, 1998 | Feb 24, 1999 | F. Hoffmann-La Roche Ag | Process for the preparation of 2,5-disubstitued pyridines |
2 TEZOSENTAN
TEZOSENTAN
180384-57-0 CAS OF FREE ACID
N-[6-(2-Hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)pyridin-4-yl]pyrimidin-4-yl]-5-propan-2-ylpyridine-2-sulfonamide
5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
| Formula | C27H27N9O6S |
|---|---|
| Mol. mass | 605.624 |
........................................................................................
Tezosentan disodium, Ro-61-0612, Veletri
5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2)
180384-58-1 of disodium salt, 180384-57-0 (free acid)
MF C27-H25-N9-O6-S.2-Na
MW 649.5975
Roche (Originator), Actelion (Licensee), Genentech (Codevelopment)
CARDIOVASCULAR DRUGS, Heart Failure Therapy, Renal Failure, Agents for, RENAL-UROLOGIC DRUGS, Treatment of Renal Diseases, Endothelin ETA Receptor Antagonists, Endothelin ETB Receptor Antagonists
Phase III
TEZOSENTAN
Tezosentan is a non-selective ETA and ETB receptor antagonist.[1] It acts as a vasodilator and was designed as a therapy for patients with acuteheart failure. Recent studies have shown however, that tezosentan does not improve dyspnea or reduce the risk of fatal or nonfatal cardiovascular events.[2]
Pulmonary disease (COPD), which may possibly be associated with pulmonary hypertension, as well as allergic and non-allergic rhinitis, provided that treatment with endothelin from a therapeutic standpoint is not contraindicated.
Tezosentan disodium is an endothelin ETB receptor antagonist in phase II clinical development for the treatment of stable, chronic pulmonary arterial hypertension. The drug was previously being evaluated for heart failure, but trials in that indication have been discontinued. The compound is being developed by Actelion.
......................................
SYNTHESIS
......................................
SYNTHESIS
Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF. refEP 0799209; JP 1998509182; WO 9619459
.........................................
SYNTHESIS PROCEDURE as in EP0979822A1
Examples
- 1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0°C, 11.7 ml (219 mmol) of concentrated sulfuric acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0°C to 5°C. The viscous yellow-brownish suspension was stirred at 0°C for 1.5 hr. Subsequently, a solution of 83 g (437 mmol) of sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0°C to 5°C and the reaction mixture was stirred at 0°C to 5°C for 30 min. The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80°C, 2000 Pa for 16 hr. There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
- Preparation of starting material:
- a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH4Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydrochloride (decomposition point 245-247°C).
- b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250°C.
- Example 1
Example 2
- Within 20 min. 61 ml (633 mmol) of POCl3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5°C to 10°C followed by stirring at 5°C to 10°C for 15 min. Then 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90°C for 25 hr. The reaction mixture was cooled down to 20°C and transferred to a new flask together with 50 ml of dichloromethane. Volatile components (i.e. excess of POCl3) was removed by evaporation from 20°C to 70°C followed by re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35°C to 40°C and 80 ml of de-ionized water were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35°C to 40°C for 30 min. followed by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20°C and was stirred for additional 2 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70°C, 2000 Pa for 16 hr. There were obtained 21.3 g (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
- 8.95 g (24 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile were suspended in 100 ml of acetone. At a temperature of 20°C, 5.04 g (25 mmol) of 5-isopropyl-pyridine-2-sulfonamide, 1 ml of de-ionized water, 10.6 g (77 mmol) of potassium carbonate and 135 mg (1.2 mmol) 1,4-diazobicyclo[2.2.2]octane were added. The mixture was stirred at 40°C for 20 hr. Thereafter, another 240 mg (1.2 mmol) of 5-isopropyl-pyridine-2-sulfonamide and 80 mg (0.7 mmol) of 1,4-diazobicyclo[2.2.2]octane were added. The reaction mixture was stirred for 24 hr at 40°C followed by cooling to 20°C. Then 50 ml of de-ionized water and 45 ml of 3 N aqueous hydrochloric acid were added slowly until pH = 1. The acetone was removed by distillation and the resulting suspension was stirred at 20°C for 1.5 hr. The solid was filtered off under suction, washed first with 100 ml of de-ionized water and thereafter with 50 ml of t-butylmethylether. Then the solid was dried at 70°C, 2000 Pa for 20 hr. There were obtained 13.2 g (102% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 87.8% (w/w).
- Example 4
- 122 g (233 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide was suspended in 450 ml of N,N-dimethyl formamide and the mixture was cooled down to 15°C. At this temperature, 35 ml of hydrazine hydrate were added dropwise within 1 hr. The resulting solution was stirred at 15°C to 20°C for 16 hr and thereafter diluted with 600 ml of de-ionized water. Then 50 ml of glacial acetic acid were added dropwise at 0°C to 5°C until pH = 5.5. 600 g of ice were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 3000 ml of water and dried at 40°C, 2000 Pa for 24 hr. There were obtained 126 g (97% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 91.8% (w/w).
- Example 6
- 20 g (35 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. The solution was kept at 15°C to 20°C and 23 ml of 6 N aqueous hydrochloric acid were added, followed by addition of a solution containing 4.8 g (7 mmol) of sodium nitrite in 20 ml de-ionized water within 10 min. The mixture was stirred at 20°C for 1 hr, then 140 ml of de-ionized water were added and the suspension was stirred at 0°C for 1 hr. The solid was filtered, firstly washed with 80 ml of de-ionized water and thereafter with 80 ml of t-butylmethylether. Then the solid was dried at 70°C and 2000 Pa for 16 hr. The crude product (23.4 g) was taken up with 117 ml of tetrahydrofuran for 1 hr. After filtration at 0°C the crystallized product was washed with 25 ml of t-butylmethylether and was then dried at 70°C, 2000 Pa for 16 hr. There were obtained 17.3 g (84% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 91.1% (w/w).
- Example 8
- Example 10
- 6.2 g of sodium hydroxide were added to 15 g (26 mmol) of 5-isopropyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amid and 75 ml of ethylene glycol. The mixture was heated to 85°C for 5 hr. Then 55 ml of de-ionized water were added and thereafter 55 ml of 3 N hydrochloric acid were added dropwise. The mixture was allowed to cool down to 20°C and was stirred for 1 hr. The solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 16.2 g (103%) of 5-isopropyl-pyridine-2-sulfonic acid 16-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 92% (w/w). 80 ml of dioxane and 80 ml of ethanol were added to this solid. At a temperature of 60°C, gaseous ammonia was introduced into the liquid until pH = 9 to 10. The resulting suspension was allowed to cool down to 20°C and was stirred at 20°C for 20 hr and thereafter at 0°C for 2.5 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 18 hr. There were obtained 14.2 g of mono ammonium salt with a HPLC purity of 96.2% (w/w). The solid was heated (reflux) in 70 ml of methanol, cooled down slowly to 20°C and stirred at 20°C for 19 hr and thereafter at 0°C for 2 hr. Then the solid was filtered off and dried at 70°C, 2000 Pa for 19 hr. There were obtained 11.5 g (66% of theory) of 5-isopropyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide sodium salt (1:2) with a HPLC purity of 98.6% (w/w).
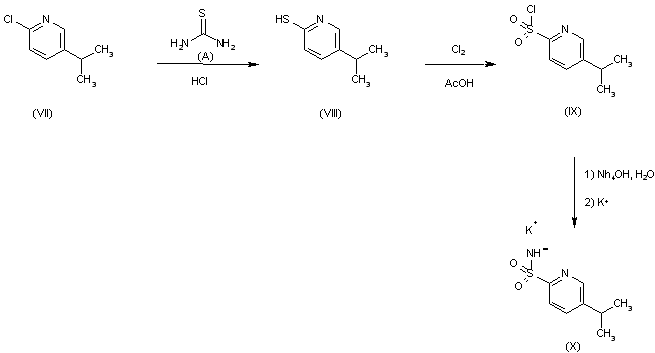
Reaction of 2-chloro-5-ispropylpyridine (VII) with thiourea (A) in aqueous HCl gives 5-isopropyl- pyridine-2-thiol (VIII), which is chlorinated with chlorine in acetic acid to yield 5-isopropylpyridine-2-sulfochloride (IX). This compound is converted into 5-isopropylpyridine-2-sulfonamide potassium salt (X).
..............................
synthesis
. Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3 for analogy only compd is different
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
Example 30 final product
In analogy to Example 3, from 5-isopropyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-isopropyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5- (2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide (tezosantan free base) as a white substance of melting point 1 98- 200°C from acetonitrile.
The corresponding disodium salt (tezosantan di sodium salt) is obtained as a white powder from this product using sodium methylate in analogy to Example 5
Example 5 for analogy only, compd is different
A solution of 47.8 g of 2-[6-(4-tert-butyl-phenylsulphonyl- amino)-5-(2-methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin- 4-yl)-pyrimidin-4-yloxy]-ethyl pyridin-2-ylcarbamate in 500 ml of abs. THF is treated dropwise with a cold solution of 2.8 g of sodium in 50 ml of methanol, whereby there forms gradually a solid precipitate which, after stirring at room temperature for 1 hour, is filtered off under suction, dried under greatly reduced pressure at 35°C for 3 days and thereafter at 50°C for 2 days. There is thus obtained the bis-sodium salt, decomposition point above 250°C.
References
- Urbanowicz, W; Sogni, P, Moreau, R, Tazi, K A, Barriere, E, Poirel, O, Martin, A, Guimont, M C, Cazals-Hatem, D, Lebrec, D (2004). "Tezosentan, an endothelin receptor antagonist, limits liver injury in endotoxin challenged cirrhotic rats". Gut (BMJ Publishing Group Ltd & British Society of Gastroenterology) 53 (12): 1844–1849. doi:10.1136/gut.2003.036517. PMC 1774327. PMID 15542526.
- "Tezosentan does not appear to improve symptoms for patients with acute heart failure". Medical Studies/Trials. news-medical.net. 7 Nov 2007. Retrieved 2007-11-24.
4 US2003/100507 A1
5 Drugs Fut 2003,28(8),754
6 WO 1996019459......
7 EP 0897914
8 WO 2011163085
9 WO 2004082637
| 10 WO 2002074034 |
11...
| 15055997 | 4-8-2004 | Discovery, modeling, and human pharmacokinetics of N-(2-acetyl-4,6-dimethylphenyl)-3-(3,4-dimethylisoxazol-5-ylsulfamoyl)thiophene-2-carboxamide (TBC3711), a second generation, ETA selective, and orally bioavailable endothelin antagonist. | Journal of medicinal chemistry |
12 ..
| 10610277 | 7-1-1999 | RO 610612 . | Drugs in R&D |
13....
| 3-27-2003 | Aqueous pharmaceutical composition comprising Tezosentan | |
| US6103902 | 8-16-2000 | Carbamoylation process |
| WO0036918 | 6-30-2000 | METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS METHODS AND COMPOSITIONS FOR TREATMENT OF CELL PROLIFERATIVE DISORDERS |
| US6063911 | 5-17-2000 | Methods and compositions for treatment of cell proliferative disorders |
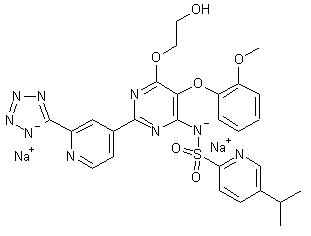
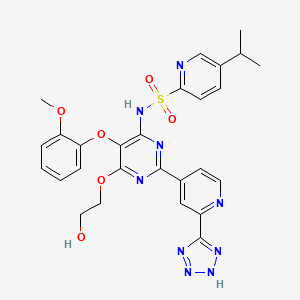
3 CLAZOSENTAN
Clazosentan
IUPAC Name: N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)
pyridin-4-yl]pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
pyridin-4-yl]pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide
VASODILATOR, Endothelin -1 - receptor antagonist
Clazosentan (Ro61-1790, AXV-034343)
- AXV 034
- AXV 034343
- AXV-034343
- AXV-343434
- Clazosentan
- Ro 61-1790
- Ro-61-1790
- UNII-3DRR0X4728
- VML 588
- VML-588
180384-56-9 cas no
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=CLAZOSENTAN in phase 3
| Formula: | C25H23N9O6S |
|---|---|
| Molecular Weight: | 577.5718 |
Endothelin type-A receptor antagonist for the treatment of vasospasm in subarachnoid hemorrhage
Selective endothelin receptor antagonist (Pivlaz)
Acteliion…… innovator
Clazosentan is a drug with orphan drug status , which since 2007, currently in Phase III clinical trials CONSCIOUS-2 ( Clazosentan to O vercome N euro logical i SC Hemia and I nfarct O cc U rring after S ubarachnoid hemorrage) is located. It is for treatment of vasospasm after subarachnoid hemorrhage are used (SAH).
Clazosentan is used by the Swiss pharmaceutical company Actelion developed. Medicinally, the disodium salt is used.Clazosentan to come under the name Pivlaz on the market.
The endothelin -1 - receptor is one of the strongest known vasoconstrictors . Clazosentan is an E -1 - receptor antagonist , for the treatment of vasospasm after subarachnoid hemorrhage is under development. After subarachnoid hemorrhage , an irritation of theblood vessels to a vasospasm and the associated to a reduced supply of brain tissue with oxygen lead. A possible consequence may be a Ischemic stroke be. Clazosentan acts this vasoconstriction contrary.
The plasma half-life of 6-10 min .
Actelion has initiated a comprehensive global phase IIb/III development program for clazosentan sodium (formerly Ro-61-1790, VML-588, …
CLAZOSENTANCLAZOSENTAN DI-NA SALT is discontinued
Clazosentan, shown below, is a well known endothelin receptor antagonist.
Since clazosentan is a known and useful pharmaceutical, it is desirable to discover novel derivatives thereof. Clazosentan is described in European Patent No. 0,799,209
IT IS DESCRIBED IN US6103902
- Paul LM van Giersbergen things Manse, J.: tolerability, pharmacokinetics, and pharmacodynamics of clazosentan, a parenteral endothelin receptor antagonist . In: European Journal of Clinical Pharmacology . 63, No. 2, February 2007, pp. 151-158. doi :10.1007/s00228-006-0117-z . PMID 16,636,870 .
- Paul LM van Giersbergen things Manse J., Gunawardena, KA: Influence of ethnic origin and sex on the pharmacokinetics of clazosentan . In: Journal of Clinical Pharmacology . 47, No. 11, November 2007, pp. 1374-1380. doi :10.1177/0091270007307337 . PMID 17,906,281
| US6004965 * | Dec 8, 1995 | Dec 21, 1999 | Hoffmann-La Roche Inc. | Sulfonamides |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
8-13-1998
|
Pyrrolidine-3-carboxylic acids as endothelin antagonists. 3. Discovery of a potent, 2-nonaryl, highly selective ETA antagonist (A-216546).
|
Journal of medicinal chemistry
|
7-23-2009
|
In silico prediction of volume of distribution in human using linear and nonlinear models on a 669 compound data set.
|
Journal of medicinal chemistry
|
………………………………………………………..
SYNTHESIS
EXAMPLE 15
a) In analogy to Example 1a), from 5-methyl-pyridine-2-sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190
b) In analogy to Example 2, from 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide.
……………………………………………..
SYNTHESIS
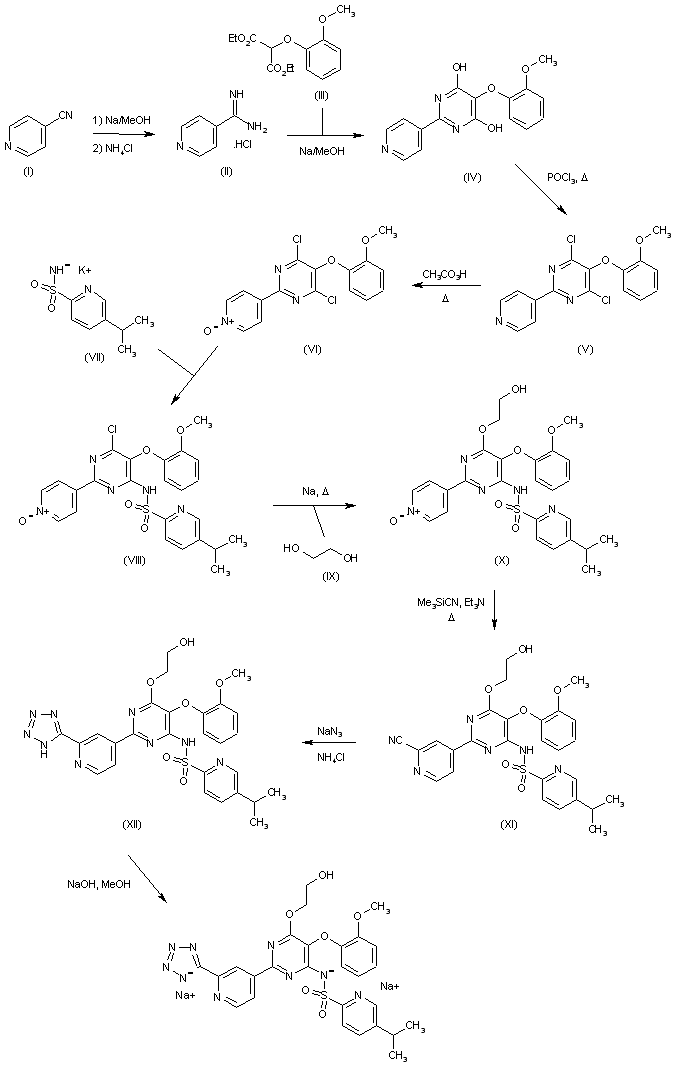
4-Cyanopyridine (I) is reacted with ammonium chloride in methanolic NaOMe to afford the amidine (II), which is cyclized with diethyl (2-methoxyphenoxy)malonate (III) producing the dihydroxypyrimidine (IV). Chlorination of (IV) in hot POCl3, followed by oxidation of the obtained dichloropyrimidine (V) with peracetic acid leads to the pyridine N-oxide (VI). Subsequent condensation of the dichloropyrimidine derivative (VI) with the potassium salt of 5-methylpyridine-2-sulfonamide (VII) yields the sulfonamido pyrimidine (VIII). The remaining chloride group of (VIII) is then displaced with the sodium alkoxide of ethylene glycol in hot DME to furnish the hydroxyethyl ether (IX). Treatment of the pyridine N-oxide (IX) with cyanotrimethylsilane and Et3N in refluxing acetonitrile gives rise to the 2-cyanopyridine (X), which is finally converted to the title tetrazole derivative by treatment with sodium azide in the presence of NH4Cl in DMF (1).
………………………………………………………….
SYNTHESIS OF DISODIUM SALT OF CLAZOSENTAN
DESCRIBED IN WO 9619459.
- III is reacted with a compound of formula V

- The reaction type is known in the art and may be performed under basic conditions for example in the presence of a coupling agent, e.g. 1,4-diazobicyclo[2.2.2]octane, together with potassium carbonate in acetone.
EXAMPLE 1
1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0 acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0 to 5 0 sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0 reaction mixture was stirred at 0 The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80 There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy -5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
Preparation of Starting Material
a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH.sub.4 Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50 4-amidino-pyridine hydrochloride (decomposition point 245-247
b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50 of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H.sub.2 O and treated little by little with 50 ml of CH.sub.3 COOH. The precipitate is filtered off under suction, washed with 400 ml of H.sub.2 O and dried at 80 obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250
EXAMPLE 2
Within 20 min. 61 ml (633 mmol) of POCl.sub.3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5 followed by stirring at 5 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90 to 20 dichloromethane. Volatile components (i.e. excess of POCl.sub.3) was removed by evaporation from 20 re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35 were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35 by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70 (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
EXAMPLE 3
12.5 g (33.5 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile and 6.06 g (35 mmol) of 5-methyl-pyridine-2-sulfonamide were added to 130 ml of acetone. 15 g of potassium carbonate and 190 mg (1.6 mmol) of 1,4-diazobicyclo[2.2.2]octane were added and the suspension was stirred at 40 de-ionized water were added followed by dropwise addition of 50 ml of 3 N hydrochloric acid (pH of the solution=1). Acetone was removed by evaporation and the suspension was stirred for 1 hr. The solid was filtered and washed with 100 ml of water. The residue was heated (reflux) in 100 ml of methanol for 1 hr followed by cooling to 20 solid was filtered and dried at 80 were obtained 16.0 g (93% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 90.3% (w/w).
EXAMPLE 5
20 g (39 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were suspended in 100 ml of N,N-dimethyl formamide and 7.6 ml (156 mmol) of hydrazine hydrate were added within 15 min. The reaction mixture was allowed to warm up slowly to 20 temperature of 15 followed by slow addition of 10.5 ml acetic acid (until pH=5.4). The resulting suspension was stirred for 2 hr at 20 additional 2 hr 0 firstly washed with 200 ml of de-ionized water and thereafter with 100 ml of t-butylmethylether. The residue was dried at 40 18 hr. There were obtained 21.7 g (102% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 81.4% (w/w).
EXAMPLE 7
20 g (37 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. To this solution was added dropwise 23 ml of 6 N aqueous hydrochloric acid at a temperature of 15 mmol) of sodium nitrite in 20 ml de-ionized water was added slowly. The reaction mixture was allowed to warm up to 20 for 1.5 hr. Then 160 ml of de-ionized water were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 100 ml of de-ionized water and dried at 50 hr. There were obtained 18.9 g (92% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 89.6% (w/w).
EXAMPLE 9
15 g (27 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide were suspended in 75 ml of ethylene glycol and 6.5 g (163 mmol) of sodium hydroxide were added. The reaction mixture was heated to 85 thereafter 55 ml of 3 N aqueous hydrochloric acid were added dropwise. The suspension was stirred at 20 off under suction, washed with 150 ml of de-ionized water and dried at 70 in 50 ml of N,N-dimethyl formamide and 40 ml of dioxane at 70 Gaseous ammonia was introduced into this solution until pH=9. The resulting suspension was allowed to cool down slowly. The suspension was stirred at 0 washed with 25 ml of dioxane and thereafter with 25 ml of ethanol. Then the solid was dried at 50 ammonium salt (10.4 g, 17.5 mmol) was suspended in 50 ml of methanol and thereafter 6.5 ml (35 mmol) of a 5.4 N sodium methylate solution were added. The solution was heated (reflux) for 3 hr, cooled down slowly to 20 filtering, washed with 10 ml of ice-cold methanol and dried at 70 C., 2000 Pa for 17 hr. There were obtained 6.9 g (41% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amidesodium salt (1:2) with a HPLC purity of 98.2% (w/w).
This application claims benefit to EP 98114978.4 filed Aug. 10, 1998.
SIMILAR SYNTHESIS OF TEZOSENTAN AND INTERMEDIATES… AN EXPERT WILL PICK UP NAMES AND INTERMEDIATES… just change the isopropyl gp in vii to methyl
Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF.
………………………………..
SYNTHESIS
Example 29
In analogy to Example 3, from 5-methyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide as a white substance of melting point 239- 241 °C from CH3CN
Exgmple, 1 5
a) In analogy to Example l a), from 5-methyl-pyridine-2- sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1 -oxy- pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl- pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy- phenoxy)-2-(l -oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190°C (from acetonitrile).
b) In analogy to Example 2, from 5-methyl-pyridine-2- sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2- (1 -oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5- methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6- (2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- ylamide
Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
for analogy
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
////////////////////////////////
////////////////////////////////
EXTRA INFO
Bosentan (Ro-470203), Atransentan (ABT627), Tezosentan (Ro-610612), Sitaxsentan (TBC-11251), Darusentan (LU-135252), Clazosentan (Ro61-1790, AXV-034343), ZD-4054, Ambrisentan (LU-208075), TAK-044, Avosentan (SPP301), and BQ-123 (Ihara et al Life Sci 1992, 50(4):247-55).
| Antagonists of Endothelin type A receptor ETA | |
| Name | Structure |
| BQ-123 |  |
| Bosentan |  |
| Atrasentan |  |
| Tezosentan |  |
| Sitaxsentan |  |
| Darusentan |  |
| Clazosentan | |
| ZD-4054 (Zibotentan) |  |
| Ambrisentan |  |
| Tak-044 |  |
| Avosentan |  |
Clazosentan
IUPAC Name: N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-[2-(2H-tetrazol-5-yl)
pyridin-4-yl]pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
pyridin-4-yl]pyrimidin-4-yl]-5-methylpyridine-2-sulfonamide
5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide
5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide
VASODILATOR, Endothelin -1 - receptor antagonist
Clazosentan (Ro61-1790, AXV-034343)
- AXV 034
- AXV 034343
- AXV-034343
- AXV-343434
- Clazosentan
- Ro 61-1790
- Ro-61-1790
- UNII-3DRR0X4728
- VML 588
- VML-588
180384-56-9 cas no
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=CLAZOSENTAN in phase 3
| Formula: | C25H23N9O6S |
|---|---|
| Molecular Weight: | 577.5718 |
Endothelin type-A receptor antagonist for the treatment of vasospasm in subarachnoid hemorrhage
Selective endothelin receptor antagonist (Pivlaz)
Acteliion…… innovator
Clazosentan is a drug with orphan drug status , which since 2007, currently in Phase III clinical trials CONSCIOUS-2 ( Clazosentan to O vercome N euro logical i SC Hemia and I nfarct O cc U rring after S ubarachnoid hemorrage) is located. It is for treatment of vasospasm after subarachnoid hemorrhage are used (SAH).
Clazosentan is used by the Swiss pharmaceutical company Actelion developed. Medicinally, the disodium salt is used.Clazosentan to come under the name Pivlaz on the market.
The endothelin -1 - receptor is one of the strongest known vasoconstrictors . Clazosentan is an E -1 - receptor antagonist , for the treatment of vasospasm after subarachnoid hemorrhage is under development. After subarachnoid hemorrhage , an irritation of theblood vessels to a vasospasm and the associated to a reduced supply of brain tissue with oxygen lead. A possible consequence may be a Ischemic stroke be. Clazosentan acts this vasoconstriction contrary.
The plasma half-life of 6-10 min .
Actelion has initiated a comprehensive global phase IIb/III development program for clazosentan sodium (formerly Ro-61-1790, VML-588, …
CLAZOSENTANCLAZOSENTAN DI-NA SALT is discontinued
Clazosentan, shown below, is a well known endothelin receptor antagonist.
Since clazosentan is a known and useful pharmaceutical, it is desirable to discover novel derivatives thereof. Clazosentan is described in European Patent No. 0,799,209
IT IS DESCRIBED IN US6103902
- Paul LM van Giersbergen things Manse, J.: tolerability, pharmacokinetics, and pharmacodynamics of clazosentan, a parenteral endothelin receptor antagonist . In: European Journal of Clinical Pharmacology . 63, No. 2, February 2007, pp. 151-158. doi :10.1007/s00228-006-0117-z . PMID 16,636,870 .
- Paul LM van Giersbergen things Manse J., Gunawardena, KA: Influence of ethnic origin and sex on the pharmacokinetics of clazosentan . In: Journal of Clinical Pharmacology . 47, No. 11, November 2007, pp. 1374-1380. doi :10.1177/0091270007307337 . PMID 17,906,281
| US6004965 * | Dec 8, 1995 | Dec 21, 1999 | Hoffmann-La Roche Inc. | Sulfonamides |
| WO1996019459A1 * | Dec 8, 1995 | Jun 27, 1996 | Volker Breu | Novel sulfonamides |
8-13-1998
|
Pyrrolidine-3-carboxylic acids as endothelin antagonists. 3. Discovery of a potent, 2-nonaryl, highly selective ETA antagonist (A-216546).
|
Journal of medicinal chemistry
|
7-23-2009
|
In silico prediction of volume of distribution in human using linear and nonlinear models on a 669 compound data set.
|
Journal of medicinal chemistry
|
………………………………………………………..
SYNTHESIS
EXAMPLE 15
a) In analogy to Example 1a), from 5-methyl-pyridine-2-sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190
b) In analogy to Example 2, from 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide.
……………………………………………..
SYNTHESIS
4-Cyanopyridine (I) is reacted with ammonium chloride in methanolic NaOMe to afford the amidine (II), which is cyclized with diethyl (2-methoxyphenoxy)malonate (III) producing the dihydroxypyrimidine (IV). Chlorination of (IV) in hot POCl3, followed by oxidation of the obtained dichloropyrimidine (V) with peracetic acid leads to the pyridine N-oxide (VI). Subsequent condensation of the dichloropyrimidine derivative (VI) with the potassium salt of 5-methylpyridine-2-sulfonamide (VII) yields the sulfonamido pyrimidine (VIII). The remaining chloride group of (VIII) is then displaced with the sodium alkoxide of ethylene glycol in hot DME to furnish the hydroxyethyl ether (IX). Treatment of the pyridine N-oxide (IX) with cyanotrimethylsilane and Et3N in refluxing acetonitrile gives rise to the 2-cyanopyridine (X), which is finally converted to the title tetrazole derivative by treatment with sodium azide in the presence of NH4Cl in DMF (1).
………………………………………………………….
SYNTHESIS OF DISODIUM SALT OF CLAZOSENTAN
DESCRIBED IN WO 9619459.
- III is reacted with a compound of formula V
- The reaction type is known in the art and may be performed under basic conditions for example in the presence of a coupling agent, e.g. 1,4-diazobicyclo[2.2.2]octane, together with potassium carbonate in acetone.
EXAMPLE 1
1360 ml of formamide were added to 136 g (437 mmol) of 5-(2-methoxy-phenoxy)-2-pyridine-4-yl-pyrimidine-4,6-diole. Then, at a temperature of 0 acid and thereafter 36.5 g (130 mmol) of iron(II)sulfate heptahydrate were added to the suspension. After that, 89 ml (874 mmol) of 30% hydrogen peroxide were added dropwise within 1 hr at a temperature of 0 to 5 0 sodium pyrosulfite in 680 ml of de-ionized water was added dropwise to the reaction mixture within 30 min. at 0 reaction mixture was stirred at 0 The suspension was then filtered under reduced pressure. The filtrate was first washed with 1750 ml of de-ionized water and thereafter with 700 ml of ethanol. Then the solid was dried at 80 There were obtained 132.4 g (91% of theory) of 4-[4,6-dihydroxy -5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide with a HPLC purity of 91.4% (w/w).
Preparation of Starting Material
a) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After 6 hr 29.5 g of NH.sub.4 Cl are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50 4-amidino-pyridine hydrochloride (decomposition point 245-247
b) 112.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 min. to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydrochloride obtained in a) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50 of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H.sub.2 O and treated little by little with 50 ml of CH.sub.3 COOH. The precipitate is filtered off under suction, washed with 400 ml of H.sub.2 O and dried at 80 obtained 5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidine-4,6-diole (or tautomer), melting point above 250
EXAMPLE 2
Within 20 min. 61 ml (633 mmol) of POCl.sub.3 were added dropwise to 34 ml (200 mmol) of diisopropyl ethylamine at 5 followed by stirring at 5 23.5 g (66 mmol) of 4-[4,6-dihydroxy-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carboxylic acid amide were added in four portions under cooling followed by stirring at 90 to 20 dichloromethane. Volatile components (i.e. excess of POCl.sub.3) was removed by evaporation from 20 re-distillation with 100 ml of toluene. After adding 250 ml of dichloromethane to the residue (88 g of a black oil) the solution was heated to 35 were added dropwise within 30 min. whereby the pH was kept constant by the subsequent addition of 28% NaOH solution (60 ml) within 5 to 6 hr. The mixture was stirred at 35 by removal of dichloromethane by distillation. The resulting suspension was allowed to cool down to 20 hr. The solid was filtered off under suction, washed with 500 ml of water and dried at 70 (86% of theory) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile with a HPLC purity of 94.3% (w/w).
EXAMPLE 3
12.5 g (33.5 mmol) of 4-[4,6-dichloro-5-(2-methoxy-phenoxy)-pyrimidine-2-yl]-pyridine-2-carbonitrile and 6.06 g (35 mmol) of 5-methyl-pyridine-2-sulfonamide were added to 130 ml of acetone. 15 g of potassium carbonate and 190 mg (1.6 mmol) of 1,4-diazobicyclo[2.2.2]octane were added and the suspension was stirred at 40 de-ionized water were added followed by dropwise addition of 50 ml of 3 N hydrochloric acid (pH of the solution=1). Acetone was removed by evaporation and the suspension was stirred for 1 hr. The solid was filtered and washed with 100 ml of water. The residue was heated (reflux) in 100 ml of methanol for 1 hr followed by cooling to 20 solid was filtered and dried at 80 were obtained 16.0 g (93% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 90.3% (w/w).
EXAMPLE 5
20 g (39 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-(2-cyano-pyridine-4-yl)-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were suspended in 100 ml of N,N-dimethyl formamide and 7.6 ml (156 mmol) of hydrazine hydrate were added within 15 min. The reaction mixture was allowed to warm up slowly to 20 temperature of 15 followed by slow addition of 10.5 ml acetic acid (until pH=5.4). The resulting suspension was stirred for 2 hr at 20 additional 2 hr 0 firstly washed with 200 ml of de-ionized water and thereafter with 100 ml of t-butylmethylether. The residue was dried at 40 18 hr. There were obtained 21.7 g (102% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide with a HPLC purity of 81.4% (w/w).
EXAMPLE 7
20 g (37 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-2-[2-(hydrazino-imino-methyl)-pyridine-4-yl]-5-(2-methoxy-phenoxy)-pyrimidine-4-yl]-amide were added to 160 ml of N,N-dimethyl formamide. To this solution was added dropwise 23 ml of 6 N aqueous hydrochloric acid at a temperature of 15 mmol) of sodium nitrite in 20 ml de-ionized water was added slowly. The reaction mixture was allowed to warm up to 20 for 1.5 hr. Then 160 ml of de-ionized water were added and the suspension was stirred for 1 hr. The solid was filtered off under suction, washed with 100 ml of de-ionized water and dried at 50 hr. There were obtained 18.9 g (92% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide with a HPLC purity of 89.6% (w/w).
EXAMPLE 9
15 g (27 mmol) of 5-methyl-pyridine-2-sulfonic acid [6-chloro-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amide were suspended in 75 ml of ethylene glycol and 6.5 g (163 mmol) of sodium hydroxide were added. The reaction mixture was heated to 85 thereafter 55 ml of 3 N aqueous hydrochloric acid were added dropwise. The suspension was stirred at 20 off under suction, washed with 150 ml of de-ionized water and dried at 70 in 50 ml of N,N-dimethyl formamide and 40 ml of dioxane at 70 Gaseous ammonia was introduced into this solution until pH=9. The resulting suspension was allowed to cool down slowly. The suspension was stirred at 0 washed with 25 ml of dioxane and thereafter with 25 ml of ethanol. Then the solid was dried at 50 ammonium salt (10.4 g, 17.5 mmol) was suspended in 50 ml of methanol and thereafter 6.5 ml (35 mmol) of a 5.4 N sodium methylate solution were added. The solution was heated (reflux) for 3 hr, cooled down slowly to 20 filtering, washed with 10 ml of ice-cold methanol and dried at 70 C., 2000 Pa for 17 hr. There were obtained 6.9 g (41% of theory) of 5-methyl-pyridine-2-sulfonic acid [6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-[2-(1H-tetrazole-5-yl)-pyridine-4-yl]-pyrimidine-4-yl]-amidesodium salt (1:2) with a HPLC purity of 98.2% (w/w).
This application claims benefit to EP 98114978.4 filed Aug. 10, 1998.
SIMILAR SYNTHESIS OF TEZOSENTAN AND INTERMEDIATES… AN EXPERT WILL PICK UP NAMES AND INTERMEDIATES… just change the isopropyl gp in vii to methyl
Reaction of 4-cyano-pyridine (I) with Na in methanol followed by treatment with ammonium chloride provides 4-amidino-pyridine hydrochloride (II), which is then converted into 5-(2-methoxyphenoxy)-2-(pyridin-4-yl)-pyrimidine-4,6-diol (IV) by condensation with diethyl malonate derivative (III) by means of Na in MeOH. By heating compound (IV) with phosphorus oxychloride (POCl3), 4,6-dichloro-5-(2-methoxyphenoxy)-2-pyridin-4-yl)pyrimidine (V) is obtained, which in turn is oxidized with peracetic acid in refluxing acetonitrile to afford N-oxide derivative (VI). Condensation of (VI) with 5-isopropylpyridine-2-sulfonamide potassium (VII) furnishes 5-isopropylpyridine-2-sulfonic acid 6-chloro-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl amide (VIII), which is then dissolved in dimethoxyethane and subjected to reaction with Na in hot ethylene glycol (IX) to provide N-[6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(1-oxy-pyridin-4-yl)-pyrimidin-4-yl]-5-isopropylpyridine-2-sulfonamide (X). Refluxing of (X) with trimethylsilylcyanide and Et3N in acetonitrile yields cyano derivative (XI), which is then converted into the tetrazole derivative (XII) by reaction with sodium azide and NH4Cl in DMF at 70 C. Finally, the disodium salt of tezosentan is obtained by treatment of (XII) with Na/MeOH in THF.
………………………………..
SYNTHESIS
Example 29
In analogy to Example 3, from 5-methyl-pyridine-2- sulphonic acid 2-(2-cyano-pyridin-4-yl)-6-(2-hydroxy-ethoxy)- 5-(2-methoxy-phenoxy)-pyrimidin-4-ylamide there is obtained 5-methyl-pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-ylamide as a white substance of melting point 239- 241 °C from CH3CN
Exgmple, 1 5
a) In analogy to Example l a), from 5-methyl-pyridine-2- sulphonic acid 6-chloro-5-(2-methoxy-phenoxy)-2-(1 -oxy- pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5-methyl- pyridine-2-sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy- phenoxy)-2-(l -oxy-pyridin-4-yl)-pyrimidin-4-ylamide, melting point 188-190°C (from acetonitrile).
b) In analogy to Example 2, from 5-methyl-pyridine-2- sulphonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2- (1 -oxy-pyridin-4-yl)-pyrimidin-4-ylamide there is obtained 5- methyl-pyridine-2-sulphonic acid 2-(2-cyano-pyridin-4-yl)-6- (2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- ylamide
Example 1
a) 200 ml of dimethoxyethane and 1 10.9 g of 4-[4-(4-tert- butyl-phenyl-sulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide are added all at once to a solution of 23.80 g of sodium in 660 ml of ethylene glycol. The solution is heated at 90°C for 20 hours while stirring, thereafter cooled, poured into 2500 ml of H2O and thereafter treated with CH3COOH to pH 5. The mixture is extracted three times with EtOAc, the organic phase is washed with H2O, dried with Na2Sθ4 and evaporated under reduced pressure. The residue is recrystall- ized from CH3CN and thereafter twice from a mixture of acetone and CH3CN. There is thus obtained 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide.
Preparation of the starting material:
b) 53.1 g of 4-cyano-pyridine (98%) are added all at once to a solution of 1.15 g of sodium in 200 ml of abs. MeOH. After
6 hours 29.5 g of NH4CI are added while stirring vigorously. The mixture is stirred at room temperature overnight. 600 ml of ether are added thereto, whereupon the precipitate is filtered off under suction and thereafter dried at 50°C under reduced pressure. There is thus obtained 4-amidino-pyridine hydro- chloride (decomposition point 245-247°C).
c) 1 12.9 g of diethyl (2-methoxyphenoxy)malonate are added dropwise within 30 minutes to a solution of 27.60 g of sodium in 400 ml of MeOH. Thereafter, 74.86 g of the amidine hydro- chloride obtained in b) are added all at once. The mixture is stirred at room temperature overnight and evaporated at 50°C under reduced pressure. The residue is treated with 500 ml of ether and filtered off under suction. The filter cake is dissolved in 1000 ml of H2O and treated little by little with 50 ml of CH3COOH. The precipitate is filtered off under suction, washed with 400 ml of H2O and dried at 80°C under reduced pressure. There is thus obtained 5-(2-methoxy-phenoxy)-2-(pyridin-4-yl)- pyrimidine-4,6-diol (or tautomer), melting point above 250°C.
d) A suspension of 1 54.6 g of 5-(2-methoxy-phenoxy)-2- (pyridin-4-yl)-pyrimidine-4,6-diol (or tautomer) in 280 ml of POCI3 is heated at 120°C in an oil bath for 24 hours while stirring vigorously. The reaction mixture changes gradually into a dark brown liquid which is evaporated under reduced pressure and thereafter taken up three times with 500 ml of toluene and evaporated. The residue is dissolved in 1000 ml of CH2CI2, treated with ice and H2O and thereafter adjusted with 3N NaOH until the aqueous phase has pH 8. The organic phase is separated and the aqueous phase is extracted twice with CH2CI2. The combined CH2CI2 extracts are dried with MgSθ4, evaporated to half of the volume, treated with 1000 ml of acetone and the CH2CI2 remaining is distilled off at normal pressure. After standing in a refrigerator for 2 hours the crystals are filtered off under suction and dried at 50°C overnight. There is thus obtained 4,6-dichloro-5-(2-methoxy-phenoxy)-2-pyridin-4-yl)- pyrimidine, melting point 1 78-1 80°C.
e) A solution of 1 7.4 g of 4,6-dichloro-5-(2-methoxy- phenoxy)-2-pyridin-4-yl)-pyrimidine in 100 ml of CH3CN is boiled at reflux for 3 hours with 1 5 ml of a 32% peracetic acid solution, thereafter cooled and stored in a refrigerator overnight. The crystals are filtered off under suction and dried at 50°C under reduced pressure. There is thus obtained 4-[4,6-dichloro- 5-(2-methoxy-phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide, melting point 189-1 90°C.
for analogy
f) A solution of 36.4 g of 4-[4,6-dichloro-5-(2-methoxy- phenoxy)-pyrimidin-2-yl]-pyridine 1 -oxide and 52.8 g of p-tert- butylphenyl-sulphonamide potassium in 1 50 ml of abs. DMF is stirred at room temperature for 24 hours. Thereafter, it is poured into a mixture of 1 500 ml of H2O and 1000 ml of ether while stirring mechanically, whereby a precipitate forms. The suspension is adjusted to pH 5 with CH3COOH, suction filtered, the crystals are washed with cold water and thereafter with ether and dried at 50°C. There is thus obtained 4-[4-(4-tert- butyl-phenylsulphonylamino)-6-chloro-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide as a colourless material of melting point 247-249°C.
Example 2
Example 2
A solution of 78.45 g of 4-[4-(4-tert-butyl-phenyl- sulphonylamino)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-2-yl]-pyridine 1 -oxide, 122.5 g of trimethylsilyl cyanide, 127.8 g of triethylamine and 1200 ml of CH3CN is boiled at reflux for 20 hours and thereafter evaporated under reduced pressure. The oily residue is taken up in 1000 ml of EtOAc and the solution is washed with CH3COOH:H2θ 9:1 and then with H2O. The EtOAc extracts are dried with Na2SO4. After evaporation of the solvent the residue is taken up in a mixture of CH3CN and CF3COOH (20:1 ), whereby a crystalline precipitate separates. There is thus obtained 4-tert-butyl-N-[2-(2-cyano-pyridin-4- yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-pyrimidin-4- yl]-benzenesulphonamide of melting point 176-1 79°C.
Example 3
A suspension of 50.0 g of 4-tert-butyl-N-[2-(2-cyano- pyridin-4-yl)-6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)- pyrimidin-4-yl]-benzenesulphonamide, 46.33 g of NH4CI and 56.47 g of NaN3 in 1600 ml of DMF is heated to 70°C for 24 hours while stirring vigorously. The majority of the solvent is distilled off under reduced pressure, the residue is dissolved in H2O, the solution is extracted four times at pH 6.5 with ether, thereafter treated with CH3COOH to pH = 4.5 and extracted with EtOAc. After working up there is obtained a residue which is treated with ether and filtered off under suction therefrom. There is thus obtained 4-tert-butyl-N-[6-(2-hydroxy-ethoxy)-5-(2- methoxy-phenoxy)-2-(2-1 H-tetrazol-5-yl-pyridin-4-yl)- pyrimidin-4-yl]-benzenesulphonamide, melting point 225-227°C.
..........................................
ATRASENTAN

Atrasentan
A-147627, (+)-A-127722, ABT-627,173937-91-2,
Endothelin ET-A antagonist
Diabetic nephropathy; End stage renal disease; Renal disease
Atrasentan failed a phase 3 trial for prostate cancer in patients unresponsive to hormone therapy.[3] A second trial confirmed this finding.[4]
It is an endothelin receptor antagonist selective for subtype A (ETA). While other drugs of this type (sitaxentan, ambrisentan) exploit the vasoconstrictive properties of endothelin and are mainly used for the treatment of pulmonary arterial hypertension, atrasentan blocks endothelin induced cell proliferation.
In April 2014, de Zeeuw et al. showed that 0.5 mg and 1.25 mg of atrasentan reduced urinary albumin by 35 and 38% respectively with modest side effects. Patients also had decreased home blood pressures (but no change in office readings) decrease total cholesterol and LDL. Patients in the 1.25 mg dose group had increased weight gain which was presumably due to increased edema and had to withdraw from the study more than the placebo or 0.5 mg dose group.[5] Reductions in proteinuria have been associated with beneficial patient outcomes in diabetic kidney disease with other interventions but is not an accepted end-point by the FDA.
The recently initiated SONAR trial[6] will determine if atrasentan reduces kidney failure in diabetic kidney disease.
Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes. For a prior filing see WO2015006219 , claiming the stable solid composition in the form of a tablet comprising atrasentan and an anti-oxidant. AbbVie (following its spin-out from Abbott), is developing atrasentan (phase III; February 2015) for treating chronic kidney disease, including diabetic nephropathy.
……………….
European Journal of Organic Chemistry
Enantioselective Synthesis of the Pyrrolidine Core of Endothelin Antagonist ABT-627 (Atrasentan) via 1,2-Oxazines
Year:2003
Volume:2003
Issue:18
page:3524-3533
………………….
http://www.google.com/patents/US20080132710
EXAMPLE 1
A mixture of bromoacetyl bromide (72.3 mL) in toluene (500 mL) at 0° C. was treated with dibutylamine (280 mL) in toluene (220 mL) while keeping the solution temperature below 10° C., stirred at 0° C. for 15 minutes, treated with 2.5% aqueous phosphoric acid (500 mL) and warmed to 25° C. The organic layer was isolated, washed with water (500 mL) and concentrated to provide the product as a solution in toluene.
EXAMPLE 25-((E)-2-nitroethenyl)-1,3-benzodioxole
3,4-methylenedioxybenzaldehyde (15.55 Kg) was treated sequentially with ammonium acetate (13.4 Kg,), acetic acid (45.2 Kg) and nitromethane (18.4 Kg), warmed to 70° C., stirred for 30 minutes, warmed to 80° C., stirred for 10 hours, cooled to 10° C. and filtered. The filtrant was washed with acetic acid (2×8 Kg) and water (2×90 Kg) and dried under a nitrogen stream then in under vacuum at 50° C. for 2 days.
EXAMPLE 3ethyl 3-(4-methoxyphenyl)-3-oxopropanoate
A mixture of potassium tert-amylate (50.8 Kg) in toluene (15.2 Kg) at 5° C. was treated with 4-methoxyacetophenone (6.755 Kg) and diethyl carbonate (6.4 Kg) in toluene over 1 hour while keeping the solution temperature below 10° C., warmed to 60° C. for 8 hours, cooled to 20° C. and treated with acetic acid (8 Kg) and water (90 Kg) over 30 minutes while keeping the solution temperature below 20° C. The organic layer was isolated, washed with 5% aqueous sodium bicarbonate (41 Kg) and concentrated at 50° C. to 14.65 Kg.
EXAMPLE 4ethyl 2-(4-methoxybenzoyl)-4-nitromethyl-3-(1,3-benzodioxol-5-yl)butyrate
A mixture of EXAMPLE 3 (7.5 Kg) in THF (56 Kg) was treated with EXAMPLE 3 (8.4 Kg), cooled to 17° C., treated with sodium ethoxide (6.4 g), stirred for 30 minutes, treated with more sodium ethoxide (6.4 g), stirred at 25° C. until HPLC shows less than 1 area % ketoester remaining and concentrated to 32.2 Kg.
EXAMPLE 5ethyl cis,cis-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate
Raney nickel (20 g), from which the water had been decanted, was treated sequentially with THF (20 mL), EXAMPLE 4 (40.82 g), and acetic acid (2.75 mL). The mixture was stirred under hydrogen (60 psi) until hydrogen uptake slowed, treated with trifluoroacetic acid, stirred under hydrogen (200 psi) until HPLC shows no residual imine and less than 2% nitrone and filtered with a methanol (100 mL) wash. The filtrate, which contained 13.3 g of EXAMPLE 5, was concentrated with THF (200 mL) addition to 100 mL, neutralized with 2N aqueous NaOH (50 mL), diluted with water (200 mL), and extracted with ethyl acetate (2×100 mL). The extract was used in the next step.
EXAMPLE 6ethyl trans,trans-2-(4-methoxyphenyl)-4-(1,3 -benzodioxol-5 -yl)pyrrolidine-3-carboxylate
Example 501E (38.1 g) was concentrated with ethanol (200 mL) addition to 100 mL, treated with sodium ethoxide (3.4 g), heated to 75° C., cooled to 25° C. when HPLC showed less than 3% of EXAMPLE 1E and concentrated. The concentrate was mixed with isopropyl acetate (400 mL), washed with water (2×150 mL) and extracted with 0.25 M phosphoric acid (2×400 mL). The extract was mixed with ethyl acetate (200 mL) and neutralized to pH 7 with sodium bicarbonate (21 g), and the organic layer was isolated.
EXAMPLE 7ethyl (2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate, (S)-(+) mandelate
EXAMPLE 501F was concentrated with acetonitrile (100 mL) addition to 50 mL, treated with (S)-(+)-mandelic acid (2.06 g), stirred until a solution formed, stirred for 16 hours, cooled to 0° C., stirred for 5 hours and filtered. The filtrant was dried at 50° C. under a nitrogen stream for 1 day. The purity of the product was determined by chiral HPLC using Chiralpak AS with 95:5:0.05 hexane/ethanol/diethylamine, a flow rate of 1 mL/min. and UV detection at 227 nm. Retention times were 15.5 minutes for the (+)-enantiomer and 21.0 minutes for the (−)-enantiomer.
EXAMPLE 8(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
A mixture of EXAMPLE 7 (20 g) in ethyl acetate (150 mL) and 5% aqueous sodium bicarbonate was stirred at 25° C. until the salt dissolved and gas evolution stopped. The organic layer was isolated and concentrated. The concentrate was treated with acetonitrile (200 mL), concentrated to 100 mL, cooled to 10° C., treated with diisopropylethylamine (11.8 mL) and EXAMPLE 1 (10.5 g), stirred for 12 hours and concentrated. The concentrate was treated with ethanol (200 mL), concentrated to 100 mL, treated with 40% aqueous NaOH (20 mL), stirred at 60° C. for 4 hours, cooled, poured into water (400 mL), washed with hexanes (2×50 mL then 2×20 mL), treated with ethyl acetate (400 mL) and adjusted to pH 5 with concentrated HCl (12 mL). The organic layer was isolated and concentrated.
………………….


US-8962675, AbbVie Inc
Granted in February 2015, this patent claims novel crystalline anhydrous S-mandelate salt of atrasentan. Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes.
Atrasentan
A-147627, (+)-A-127722, ABT-627,173937-91-2,
Endothelin ET-A antagonist
Diabetic nephropathy; End stage renal disease; Renal disease
1-(N,N-Dibutylcarbamoylmethyl)-2(R)-(4-methoxyphenyl)-4(S)-(3,4-methylenedioxyphenyl)pyrrolidine-3(R)-carboxylic acid
(2R,3R,4S)-(+)-2-(4-Methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)-pyrrolidine-3-carboxylic acid
C29H38N2O6, 510.631
Atrasentan is an experimental drug that is being studied for the treatment of various types of cancer,[1] including non-small cell lung cancer.[2] It is also being investigated as a therapy for diabetic kidney disease.Atrasentan failed a phase 3 trial for prostate cancer in patients unresponsive to hormone therapy.[3] A second trial confirmed this finding.[4]
It is an endothelin receptor antagonist selective for subtype A (ETA). While other drugs of this type (sitaxentan, ambrisentan) exploit the vasoconstrictive properties of endothelin and are mainly used for the treatment of pulmonary arterial hypertension, atrasentan blocks endothelin induced cell proliferation.
In April 2014, de Zeeuw et al. showed that 0.5 mg and 1.25 mg of atrasentan reduced urinary albumin by 35 and 38% respectively with modest side effects. Patients also had decreased home blood pressures (but no change in office readings) decrease total cholesterol and LDL. Patients in the 1.25 mg dose group had increased weight gain which was presumably due to increased edema and had to withdraw from the study more than the placebo or 0.5 mg dose group.[5] Reductions in proteinuria have been associated with beneficial patient outcomes in diabetic kidney disease with other interventions but is not an accepted end-point by the FDA.
The recently initiated SONAR trial[6] will determine if atrasentan reduces kidney failure in diabetic kidney disease.
Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes. For a prior filing see WO2015006219 , claiming the stable solid composition in the form of a tablet comprising atrasentan and an anti-oxidant. AbbVie (following its spin-out from Abbott), is developing atrasentan (phase III; February 2015) for treating chronic kidney disease, including diabetic nephropathy.
……………….
European Journal of Organic Chemistry
Enantioselective Synthesis of the Pyrrolidine Core of Endothelin Antagonist ABT-627 (Atrasentan) via 1,2-Oxazines
Year:2003
Volume:2003
Issue:18
page:3524-3533
………………….
http://www.google.com/patents/US20080132710
EXAMPLE 1
A mixture of bromoacetyl bromide (72.3 mL) in toluene (500 mL) at 0° C. was treated with dibutylamine (280 mL) in toluene (220 mL) while keeping the solution temperature below 10° C., stirred at 0° C. for 15 minutes, treated with 2.5% aqueous phosphoric acid (500 mL) and warmed to 25° C. The organic layer was isolated, washed with water (500 mL) and concentrated to provide the product as a solution in toluene.
EXAMPLE 25-((E)-2-nitroethenyl)-1,3-benzodioxole
3,4-methylenedioxybenzaldehyde (15.55 Kg) was treated sequentially with ammonium acetate (13.4 Kg,), acetic acid (45.2 Kg) and nitromethane (18.4 Kg), warmed to 70° C., stirred for 30 minutes, warmed to 80° C., stirred for 10 hours, cooled to 10° C. and filtered. The filtrant was washed with acetic acid (2×8 Kg) and water (2×90 Kg) and dried under a nitrogen stream then in under vacuum at 50° C. for 2 days.
EXAMPLE 3ethyl 3-(4-methoxyphenyl)-3-oxopropanoate
A mixture of potassium tert-amylate (50.8 Kg) in toluene (15.2 Kg) at 5° C. was treated with 4-methoxyacetophenone (6.755 Kg) and diethyl carbonate (6.4 Kg) in toluene over 1 hour while keeping the solution temperature below 10° C., warmed to 60° C. for 8 hours, cooled to 20° C. and treated with acetic acid (8 Kg) and water (90 Kg) over 30 minutes while keeping the solution temperature below 20° C. The organic layer was isolated, washed with 5% aqueous sodium bicarbonate (41 Kg) and concentrated at 50° C. to 14.65 Kg.
EXAMPLE 4ethyl 2-(4-methoxybenzoyl)-4-nitromethyl-3-(1,3-benzodioxol-5-yl)butyrate
A mixture of EXAMPLE 3 (7.5 Kg) in THF (56 Kg) was treated with EXAMPLE 3 (8.4 Kg), cooled to 17° C., treated with sodium ethoxide (6.4 g), stirred for 30 minutes, treated with more sodium ethoxide (6.4 g), stirred at 25° C. until HPLC shows less than 1 area % ketoester remaining and concentrated to 32.2 Kg.
EXAMPLE 5ethyl cis,cis-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate
Raney nickel (20 g), from which the water had been decanted, was treated sequentially with THF (20 mL), EXAMPLE 4 (40.82 g), and acetic acid (2.75 mL). The mixture was stirred under hydrogen (60 psi) until hydrogen uptake slowed, treated with trifluoroacetic acid, stirred under hydrogen (200 psi) until HPLC shows no residual imine and less than 2% nitrone and filtered with a methanol (100 mL) wash. The filtrate, which contained 13.3 g of EXAMPLE 5, was concentrated with THF (200 mL) addition to 100 mL, neutralized with 2N aqueous NaOH (50 mL), diluted with water (200 mL), and extracted with ethyl acetate (2×100 mL). The extract was used in the next step.
EXAMPLE 6ethyl trans,trans-2-(4-methoxyphenyl)-4-(1,3 -benzodioxol-5 -yl)pyrrolidine-3-carboxylate
Example 501E (38.1 g) was concentrated with ethanol (200 mL) addition to 100 mL, treated with sodium ethoxide (3.4 g), heated to 75° C., cooled to 25° C. when HPLC showed less than 3% of EXAMPLE 1E and concentrated. The concentrate was mixed with isopropyl acetate (400 mL), washed with water (2×150 mL) and extracted with 0.25 M phosphoric acid (2×400 mL). The extract was mixed with ethyl acetate (200 mL) and neutralized to pH 7 with sodium bicarbonate (21 g), and the organic layer was isolated.
EXAMPLE 7ethyl (2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)pyrrolidine-3-carboxylate, (S)-(+) mandelate
EXAMPLE 501F was concentrated with acetonitrile (100 mL) addition to 50 mL, treated with (S)-(+)-mandelic acid (2.06 g), stirred until a solution formed, stirred for 16 hours, cooled to 0° C., stirred for 5 hours and filtered. The filtrant was dried at 50° C. under a nitrogen stream for 1 day. The purity of the product was determined by chiral HPLC using Chiralpak AS with 95:5:0.05 hexane/ethanol/diethylamine, a flow rate of 1 mL/min. and UV detection at 227 nm. Retention times were 15.5 minutes for the (+)-enantiomer and 21.0 minutes for the (−)-enantiomer.
EXAMPLE 8(2R,3R,4S)-(+)-2-(4-methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic acid
A mixture of EXAMPLE 7 (20 g) in ethyl acetate (150 mL) and 5% aqueous sodium bicarbonate was stirred at 25° C. until the salt dissolved and gas evolution stopped. The organic layer was isolated and concentrated. The concentrate was treated with acetonitrile (200 mL), concentrated to 100 mL, cooled to 10° C., treated with diisopropylethylamine (11.8 mL) and EXAMPLE 1 (10.5 g), stirred for 12 hours and concentrated. The concentrate was treated with ethanol (200 mL), concentrated to 100 mL, treated with 40% aqueous NaOH (20 mL), stirred at 60° C. for 4 hours, cooled, poured into water (400 mL), washed with hexanes (2×50 mL then 2×20 mL), treated with ethyl acetate (400 mL) and adjusted to pH 5 with concentrated HCl (12 mL). The organic layer was isolated and concentrated.
………………….
The Michael reaction between 3,4-(methylenedioxy)-beta-nitrostyrene
(I) and ethyl (4-methoxybenzoyl)acetate (II) in the presence of DBU
gave adduct (III) as a mixture of isomers. Hydrogenation of this nitro
ketone over Raney-Ni afforded, after spontaneous cyclization of the
resulting amino ketone, the pyrroline (IV). Further reduction of the
imine with NaBH3CN yielded a mixture of three pyrrolidine isomers. The
desired trans-trans isomer (VI) could not be separated from the
cis-trans isomer by column chromatography. However, the pure cis-cis
compound (V) was isomerized to (VI) with NaOEt in refluxing EtOH. The
protection of the amine as the tert-butyl carbamate with Boc2O, and
saponification of the ester function provided the racemic acid (VII).
Resolution of (VII) was achieved by conversion to the mixed anhydride
(VIII) with pivaloyl chloride, followed by condensation with the lithium
salt of (S)-4-benzyl-2-oxazolidinone (IX), and chromatographic
separation of the resulting diastereomeric imides. Alternatively,
racemic (VII) could be resolved by crystallization of its salt with
(R)-a-methylbenzylamine. Removal of the Boc group from the appropriate
isomer (X) with HCl in dioxan, followed by alkylation with
N,N-dibutylbromoacetamide (XI) in the presence of i-Pr2NEt furnished the
pyrrolidinylacetamide (XII). Finally, hydrolysis of the imide with
lithium hydroperoxide provided the target acid.
J Med Chem1996,39,(5):1039
Cyclization of 5-(2-nitrovinyl)-1,3-benzodioxole (I) with ethyl
2-(4-methoxybenzoyl)acetate (II) by means of DBU in THF gives the
4-nitrobutyrate (III), which is reduced with H2 over Ni in ethanol to
the corresponding amine, which undergoes immediate cyclization to give
the pyrroline carboxylate (IV). Reduction of pyrroline (IV) with NaCNBH3
in THF affords the expected pyrrolidine as a mixture of the
(trans,trans)-(V), (cis,cis)-(VI) and (cis,trans)-(VII) isomers. Using
chromatography on silica gel, only the (cis,cis)-isomer (VI) is
separated and completely isomerized to the (trans,trans)-isomer (V) by
treatment with NaOEt in refluxing ethanol. Pure (trans,trans)-isomer (V)
or the remaining mixture of (trans,trans)-(V) and (cis,trans)-(VII) is
N-protected with Boc2O in dichloromethane to provide a mixture of
carbamates. Then hydrolysis of the esters is performed with NaOH in
ethanol/water at room temperature, and under these conditions only the
(trans,trans)-isomer hydrolyzes, giving the racemic (trans,trans)-acid
(VIII). Unreacted (cis,trans)-ester (VII) is easily removed by
conventional methods. Condensation of the racemic acid (VIII) with the
lithium salt of the chiral oxazolidinone (IX) by means of pivaloyl
chloride yields the corresponding amide as a diastereomeric mixture of
(X) and (XI) that are separated by chromatography. The desired isomer
(XI) is deprotected with HCl in dioxane to afford the chiral pyrrolidine
(XII), which is condensed with 2-bromo-N,N-dibutylacetamide (XIII) by
means of diisopropylamine in acetonitrile to give the adduct (XIV).
Finally, the chiral auxiliary of (XIV) is eliminated by means of LiOOH
(LiOH + H2O2) in water.
J Med Chem1996,39,(5):1039
………………………………………………………..
EXAMPLE
95D(2R,3R,4S)-(+)-2-(4-Methoxyphenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)aminocarbonylmethyl)pyrrolidine-3-carboxylic
acidTo the resulting compound from Example 95C (131 mg, 0.355 mmol) was
added, diisopropylethylamine (137 mg, 185 μL, 1.06 mmol), acetonitrile
(2 mL), N,N-di-(n-butyl)bromoacetamide (133 mg, 0.531 mmol), and the
mixture was heated at 50° C. for 1.5 hours. The reaction mixture was
concentrated to a solid, dried under high vacuum, and purified by
chromatography on silica gel eluting with 1:3 ethyl acetate-hexane to
give pure ester as a colorless oil. 1 H NMR (CDCl3,
300MHz) δ 0.81 (t, J=7 Hz, 3H), 0.88 (t, J=7 Hz, 3H), 1.10 (t, J=7 Hz,
3H), 1.00-1.52 (m, 8H), 2.78 (d, J=14 Hz,1H), 2.89-3.10 (m, 4H),
3.23-3.61 (m, 5H), 3.71 (d, J=9 Hz, 1H), 3.80 (s, 3H), 4.04 (q, J=7 Hz,
2H), 5.94 (dd, J=1.5 Hz, 2H), 6.74 (d, J=9 Hz, 1H), 6.83-6.90 (m, 3H),
7.03 (d, J=2 Hz, 1H), 7.30 (d, J=9 Hz, 2H). MS (DCl/NH3) m/e 539 (M+H)+.To
the ethyl ester dissolved in 7 mL of ethanol was added a solution of
lithium hydroxide (45 mg, 1.06 mmol) in water (2.5 mL). The mixture was
stirred for 1 hour at ambient temperature and then warmed slowly to 40°
C. over 2.5 hours at which point all of the starting material had been
consumed. The reaction mixture was concentrated to remove the ethanol,
diluted with 60 mL water and extracted with ether (3×40 mL). The aqueous
solution was treated with 1N aqueous hydrochloric acid until cloudy,
and the pH was then adjusted to ˜4-5 with 10% aqueous citric acid. This
mixture was extracted with 1:19 ethanol-methylene chloride (3×50 mL).
The combined extracts were dried (Na2 SO4), filtered, concentrated and dried under high vacuum to give the title compound as a white foam (150 mg, 83%). 1 H NMR (CDCl3,
300MHz) δ 0.80 (t, J=7 Hz, 3H), 0.88 (t, J=7 Hz, 3H), 1.08 (m, 2H),
1.28 (m, 3H), 1.44 (m, 3H), 2.70-3.77 (svr br m, 12H), 3.79 (s, 3H),
5.95 (m, 2H), 6.75 (d, J=8 Hz, 1H), 6.87 (br d, J=8 Hz, 3H), 7.05 (br
s,1H),7.33 (v br s, 2H). MS (DCl/NH3) m/e 511 (M+H)+. α!22 =+74.42°. Anal calcd for C29 H38 N2 O6.0.5 H2 O: C ,67.03; H, 7.56; N, 5.39. Found: C, 67.03; H, 7.59; N, 5.33.
References
1- “Atrasentan”. NCI Dictionary of Cancer Terms. National Institute of Cancer.
- 2
- Chiappori, Alberto A.; Haura, Eric; Rodriguez, Francisco A.; Boulware, David; Kapoor, Rachna; Neuger, Anthony M.; Lush, Richard; Padilla, Barbara; Burton, Michelle; Williams, Charles; Simon, George; Antonia, Scott; Sullivan, Daniel M.; Bepler, Gerold (March 2008). “Phase I/II Study of Atrasentan, an Endothelin A Receptor Antagonist, in Combination with Paclitaxel and Carboplatin as First-Line Therapy in Advanced Non–Small Cell Lung Cancer”. Clinical Cancer Research 14 (5): 1464–9. doi:10.1158/1078-0432.CCR-07-1508. PMID 18316570.
- 3
- “Addition of experimental drug to standard chemotherapy for advanced prostate cancer shows no benefit in phase 3 clinical trial” (Press release). National Cancer Institute. April 21, 2011. Retrieved October 18, 2014.
- 4
- Quinn, David I; Tangen, Catherine M; Hussain, Maha; Lara, Primo N; Goldkorn, Amir; Moinpour, Carol M; Garzotto, Mark G; Mack, Philip C; Carducci, Michael A; Monk, J Paul; Twardowski, Przemyslaw W; Van Veldhuizen, Peter J; Agarwal, Neeraj; Higano, Celestia S; Vogelzang, Nicholas J; Thompson, Ian M (August 2013). “Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial”. The Lancet Oncology 14 (9): 893–900. doi:10.1016/S1470-2045(13)70294-8. PMID 23871417.
- 5
- de Zeeuw, Dick; Coll, Blai; Andress, Dennis; Brennan, John J.; Tang, Hui; Houser, Mark; Correa-Rotter, Ricardo; Kohan, Donald; Lambers Heerspink, Hiddo J.; Makino, Hirofumi; Perkovic, Vlado; Pritchett, Yili; Remuzzi, Giuseppe; Tobe, Sheldon W.; Toto, Robert; Viberti, Giancarlo; Parving, Hans-Henrik (May 2014). “The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy”. Journal of the American Society of Nephrology 25 (5): 1083–93. doi:10.1681/ASN.2013080830. PMID 24722445.
- 6
US-8962675, AbbVie Inc
Granted in February 2015, this patent claims novel crystalline anhydrous S-mandelate salt of atrasentan. Useful for treating nephropathy and chronic kidney disease associated with Type II diabetes.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2R,3R,4S)-4-(1,3-Benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)pyrrolidine-3-carboxylic acid | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 173937-91-2 |
| ATC code | None |
| PubChem | CID 159594 |
| ChemSpider | 140321 |
| UNII | V6D7VK2215 |
| ChEMBL | CHEMBL9194 |
| Chemical data | |
| Formula | C29H38N2O6 |
| Molecular mass | 510.621 g/mol |
Share this:
.................................................
5
AMBRISENTAN
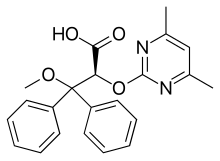




 .......................
.......................
6
ZIBOTENTAN



......................
7
Ambrisentan
(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy- 3,3-diphenylpropanoic acid
177036-94-1 cas no
Ambrisentan (U.S. trade name Letairis; E.U. trade name Volibris; India trade namepulmonext by MSN labs ) is a drug indicated for use in the treatment of pulmonary hypertension.
It functions as an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA).[1] Once daily oral ambrisentan 2.5 to 10 mg/day significantly improved exercise capacity (6-minute walk distance) compared with placebo in two double-blind, multicenter trials (ARIES-1 & ARIES-2).[2]
Ambrisentan was approved for sale by the U.S. Food and Drug Administration (FDA) on June 15, 2007 for the once-daily treatment of pulmonary arterial hypertension.[3][4][5] It was later approved by the European Medicines Agency for use in the EU on April 2008.[6]Ambrisentan had previously been designated an orphan drug by both the FDA and the European Commission, in August 2004 and May 2005 respectively.[7]
Ambrisentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group 1) in patients with WHO class II or III symptoms to improve exercise capacity and delay clinical worsening.
The LETAIRIS Education and Access Program (LEAP) is a program to help physicians and patients learn about the risks of LETAIRIS, including the serious risks of liver injury and birth defects.
LEAP works by:
- Providing information to prescribers on the risks of LETAIRIS
- Providing comprehensive education to patients and assistance with obtaining LETAIRIS
- Requiring enrollment of both prescriber and patient in LEAP
- Controlling dispensing through a specialized distribution network (specialty pharmacies)
- Letairis website run by Gilead Sciences
- Prescribing information
- Information on the LETAIRIS Education and Access Program (LEAP)
- Vatter H, Seifert V (2006). “Ambrisentan, a non-peptide endothelin receptor antagonist”. Cardiovasc Drug Rev 24 (1): 63–76.doi:10.1111/j.1527-3466.2006.00063.x. PMID 16939634.
- Frampton JE. Ambrisentan. American Journal of Cardiovascular Drugs August 1, 2011; 11 (4): 215-226.Link text
- Pollack, Andrew (2007-06-16). “Gilead’s Drug Is Approved to Treat a Rare Disease”. New York Times. Archived from the original on 20 June 2007. Retrieved 2007-05-25.
- “U.S. Food and Drug Administration Approves Gilead’s Letairis Treatment of Pulmonary Arterial Hypertension” (Press release).Gilead Sciences. 2007-06-15. Retrieved 2007-06-16.
- “FDA Approves New Orphan Drug for Treatment of Pulmonary Arterial Hypertension” (Press release). Food and Drug Administration. 2007-06-15. Archived from the original on 23 June 2007. Retrieved 2007-06-22.
- “GlaxoSmithKline’s Volibris (ambrisentan) receives authorisation from the European Commission for the treatment of Functional Class II and III Pulmonary Arterial Hypertension” (Press release). GlaxoSmithKline. 2008-04-25. Archived from the original on 30 April 2008. Retrieved 2008-04-29.
- Waknine, Yael (2005-05-09). “International Approvals: Ambrisentan, Oral-lyn, Risperdal”. Medscape. Retrieved 2007-06-16.
Patent EP2547663A1
6
ZIBOTENTAN
186497-07-4, ZD4054, ZD-4054, Zd 4054, ZD4054, Zibotentan
Molecular Formula:C19H16N6O4S
Molecular Weight:424.43314 g/mol
N-(3-methoxy-5-methylpyrazin-2-yl)-2-[4-(1,3,4-oxadiazol-2-yl)phenyl]pyridine-3-sulfonamide
Oncolytic Drugs, Prostate Cancer Therapy, Solid Tumors Therapy, Antimitotic Drugs, Endothelin ETA Receptor Antagonists
Zibotentan (INN) (earlier code name ZD4054) is an anti-cancer candidate.[1] It is an endothelin receptor antagonist.[2]
It failed a phase III clinical trial for prostate cancer[3] but other trials are planned.[4] Tolerability of zibotentan plus docetaxel has been evaluated.[5]
SYN
Bromination of 2-amino-5-methylpyrazine (I) with Br2 in CHCl3 affords the bromopyrazine (II). Subsequent bromide displacement in (II) by means of sodium methoxide gives rise to the methoxypyrazine (III). The amino group of (III) is then protected by acylation with isobutyl chloroformate, to produce carbamate (IV). Diazotization of 3-amino-2-chloropyridine (V), followed by treatment with sulfur dioxide in the presence of CuCl furnishes sulfonyl chloride (VI). Carbamate (IV) is then acylated by means of NaH and sulfonyl chloride (VI) in DMF to furnish the N-sulfonyl carbamate (VII). Esterification of 4-carboxyphenylboronic acid (VIII) with H2SO4 in MeOH gives 4-(methoxycarbonyl)phenylboronic acid (IX). Mitsunobu coupling between boronic acid (IX) and chloropyridine (VII) furnishes adduct (X). Methyl ester (X) is converted into hydrazide (XI) by treatment with hydrazine hydrate in refluxing methanol. Then, cyclization of the acyl hydrazide (XI) with boiling triethyl orthoformate gives rise to the target oxadiazole derivative.
Example 36
Hydrazine hydrate (1.2 ml) was added to a solution of N-(isobutoxycarbonyl)-2- (4-memoxycarbonylphenyl)-N-(3-metJ oxy-5-methylpyrazin-2-yl)pyridine-3-sulphonamide (1.54 g) in methanol (15 ml) and the mixture was heated and stiπed under reflux for 24 hours then cooled. The solid was collected and dried under reduced pressure to give the free sulphonamido-acylhydrazide (0.857 g); 1H NMR (cVDMSO): 2.2 (s, 3H), 3.7 (s, 3H), 6.7 (br s, 2H), 7.3 (s, IH), 7.5 (m, 3H), 7.8 (d, 2H), 8.4 (d, IH), 8.75 (dd, IH), 9.8 (br s, IH). A solution of this acylhydrazide (207 mg) in triethylorthoformate (5 ml) was heated under reflux for 17 hours then cooled. The resultant solid was collected and purified by chromatography on a silica gel Mega Bond Elut column, eluting with 0-10% methanol/dichloromethane to give N-(3-methoxy-5-mef ylpyrazin-2-yl)-2-(4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3- sulphonamide (39 mg) as a solid; 1H NMR (DMSO-do): 2.2 (br s, 3H), 3.8 (s, 3H), 7.4 (br s, IH), 7.6-7.8 (m, 3H), 8.0 (m, 2H), 8.5 (dd, IH), 8.9 (dd, IH), 9.4 (s, IH); mass spectrum (+ve ESP): 425 (M+H)+.
………………………….
N-(3-methoxy-5-methylpyrazin-2-yl)-2- (4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3-sulphonamide (hereafter “Compound (I)). More specifically the invention relates to the ethanolamine salt of Compound (I) (hereafter “Compound (I) ethanolamine salt), and to pharmaceutical compositions containing it. The invention further relates to the use of Compound (I) ethanolamine salt in the manufacture of medicament for use in treating cancer and to methods of treating cancer in a warm blooded animal such as man using this salt. The invention further relates to the use of Compound (I) ethanolamine salt in producing Compound (I) during manufacture.
Compound (I) is an endothelin antagonist. The endothelins are a family of endogenous 21 amino acid peptides comprising three isoforms, endothelin-1 (ET-I), endothelin-2 and endothelin-3. The endothelins are formed by cleavage of the Trp2I-Val22 bond of their corresponding proendothelins by an endothelin converting enzyme. The endothelins are among the most potent vasoconstrictors known and have a characteristic long duration of action. They exhibit a wide range of other activities including cell proliferation and mitogenesis, extravasation and chemotaxis, and also interact with a number of other vasoactive agents.
The endothelins are released from a range of tissue and cell sources including vascular endothelium, vascular smooth muscle, kidney, liver, uterus, airways, intestine and leukocytes. Release can be stimulated by hypoxia, shear stress, physical injury and a wide range of hormones and cytokines. Elevated endothelin levels have been found in a number of disease states in man including cancers.
Recently, endothelin A receptor antagonists have been identified as potentially of value in the treatment of cancer (Cancer Research, 56, 663-668, February 15th, 1996 and Nature Medicine, Volume 1, Number 9, September 1999, 944-949).
Cancer affects an estimated 10 million people worldwide. This figure includes incidence, prevalence and mortality. More than 4.4 million cancer cases are reported from Asia, including 2.5 million cases from Eastern Asia, which has the highest rate of incidence in the world. By comparison, Europe has 2.8 million cases, North America 1.4 million cases, and Africa 627,000 cases. In the UK and US, for example, more than one in three people will develop cancer at some point in their life, Cancer mortality in the U.S. is estimated to account for about 600,000 a year, about one in every four deaths, second only to heart disease in percent of all deaths, and second to accidents as a cause of death of children 1-14 years of age. The estimated cancer incidence in the U.S. is now about 1,380,000 new cases annually, exclusive of about 900,000 cases of non-melanotic (basal and squamous cell) skin cancer.
Cancer is also a major cause of morbidity in the UK with nearly 260,000 new cases (excluding non-melanoma skin cancer) registered in 1997. Cancer is a disease that affects mainly older people, with 65% of cases occurring in those over 65. Since the average life expectancy in the UK has almost doubled since the mid nineteenth century, the population at risk of cancer has grown. Death rates from other causes of death, such as heart disease, have fallen in recent years while deaths from cancer have remained relatively stable. The result is that 1 in 3 people will be diagnosed with cancer during their lifetime and 1 in 4 people will die from cancer. In people under the age of 75, deaths from cancer outnumber deaths from diseases of the circulatory system, including ischaemic heart disease and stroke. In 2000, there were 151,200 deaths from cancer. Over one fifth (22 per cent) of these were from lung cancer, and a quarter (26 per cent) from cancers of the large bowel, breast and prostate.
Worldwide, the incidence and mortality rates of certain types of cancer (of stomach, breast, prostate, skin, and so on) have wide geographical differences which are attributed to racial, cultural, and especially environmental influences. There are over 200 different types of cancer but the four major types, lung, breast, prostate and colorectal, account for over half of all cases diagnosed in the UK and US. Prostate cancer is the fourth most common malignancy among men worldwide, with an estimated 400,000 new cases diagnosed annually, accounting for 3.9 percent of all new cancer cases. Current options for treating cancers include surgical resection, external beam radiation therapy and / or systemic chemotherapy. These are partially successful in some forms of cancer, but are not successful in others. There is a clear need for new therapeutic treatments. Compound (I) is exemplified and described in WO96/40681 as Example 36. WO96/40681 claims the endothelin receptors described therein for the treatment of cardiovascular diseases. The use of Compound (I) in the treatment of cancers and pain is described in WO04/018044. Compound (I) has the following structure:
Compound (I)
In WO04/018044 an endothelin human receptor binding assay is described. The pICjo (negative log of the concentration of compound required to displace 50% of the ligand) for Compound (I) at the ETA receptor was 8.27 [8.23 – 8.32] (n=4). Compound (I) is thus an excellent endothelin antagonist.
WO96/40681 and WO04/018044 disclose, in general terms, certain pharmaceutically acceptable salts of the compounds disclosed therein. Specifically it is stated that suitable pharmaceutically-acceptable salts include, for example, salts with alkali metal (such as sodium, potassium or lithium), alkaline earth metals (such as calcium or magnesium), ammonium salts, and salts with organic bases affording physiologically acceptable cations, such as salts with methylamine, dimethylamine, trimethylamine, piperidine and morpholine. In addition, it was stated that suitable pharmaceutically-acceptable salts include, pharmaceutically-acceptable acid- addition salts with hydrogen halides, sulphuric acid, phosphoric acid and with organic acids such as citric acid, maleic acid, methanesulphonic acid and p-toluenesulphonic acid.
Example 2 Formation of Compound (I) using ethanolamine
The above organic layer from Example 1 was adjusted to 42°C and isopropyl alcohol (114 ml), water (170ml) and ethanolamine (28.2 ml) were added and stirred at 42°C for 90 mins. The reaction mixture was allowed to cool to 2O0C and the lower aqueous phase separated and filtered through a 1 μm filter. The aqueous phase was then charged over 40min to a stirred solution of acetic acid (141 g) and water (33.5 g) at 500C and then cooled to 2O0C over 60 mins. The product was isolated by filtration and washed with a mixture of isopropyl alcohol (48.5 ml) and water (48.5 ml) and then isopropyl alcohol (48.5 ml). The product was dried overnight in a vacuum oven at 55°C. Weight 43.08g, Strength = 100%, 86.7%yield. 1H NMR (400 MHz5 DMSOd6) 9.87 (IH, s), 9.14 (IH, s), 8.81 (lH,d), 8.52 (IH, d), 7.98 (2H, d), 7.65 (2H, d), 7.62 (IH, dd), 7.41 (IH, bs), 3.80 (3H, s), 2.23 (3H, s). Mass Spectra MH+ 425.1036 (Ci9Hi7N6O4S calculated 425.1032).
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Substituted pyrazin-2-yl-sulphonamide-(3-pyridyl) compounds and uses thereof [US6060475] | 2000-05-09 | |
| COMPOSITION 064 [US8168221] | 2009-04-16 | 2012-05-01 |
| THERAPEUTIC TREATMENT-014 [US2009062246] | 2009-03-05 | |
| Ethanolamine Salt of N- (3-Methoxy-5-Methylpyrazin-2Yl) -2- (4-[1, 3, 4-Oxadiazole-2-Yl] Phenyl) Pyridine-3-Sulphonamide [US2008221124] | 2008-09-11 | |
| N-HETEROARYL-PYRIDINESULFONAMIDE DERIVATIVES AND THEIR USE AS ENDOTHELIN ANTAGONISTS [WO9640681] | 1996-12-19 |
| ZIBOTENTAN | |
|---|---|
 | |
| IDENTIFIERS | |
| CAS number | 186497-07-4 |
| PubChem | 9910224 |
| ChemSpider | 8085875 |
| UNII | 8054MM4902 |
| Jmol-3D images | Image 1 |
| PROPERTIES | |
| Molecular formula | C19H16N6O4S |
| Molar mass | 424.43 g mol−1 |
References
- James and Growcott (2009). “Drugs of the Future”.
- Jump up^ Tomkinson H, Kemp J, Oliver S, Swaisland H, Taboada M, Morris T (2011). “Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: two open-label comparative studies”. BMC Clin Pharmacol 11: 3. doi:10.1186/1472-6904-11-3.PMC 3070638. PMID 21414193.
- http://www.fiercebiotech.com/story/azs-zibotentan-flunks-late-stage-prostate-cancer-trial/2010-09-27
- http://www.genengnews.com/gen-news-highlights/pfizer-astrazeneca-and-actelion-separately-report-phase-iii-trial-failures/81243985/
- Jump up^ Trump DL, Payne H, Miller K, et al. (September 2011). “Preliminary study of the specific endothelin a receptor antagonist zibotentan in combination with docetaxel in patients with metastatic castration-resistant prostate cancer”. Prostate 71(12): 1264–75.doi:10.1002/pros.21342. PMID 21271613.
External links
......................
7
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Bosentan (Ro-470203), Atransentan (ABT627), Tezosentan (Ro-610612), Sitaxsentan (TBC-11251), Darusentan (LU-135252), Clazosentan (Ro61-1790, AXV-034343), ZD-4054, Ambrisentan (LU-208075), TAK-044, Avosentan (SPP301), and BQ-123 (Ihara et al Life Sci 1992, 50(4):247-55).
| Antagonists of Endothelin type A receptor ETA | |
| Name | Structure |
| BQ-123 | |
| Bosentan | |
| Atrasentan | |
| Tezosentan | |
| Sitaxsentan | |
| Darusentan | |
| Clazosentan | |
| ZD-4054 (Zibotentan) | |
| Ambrisentan | |
| Tak-044 | |
| Avosentan | |
No comments:
Post a Comment