rolimus series
1 SIROLIMUS
2 TEMSIROLIMUS
3 EVEROLIMUS
4 RIDAFOROLIMUS
5 ZOTAROLIMUS
6 BIOLIMUS
7 PIMECROLIMUS
8TACROLIMUS
9
WILL BE UPDATED WATCH OUT

..........................................
 amcrasto@gmail.com .
amcrasto@gmail.com .

..........................................
1 SIROLIMUS (RAPAMYCIN)

 sirolimus
sirolimus
 rapamycin
rapamycin
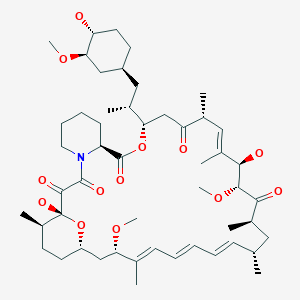 SIROLIMUS
SIROLIMUS






First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).



mTOR inhibitor
temsirolimus (CCI-779), everolimus (RAD001), deforolimus (AP23573), AP21967, biolimus, AP23102, zotarolimus (ABT 578), sirolimus (Rapamune), and tacrolimus (Prograf).
- Sirolimus
- Everolimus
- Ridaforolimus
- Temsirolimus
- Umirolimus
- Zotarolimus
1 SIROLIMUS
2 TEMSIROLIMUS
3 EVEROLIMUS
4 RIDAFOROLIMUS
5 ZOTAROLIMUS
6 BIOLIMUS
7 PIMECROLIMUS
8TACROLIMUS
9
WILL BE UPDATED WATCH OUT
..........................................
amcrasto@gmail.com .
DR ANTHONY CRASTO
..........................................
1 SIROLIMUS (RAPAMYCIN)
Rapamycin (Sirolimus)
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25, 26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone
Wyeth Pharmaceuticals (Originator)
M.Wt:914.18
Formula:C51H79NO13
53123-88-9 cas no
Antifungal and immunosuppressant. Specific inhibitor of mTOR (mammalian target of Rapamycin). Complexes with FKBP-12 and binds mTOR inhibiting its activity. Inhibits interleukin-2-induced phosphorylation and activation of p70 S6 kinase. Induces autophagy in yeast and mammalian cell lines.
Rapamycin is a triene macrolide antibiotic, which demonstrates anti-fungal, anti-inflammatory, anti-tumor and immunosuppressive properties. Rapamycin has been shown to block T-cell activation and proliferation, as well as, the activation of p70 S6 kinase and exhibits strong binding to FK-506 binding proteins. Rapamycin also inhibits the activity of the protein, mTOR, (mammalian target of rapamycin) which functions in a signaling pathway to promote tumor growth. Rapamycin binds to a receptor protein (FKBP12) and the rapamycin/FKB12 complex then binds to mTOR and prevents interaction of mTOR with target proteins in this signaling pathway. Rapamycin name is derived from the native word for Easter Island, Rapi Nui.
- (-)-Rapamycin
- Antibiotic AY 22989
- AY 22989
- AY-22989
- CCRIS 9024
- HSDB 7284
- NSC 226080
- Rapammune
- Rapamune
- Rapamycin
- SILA 9268A
- Sirolimus
- UNII-W36ZG6FT64
- WY-090217
- A 8167
A macrolide compound obtained from Streptomyces hygroscopicus that acts by selectively blocking the transcriptional activation of cytokines thereby inhibiting cytokine production. It is bioactive only when bound to IMMUNOPHILINS. Sirolimus is a potent immunosuppressant and possesses both antifungal and antineoplastic properties.
Sirolimus (INN/USAN), also known as rapamycin, is an immunosuppressant drug used to prevent rejection in organ transplantation; it is especially useful in kidney transplants. It prevents activation of T cells and B cells by inhibiting their response to interleukin-2 (IL-2). Sirolimus is also used as a coronary stent coating. Sirolimus works, in part, by eliminating old and abnormal white blood cells.[citation needed] Sirolimus is effective in mice with autoimmunity and in children with a rare condition called autoimmune lymphoproliferative syndrome (ALPS).
A macrolide, sirolimus was discovered by Brazilian researchers as a product of the bacterium Streptomyces hygroscopicus in a soil sample fromEaster Island[1] — an island also known as Rapa Nui.[2] It was approved by the FDA in September 1999 and is marketed under the trade nameRapamune by Pfizer (formerly by Wyeth).
Sirolimus was originally developed as an antifungal agent. However, this use was abandoned when it was discovered to have potent immunosuppressive and antiproliferative properties. It has since been shown to prolong the life of mice and might also be useful in the treatment of certain cancers.
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response tointerleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12(FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits themammalian target of rapamycin (mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
rapamycin
Unlike the similarly named tacrolimus, sirolimus is not a calcineurin inhibitor, but it has a similar suppressive effect on the immune system. Sirolimus inhibits the response to interleukin-2 (IL-2), and thereby blocks activation of T and B cells. In contrast, tacrolimus inhibits the secretion of IL-2.
The mode of action of sirolimus is to bind the cytosolic protein FK-binding protein 12 (FKBP12) in a manner similar to tacrolimus. Unlike the tacrolimus-FKBP12 complex which inhibits calcineurin (PP2B), the sirolimus-FKBP12 complex inhibits the mammalian target of rapamycin(mTOR, rapamycin being an older name for sirolimus) pathway by directly binding the mTOR Complex1 (mTORC1).
mTOR has also been called FRAP (FKBP-rapamycin associated protein), RAFT (rapamycin and FKBP target), RAPT1, or SEP. The earlier names FRAP and RAFT were coined to reflect the fact that sirolimus must bind FKBP12 first, and only the FKBP12-sirolimus complex can bind mTOR. However, mTOR is now the widely accepted name, since Tor was first discovered via genetic and molecular studies of sirolimus-resistant mutants of Saccharomyces cerevisiae that identified FKBP12, Tor1, and Tor2 as the targets of sirolimus and provided robust support that the FKBP12-sirolimus complex binds to and inhibits Tor1 and Tor2.
SIROLIMUS
Rapamycin and its preparation are described in US Patent No. 3,929,992, issued December 30, 1975. Alternatively, rapamycin may be purchased commercially [Rapamune®, Wyeth].
Rapamycin (Sirolimus) is a 31-member natural macrocyclic lactone [C51H79N1O13; MWt=914.2] produced by Streptomyces hygroscopicus and found in the 1970s (U.S. Pat. No. 3,929,992; 3,993,749). Rapamycin (structure shown below) was approved by the Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection in 1999.
Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP-12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., IL2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S. Pat. No. 5,286,730], bowel disorders [U.S. Pat. No. 5,286,731], smooth muscle cell proliferation and intimal thickening following vascular injury [U.S. Pat. Nos. 5,288,711 and 5,516,781], adult T-cell leukemia/lymphoma [European Patent Application 525,960 A1], ocular inflammation [U.S. Pat. No. 5,387,589], malignant carcinomas [U.S. Pat. No. 5,206,018], cardiac inflammatory disease [U.S. Pat. No. 5,496,832], anemia [U.S. Pat. No. 5,561,138] and increase neurite outgrowth [Parker, E. M. et al, Neuropharmacology 39, 1913-1919, 2000].
Although rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803). Some of the analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No. 5,362,718), as well as alkyl, aryl, alkenyl, and alkynyl analogues (U.S. Pat. Nos. 5,665,772; 5,258,389; 6,384,046; WO 97/35575).
.................................................
Synthesis
PREPARATION
CUT PASTE FROM TEXT
In one embodiment of this invention rapamycin is prepared in the followingmanner: 4
A suitable fermenter is charged with production meis reached in the fermentation mixture after 2-8 days,
usually after about 5 days, as determined by the cup plate method and Candida albicans as the test organism. The mycelium is harvested by filtration with diatomaceous earth. Rapamycin is then extracted from the mycelium with a water-miscible solvent, for example a lower alkanol, preferably methanol or ethanol. The latter extract is then concentrated, preferably under reduced pressure, and the resulting aqueous phase is extracted with a water-immiscible solvent. A preferred water-immiscible solvent for this purpose is methylene dichloride although chloroform, carbon tetrachloride, benzene, n-butanol and the like may also be used. The latter extract is concentrated, preferably under reduced pressure, to afford the crude product as an oil.
The product may be purified further by a variety of methods. Among the preferred methods of purification is to dissolve the crude product in a substantially nonpolar, first solvent, for example petroleum ether or hexane, and to treat the resulting solution with a suit able absorbent, for example charcoal or silica gel, so that the antibiotic becomes absorbed on the absorbant. The absorbant is then separated and washed or eluted with a second solvent more polar than the first solvent, for example ethyl acetate, methylene dichloride, or a mixture of methylene dichloride and ether (preferred). Thereafter, concentration of the wash solution or eluate affords substantially pure rapamycin. Further purification is obtained by partial precipitation with a nonpolar solvent, for example, petroleum ether, hexane, pentane and the like, from a solution of the rapamycin in a more polar solvent, for example, ether, ethyl acetate, benzene and the like. Still-further purification is obtained by column chromatography, preferably employing silica gel, and by crystallization of the rapamycin from ether.
In another preferred embodiment of this invention a first stage inoculum of S treptomyces hygroscopicus NRRL 5491 is prepared in small batches in a medium containing soybean flour, glucose, ammonium sulfate, and calcium carbonate incubated at about 25C at pH 7.l-7.3 for 24 hrs. with agitation, preferably on a gyrotary shaker. The growth thus obtained is used to inoculate a number of somewhat larger batches of the same medium as described above which are incubated at about 25C and pH 7.1-7.3 for 18 hrs. with agitation, preferably on a reciprocating'shaker, to obtain a sec- "ond stagc inoculum which is used to inoculate the production stage fermenters.
6 5.86'.2.-The fermenters are inoculated with the second stage inoculum described above and incubated at about 25C with' agitationand aeration while controlling and 'mai'ntaining the mixture at approximately pH 6.0 by
addition offa base, for example, sodium hydroxide, potassium hydroxide or preferably ammonium hydroxide, as required from time to time. Addition of a source -of assimilable carbon, preferably glucose, is started when theconcentrationof the latter in the broth has dropped to about 0.5% wt/vol, normally about 48 hrs after. the start of fermentation, and is maintained until the end ofthe particular run. In this manner a fermentation broth containing about 60 ug/ml of rapamycin as determined by the assay method described above is obtained in 45 days, when fermentation is stopped.
' Filtration of the'mycelium, mixing the latter with a watef-miscible 'lower' alkanol, preferably methanol, followed by extraction with a halogenated aliphatic hydrocarbon, preferably trichloroethane, and evaporation of the solvents yields a first oily residue. This first oily residue is dissolved in a lower aliphatic ketone, preferably acetone, filtered from insoluble impurities, the filtrate evaporated to yield a second oily residue which is extractedjwith a water-miscible lower alkanol,
preferably methanol, and the latter extract is evaporated to yield crude rapamycin as a third oily residue. This third oily residue is dissolved in a mixture of a lower aliphatic ketone and a lower aliphatic hydrocarbon, preferably acetone-hexane, an absorbent such as charcoal or preferably silica gel is added to adsorb the rapamycin, the latter is eluted from the adsorbate with a similar but more polar solvent mixture, for example a mixture as above but containing a higher proportion of the aliphatic ketone, the eluates are evaporated and the residue is crystallized from diethyl ether, to yield pure crystalline rapamycin. In this manner a total of 45-5 8% of the rapamycin initially present in the fermentation mixture is recovered as pure crystalline rapamycin.
CHARACTERIZATION solvent systems; for example, ether-hexane 40:60 (Rf 0.42), 'isopropyl alcoholvbenzene 15:85 (Rf= 0.5) and ethanol-benzene 20:80 (Rf f 0.43);
d. rapamycin obtained from four successive fermentation batchesgave the following values on repeated The production stage fermenters are equipped with 7 devices for controlling and maintaining pH at a predetermined level and for continuous metered addition of elemental analyses:
AVER- e. rapamycin exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum ethanol):
f. the infrared absorption spectrum of rapamycin in chloroform is reproduced in FIG. 1 and shows characteristic absorption bands at 3560, 3430, 1730, 1705 and 1630-1610 cm;
Further infrared absorption bands are characterized by the following data given in reciprocal centimeters with (s) denoting a strong, (m) denoting a medium, and (w) denoting a weak intensity band. This classification is arbitrarily selected in such a manner that a band is denoted as strong (s) if its peak absorption is more than two-thirds of the background in the same region; medium (m) if its peak is between one-third and twothirds of the background in the same region; and weak (w) if its peak is less than one-third of the background in the same region.
2990 cm (m) 1158 cm" (m) 2955 cm (s) 1129 cm (s) 2919 cm (s) 1080 cm (s) 2858 cm (s) 1060 cm (s) 2815 cm (m) 1040 cm (m) 1440 cm (s) 1020 crn' (m) 1365 cm (m) 978 cm" (s) 1316 cm (in) 905 cm (m) 1272 cm (m) 888 cm" (w) 1178 cm (s) 866 cm- (w) g. the nuclear magnetic resonance spectrum of rapamycinin deuterochloroform is reproduced in FIG. 2; SEE PATENT
CLAIMS
l. Rapamycin, an antibiotic which a. is a colourless, crystalline compound with a melting point of 183 to l8SC, after recrystallization from ether;
b. is soluble in ether, chloroform, acetone, methanol and dimethylformamide, very sparingly soluble in hexane and petroleum ether and substantially insoluble in water;
c. shows a uniform spot on thin layer plates of silica gel",
d. has a characteristic elemental analysis of about C,
e. exhibits the following characteristic absorption maxima in its ultraviolet absorption spectrum (95% ff has 'a characteristic infrared absorption spectrum shown in accompanying FIG. 1; SEE PATENT
.....................................................
Rapamycin synthetic studies. 1. Construction of the C(27)-C(42) subunit. Tetrahedron Lett 1994, 35, 28, 4907
A partial synthesis of rapamycin has been reported: The condensation of sulfone (I) with epoxide (II) by means of butyllithium followed by desulfonation with Na/Hg gives the partially protected diol (III), which is treated with methanesulfonyl chloride and NaH to afford the epoxide (IV). Ring opening of epoxide (IV) with LiI and BF3.Et2O followed by protection of the resulting alcohol with PMBOC(NH)CCl3 yields the primary iodo compound (V). The condensation of (V) with the fully protected dihydroxyaldehyde (VI) (see later) by means of butyllithium in THF/HMPT gives the fully protected trihydroxyketone (VII), which is hydrolyzed with camphorsulfonic acid (CSA) to the corresponding gemdiol and reprotected with pivaloyl chloride (the primary alcohol) and tert-butyldimethylsilyl trifluoromethanesulfonate (the secondary alcohol), yielding a new fully protected trihydroxyketone (VIII). Elimination of the pivaloyl group with DIBAL and the dithiane group with MeI/CaCO3 affords the hydroxyketone (IX), which is finally oxidized with oxalyl chloride to the ketoaldehyde (X), the C(27)-C(42) fragment [the C(12)-C(15) fragment with the C(12)-substituent based on the IUPAC nomenclature recommendations]. The fully protected dihydroxyaldehyde (VI) is obtained as follows: The reaction of methyl 3-hydroxy-2(R)-methylpropionate (XI) with BPSCl followed by reduction with LiBH4 to the corresponding alcohol and oxidation with oxalyl chloride gives the aldehyde (XII), which is protected with propane-1,3-dithiol and BF3.Et2O to afford the dithiane compound (XIII). Elimination of the silyl group with TBAF followed by esterification with tosyl chloride, reaction with NaI and, finally, with sodium phenylsulfinate gives the sulfone (XIV), which is condensed with the partially protected dihydroxyaldehyde (XV), oxidized with oxalyl chloride and desulfonated with Al/Hg to afford the dithianyl ketone (XVI). The reaction of (XVI) with lithium hexamethyldisilylazane gives the corresponding enolate, which is treated with dimethyllithium cuprate to yield the fully protected unsaturated dihydroxyaldehyde (VI).
...................................................
.................................
The Ley Synthesis of RapamycinRapamycin (3) is used clinically as an immunosuppressive agent. The synthesis of 3 (Angew. Chem. Int. Ed. 2007, 46, 591. DOI: 10.1002/anie.200604053) by Steven V. Ley of the University of Cambridge was based on the assembly and subsequent coupling of the iododiene 1 and the stannyl alkene 2.    |
.....................................
Total Synthesis of Rapamycin
Angewandte Chemie International Edition
Volume 46, Issue 4, pages 591–597, January 15, 2007
PREVIEW THIS ARTICLE WITH READCUBE
..........................
Ley, Maddess, Tackett, Watanabe, Brennan, Spilling, Scott and Osborn. ACIEE, 2006, EarlyView. DOI:10.1002/anie.200604053.
It’s been in the works for quite a while, but Steve Ley’s synthesis of Rapamycin has just been published. This complex beast has a multitude of biological activities, including an interesting immunosuppressive profile, resulting in clinical usage following organ transplantation. So, unsurprisingly, it’s been the target of many projects, with complete total syntheses published by Smith, Danishefsky, Schreiber and KCN.
So what makes this one different? Well, it does have one of the most interesting macrocyclisations I’ve seen since Jamison’s paper, and a very nice demonstration of the BDA-aldol methodology. The overall strategy is also impressive, so on with the retro:
First stop is the BDA-aldol; this type of chemistry is interesting, because the protecting group for the diol is also the stereo-directing group. The stereochemistry for this comes from a glycolic acid, and has been usedin this manner by the group before. The result is as impressive as ever, with a high yield, and presumably a very high d.r. (no mention of actual numbers).
The rest of the fragment synthesis was completed in a succinct and competent manner, but using relatively well known chemistry. However, I was especially impressed with the macrocyclisation I mentioned:
Tethering the free ends of the linear precursor with a simple etherification/esterification onto catechol gave then a macrocycle holding the desired reaction centres together. Treatment of this with base then induces a Dieckmann-condensation type cyclisation to deliver the desired macrocycle. Of course, at this stage, only a few more steps were required to complete the molecule, and end an era of the Wiffen Lab.
....................................
Drugs Fut 1999, 24(1): 22
DOI: 10.1358/dof.1999.024.01.474036
REFERENCES
- Vézina C, Kudelski A, Sehgal SN (October 1975). "Rapamycin (AY-22,989), a new antifungal antibiotic". J. Antibiot. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Pritchard DI (2005). "Sourcing a chemical succession for cyclosporin from parasites and human pathogens". Drug Discovery Today 10 (10): 688–691. doi:10.1016/S1359-6446(05)03395-7. PMID 15896681.
3. Creating diverse target-binding surfaces on FKBP12: synthesis and evaluation of a rapamycin analogue library.
Wu X, Wang L, Han Y, Regan N, Li PK, Villalona MA, Hu X, Briesewitz R, Pei D.
ACS Comb Sci. 2011 Sep 12;13(5):486-95. doi: 10.1021/co200057n. Epub 2011 Jul 28.
ACS Comb Sci. 2011 Sep 12;13(5):486-95. doi: 10.1021/co200057n. Epub 2011 Jul 28.
4. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth.
Gibbons JJ, Abraham RT, Yu K.
Semin Oncol. 2009 Dec;36 Suppl 3:S3-S17. doi: 10.1053/j.seminoncol.2009.10.011. Review.
Semin Oncol. 2009 Dec;36 Suppl 3:S3-S17. doi: 10.1053/j.seminoncol.2009.10.011. Review.
5. Hybrid inhibitors of phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR): design, synthesis, and superior antitumor activity of novel wortmannin-rapamycin conjugates.
Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
J Med Chem. 2010 Jan 14;53(1):452-9. doi: 10.1021/jm901427g.
6. Fluorescent probes to characterise FK506-binding proteins.
Kozany C, März A, Kress C, Hausch F.
Chembiochem. 2009 May 25;10(8):1402-10. doi: 10.1002/cbic.200800806.
Chembiochem. 2009 May 25;10(8):1402-10. doi: 10.1002/cbic.200800806.
7. Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs.
Graziani EI.
Nat Prod Rep. 2009 May;26(5):602-9. doi: 10.1039/b804602f. Epub 2009 Mar 5. Review.
Nat Prod Rep. 2009 May;26(5):602-9. doi: 10.1039/b804602f. Epub 2009 Mar 5. Review.
8 Total synthesis of rapamycin.
Ley SV, Tackett MN, Maddess ML, Anderson JC, Brennan PE, Cappi MW, Heer JP, Helgen C, Kori M, Kouklovsky C, Marsden SP, Norman J, Osborn DP, Palomero MA, Pavey JB, Pinel C, Robinson LA, Schnaubelt J, Scott JS, Spilling CD, Watanabe H, Wesson KE, Willis MC.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
Chemistry. 2009;15(12):2874-914. doi: 10.1002/chem.200801656.
9 Highly diastereoselective desymmetrisation of cyclic meso-anhydrides and derivatisation for use in natural product synthesis.
Evans AC, Longbottom DA, Matsuoka M, Davies JE, Turner R, Franckevicius V, Ley SV.
Org Biomol Chem. 2009 Feb 21;7(4):747-60. doi: 10.1039/b813494d. Epub 2009 Jan 6.
Org Biomol Chem. 2009 Feb 21;7(4):747-60. doi: 10.1039/b813494d. Epub 2009 Jan 6.
10 Total synthesis studies on macrocyclic pipecolic acid natural products: FK506, the antascomicins and rapamycin.
Maddess ML, Tackett MN, Ley SV.
Prog Drug Res. 2008;66:13, 15-186. Review.
Prog Drug Res. 2008;66:13, 15-186. Review.
11 Determination of sirolimus in rabbit arteries using liquid chromatography separation and tandem mass spectrometric detection.
Zhang J, Rodila R, Watson P, Ji Q, El-Shourbagy TA.
Biomed Chromatogr. 2007 Oct;21(10):1036-44.
Biomed Chromatogr. 2007 Oct;21(10):1036-44.
12 Saccharomyces cerevisiae FKBP12 binds Arabidopsis thaliana TOR and its expression in plants leads to rapamycin susceptibility.
Sormani R, Yao L, Menand B, Ennar N, Lecampion C, Meyer C, Robaglia C.
BMC Plant Biol. 2007 Jun 1;7:26.
BMC Plant Biol. 2007 Jun 1;7:26.
13 Total synthesis of rapamycin.
Maddess ML, Tackett MN, Watanabe H, Brennan PE, Spilling CD, Scott JS, Osborn DP, Ley SV.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
Angew Chem Int Ed Engl. 2007;46(4):591-7. No abstract available.
14 Drug evaluation: AP-23573--an mTOR inhibitor for the treatment of cancer.
Elit L.
IDrugs. 2006 Sep;9(9):636-44.
IDrugs. 2006 Sep;9(9):636-44.
15 lipase-catalyzed regioselective esterification of rapamycin: synthesis of temsirolimus (CCI-779).
Gu J, Ruppen ME, Cai P.
Org Lett. 2005 Sep 1;7(18):3945-8.
Org Lett. 2005 Sep 1;7(18):3945-8.
17 Everolimus. Novartis.

 A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
A plaque, written in Brazilian Portuguese, commemorating the discovery of sirolimus on Easter Island, near Rano Kau
...................................................................
amcrasto@gmail.com .
....................................................................
2 TEMSIROLIMUS

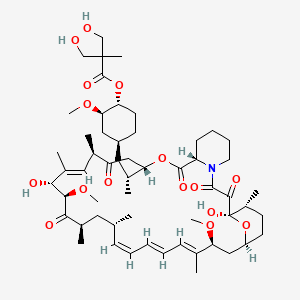 TEMSIROLIMUS
TEMSIROLIMUS




 Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS "Amano" (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS "Amano" (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.













 TEMSIROLIMUS
TEMSIROLIMUS
..........................................................................
amcrasto@gmail.com  .
.
Dumont FJ.
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
 SIROLIMUS
SIROLIMUS
FEMALE FERTILITY
http://amcrasto.theeurekamoments.com/2013/02/11/immunosuppressant-drug-rapamycin-helps-preserving-female-fertility/
PATENTS
Curr Opin Investig Drugs. 2001 Sep;2(9):1220-34. Review.
18 Kuo et al (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358 70. PMID:1614535.
19 Huang et al (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol.Ther. 2 221. PMID:12878853.
20 Kobayashi et al (2007) Rapamycin, a specific inhibitor of the mammalian target of rapamycin, suppresses lymphangiogenesis and lymphatic metastasis. Cancer Sci. 98 726. PMID: 17425689.
21 Fleming et al (2011) Chemical modulators of autophagy as biological probes and potential therapeutics. 7 9. PMID:21164513.
22 J Am Chem Soc1993,115,(10):4419
23 Tetrahedron Lett1994,35,(28):4911
24 Chemistry (Weinheim)1995,1,(5):318
24
SIROLIMUSFEMALE FERTILITY
http://amcrasto.theeurekamoments.com/2013/02/11/immunosuppressant-drug-rapamycin-helps-preserving-female-fertility/
PATENTS
| Canada | 2293793 | APPROVED2006-07-11 | EXP 2018-06-11 |
| Canada | 2103571 | 2003-04-29 | 2012-02-21 |
| United States | 5989591 | 1998-09-11 | 2018-09-11 |
| United States | 5212155 | 1993-05-18 | 2010-05-18 |
| WO1998054308A2 * | May 28, 1998 | Dec 3, 1998 | Biotica Tech Ltd | Polyketides and their synthesis and use |
| EP0589703A1 * | Sep 23, 1993 | Mar 30, 1994 | American Home Products Corporation | Proline derivative of rapamycin, production and application thereof |
| US20010039338 * | Jun 7, 2001 | Nov 8, 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO2007067560A2 * | Dec 6, 2006 | Jun 14, 2007 | Clifford William Coughlin | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| WO2012131019A1 | Mar 30, 2012 | Oct 4, 2012 | Sandoz Ag | Regioselective acylation of rapamycin at the c-42 position |
| US7622578 | Dec 6, 2006 | Nov 24, 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US3929992 | Apr 12, 1974 | Dec 30, 1975 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| US5646160 | May 26, 1995 | Jul 8, 1997 | American Home Products Corporation | Method of treating hyperproliferative vascular disease with rapamycin and mycophenolic acid |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5728710 | Jul 16, 1993 | Mar 17, 1998 | Smithkline Beecham Corporation | Rapamycin derivatives |
| US5957975 | Dec 15, 1997 | Sep 28, 1999 | The Centre National De La Recherche Scientifique | Stent having a programmed pattern of in vivo degradation |
| US5985890 | Jun 5, 1996 | Nov 16, 1999 | Novartis Ag | Rapamycin derivatives |
| US6001998 | Oct 13, 1995 | Dec 14, 1999 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6015815 | Sep 24, 1998 | Jan 18, 2000 | Abbott Laboratories | Tetrazole-containing rapamycin analogs with shortened half-lives |
| US6187568 | Aug 20, 1999 | Feb 13, 2001 | Pfizer Inc | Macrocyclic lactone compounds and their production process |
| US6273913 | Apr 16, 1998 | Aug 14, 2001 | Cordis Corporation | Modified stent useful for delivery of drugs along stent strut |
| US6585764 | Jun 4, 2001 | Jul 1, 2003 | Cordis Corporation | Stent with therapeutically active dosage of rapamycin coated thereon |
| US6641611 | Nov 26, 2001 | Nov 4, 2003 | Swaminathan Jayaraman | Therapeutic coating for an intravascular implant |
| US6805703 | Sep 18, 2001 | Oct 19, 2004 | Scimed Life Systems, Inc. | Protective membrane for reconfiguring a workpiece |
| US7025734 | Sep 28, 2001 | Apr 11, 2006 | Advanced Cardiovascular Systmes, Inc. | Guidewire with chemical sensing capabilities |
| US7056942 | Jan 16, 2004 | Jun 6, 2006 | Teva Pharmaceutical Industries Ltd. | Carvedilol |
| US7820812 * | Jul 23, 2007 | Oct 26, 2010 | Abbott Laboratories | Methods of manufacturing crystalline forms of rapamycin analogs |
| US20010027340 | Jun 4, 2001 | Oct 4, 2001 | Carol Wright | Stent with therapeutically active dosage of rapamycin coated thereon |
| US20010029351 | May 7, 2001 | Oct 11, 2001 | Robert Falotico | Drug combinations and delivery devices for the prevention and treatment of vascular disease |
| US20020005206 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Antiproliferative drug and delivery device |
| US20020007213 | May 7, 2001 | Jan 17, 2002 | Robert Falotico | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| US20020082680 | Sep 7, 2001 | Jun 27, 2002 | Shanley John F. | Expandable medical device for delivery of beneficial agent |
| US20020123505 | Sep 10, 2001 | Sep 5, 2002 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20030129215 | Sep 6, 2002 | Jul 10, 2003 | T-Ram, Inc. | Medical devices containing rapamycin analogs |
| US20040072857 | Jul 2, 2003 | Apr 15, 2004 | Jacob Waugh | Polymerized and modified rapamycins and their use in coating medical prostheses |
| US20050033417 | Jul 1, 2004 | Feb 10, 2005 | John Borges | Coating for controlled release of a therapeutic agent |
| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050152842 | Dec 22, 2004 | Jul 14, 2005 | Chun Li | Poly (L-glutamic acid) paramagnetic material complex and use as a biodegradable MRI contrast agent |
| US20050175660 | Oct 29, 2004 | Aug 11, 2005 | Mollison Karl W. | Medical devices containing rapamycin analogs |
| US20050208095 | Nov 22, 2004 | Sep 22, 2005 | Angiotech International Ag | Polymer compositions and methods for their use |
| US20050209244 | Feb 27, 2003 | Sep 22, 2005 | Prescott Margaret F | N{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]-2-methylphenyl}-4-(3-pyridyl)-2-pyrimidine-amine coated stents |
| US20050239178 | Apr 25, 2005 | Oct 27, 2005 | Wyeth | Labeling of rapamycin using rapamycin-specific methylases |
| US20060094744 | Sep 28, 2005 | May 4, 2006 | Maryanoff Cynthia A | Pharmaceutical dosage forms of stable amorphous rapamycin like compounds |
| US20060229711 | Apr 4, 2006 | Oct 12, 2006 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697 | Nov 1, 2005 | Jan 18, 2007 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
| US20070059336 | Feb 27, 2006 | Mar 15, 2007 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US20070207186 | Mar 3, 2007 | Sep 6, 2007 | Scanlon John J | Tear and abrasion resistant expanded material and reinforcement |
| US20080086198 | May 24, 2007 | Apr 10, 2008 | Gary Owens | Nanoporous stents with enhanced cellular adhesion and reduced neointimal formation |
| EP1236478A1 | Feb 27, 2002 | Sep 4, 2002 | Medtronic Ave, Inc. | Peroxisome proliferator-activated receptor gamma ligand eluting medical device |
| EP1588727A1 | Apr 20, 2005 | Oct 26, 2005 | Cordis Corporation | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO1993016189A1 | Feb 11, 1993 | Aug 19, 1993 | Pfizer | Novel macrocyclic lactones and a productive strain thereof |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO1996041807A1 | Jun 5, 1996 | Dec 27, 1996 | Sylvain Cottens | Rapamycin derivatives |
| WO1998007415A2 | Aug 18, 1997 | Feb 26, 1998 | Ciba Geigy Ag | Methods for prevention of cellular proliferation and restenosis |
| WO2001087263A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087342A2 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087372A1 | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| WO2001087373A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087374A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery systems for treatment of vascular disease |
| WO2001087375A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Delivery devices for treatment of vascular disease |
| WO2001087376A1 | May 14, 2001 | Nov 22, 2001 | Cordis Corp | Drug/drug delivery systems for the prevention and treatment of vascular disease |
| WO2002056790A2 | Dec 18, 2001 | Jul 25, 2002 | Avantec Vascular Corp | Delivery of therapeutic capable agents |
| WO2002065947A2 | Feb 18, 2002 | Aug 29, 2002 | Jomed Gmbh | Implants with fk506 for prophylaxis and treatment of restonoses |
| WO2003064383A2 | Feb 3, 2003 | Aug 7, 2003 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| WO2006116716A2 | Apr 27, 2006 | Nov 2, 2006 | William A Dunn | Materials and methods for enhanced degradation of mutant proteins associated with human disease |

...................................................................
amcrasto@gmail.com .
DR ANTHONY CRASTO
....................................................................
2 TEMSIROLIMUS
TEMSIROLIMUS
Proline CCI-779
Torisel, NCGC00167518-01
LAUNCHED 2007
PFIZER
- CCI 779
- CCI-779
- HSDB 7931
- Temsirolimus
- Torisel
- UNII-624KN6GM2T
- WAY-CCI 779
Inhibits mTOR protein
For the treatment of renal cell carcinoma (RCC). Also investigated for use/treatment in breast cancer, lymphoma (unspecified), rheumatoid arthritis, and multiple myeloma.
An ester analog of rapamycin. Temsirolimus binds to and inhibits the mammalian target of rapamycin (mTOR), resulting in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle. mTOR is a serine/threonine kinase which plays a role in the PI3K/AKT pathway that is upregulated in some tumors
(1R,2R,4S)-4-{(2R)-2-[(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-1,5,11,28,29-pentaoxo-1,4,5,6,9,10,11,12,13,14,21,22,23,24,25,26,27,28,29,31,32,33,34,34a-tetracosahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontin-3-yl]propyl}-2-methoxycyclohexyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate
cas 162635-04-3
Temsirolimus is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by Wyeth Pharmaceuticals and approved by the FDA in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
Molecular Formula: C56H87NO16
Molecular Weight: 1030.28708
Temsirolimus (CCI-779) is an intravenous drug for the treatment of renal cell carcinoma (RCC), developed by WyethPharmaceuticals and approved by the U.S. Food and Drug Administration (FDA) in late May 2007, and was also approved by the European Medicines Agency (EMEA) on November 2007. It is a derivative of sirolimus and is sold as Torisel.
TEMSIROLIMUS
Temsirolimus is a specific inhibitor of mTOR and interferes with the synthesis of proteins that regulate proliferation, growth, and survival of tumor cells. Treatment with temsirolimus leads to cell cycle arrest in the G1 phase, and also inhibits tumor angiogenesis by reducing synthesis of VEGF.
The product had been under development by Wyeth Pharmaceutical for the treatment of pancreas cancer and metastatic breast cancer, multiple sclerosis (MS) and rheumatoid arthritis (RA); however, no recent development for these indications has been reported. Pfizer had been developing the compound for the treatment of sarcoma.
Temsirolimus holds orphan drug designation in both the U.S. and the E.U. for the treatment of renal cell carcinoma. Orphan drug designation was received in the U.S. in 2006 for the treatment of mantle-cell lymphoma.
mTOR (mammalian target of rapamycin) is a kinase enzyme inside the cell that collects and interprets the numerous and varied growth and survival signals received by tumor cells. When the kinase activity of mTOR is activated, its downstream effectors, the synthesis of cell cycle proteins such as cyclin D and hypoxia-inducible factor-1a (HIF-1a) are increased. HIF-1a then stimulates VEGF. Whether or not mTOR kinase is activated, determines whether the tumor cell produces key proteins needed for proliferation, growth, survival, and angiogenesis.
mTOR is activated in tumor cells by various mechanisms including growth factor surface receptor tyrosine kinases, oncogenes, and loss of tumor suppressor genes. These activating factors are known to be important for malignant transformation and progression.mTOR is particularly important in the biology of renal cancer (RCC) owing to its function in regulating HIF-1a levels. Mutation or loss of the von Hippel Lindau tumor-suppressor gene is common in RCC and is manifested by reduced degradation of HIF-1a. In RCC tumors, activated mTOR further exacerbates accumulation of HIF-1a by increasing synthesis of this transcription factor and its angiogenic target gene products.
Rapamycin 42-ester with 3-hydroxy-2-(hydroxymethyl)-2-methylpropionic acid (CCl-779) is an ester of rapamycin which has demonstrated significant inhibitory effects on tumor growth in both in vitro and in vivo models.
CCl-779 may delay the time to progression of tumors or time to tumor recurrence which is more typical of cytostatic rather than cytotoxic agents. CCl-779 is considered to have a mechanism of action that is similar to that of sirolimus. CCl-779 binds to and forms a complex with the cytoplasmic protein FKBP, which inhibits an enzyme, mTOR (mammalian target of rapamycin, also known as FKBP12-rapamycin associated protein [FRAP]). Inhibition of mTOR's kinase activity inhibits a variety of signal transduction pathways, including cytokine-stimulated cell proliferation, translation of mRNAs for several key proteins that regulate the G1 phase of the cell cycle, and IL-2-induced transcription, leading to inhibition of progression of the cell cycle from G1 to S. The mechanism of action of CCl-779 that results in the G1-S phase block is novel for an anticancer drug.
The preparation and use of hydroxyesters of rapamycin, including CCl-779, are disclosed in U.S. Pat. No. 5,362,718. A regiospecific synthesis of CCl-779 is described in U.S. Pat. No. 6,277,983.
CCl-779 can be synthesized by the non-regioselective acylation of rapamycin, as described in U.S. Pat. No. 5,362,718. The synthesis, however, is complicated by mixtures of the desired 42-ester, with 31-esterified rapamycin, as well as 31, 42-diesterified rapamycin and unreacted rapamycin.
CCl-779 can also be prepared by the acylation of the 31-silyl ether of rapamycin with a ketal of bis-(hydroxymethyl)propionic acid, followed by removal of the 31-silyl ether and ketal protecting group from the bis-(hydroxymethyl) propionic acid, as described in U.S. Pat. No. 6,277,983. However, the crude 42-monoester produced from this regioselective synthesis requires further purification by column chromatography to remove residual amounts of diester by-products and unreacted rapamycin starting material.
Temsirolimus (CCI-779), an mTOR kinase Inhibitor of formula (I) is an antineoplastic agent indicated for the treatment of advanced renal cell carcinoma.Temsirolimus is a Rapamycin 42 ester with [3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid and was first disclosed by Skotnicki et al in US Patent No. 5,362,718.
Several processes for the preparation of Temsirolimus have been reported in the literature such as those described in US 5,362,718; US 6,277,983 and US 7, 153,957.
US Patent No 5,362,718 discloses a process for the preparation of different rapamycin 42 esters including Temsirolimus as per the scheme given below (Scheme-I).
Scheme-I: Synthesis of Temsirolimus as disclosed in US Patent No. 5,362,718
The process is non-regioselective and hence results in 31-estehfied rapamycin, 31 , 42 diesterified rapamycin and unreacted rapamycin along with the desired rapamycin-42 ester.
US Patent No. 6,277,983 reports a process for the preparation of Temsirolimus by using 31 , 42 bis silyl intermediates as per the scheme shown below (Scheme-ll).
Scheme-ll: Synthesis of Temsirolimus as disclosed in US Patent No. 6,277,983 US Patent No. 7, 153,957 reports a process for the preparation of Temsirolimusby using boronate intermediate as per the scheme shown below (Scheme-Ill).
Scheme-Ill: Synthesis of Temsirolimus as disclosed in US Patent No. 7, 153,957
Temsirolimus synthesis by Sirolimus (sirolimus, also known as rapamycin Rapamycin) esterification from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites.Sirolimus 31 and 42 have two alcohol, but 42 slightly smaller steric hindrance. Protected with trimethylsilyl 31 and 42 of the secondary alcohol to give intermediate 1 , 42 for selective removal of sulfuric acid trimethylsilyl obtain 2 , 2 with an acid chloride 3 and a carboxylic acid4 formed by esterification of acid anhydride reaction of 5 under acidic conditions after removal of the 31-bit trimethylsilyl get 6 , 6 with an alcohol 7 boronate protection is removed Temsirolimus. This synthetic route as 31 and 42 to protect the hydroxyl group appear more cumbersome. Later, the development of an enzyme-catalyzed synthesis route (OL2005, 3945). Lipase PS "Amano" (Burkholderia cepacia) of the catalyst, sirolimus and ester 8 reaction of compound 9 .Good selectivity for the enzyme, so that the esterification reaction occurs only in 42, and slightly larger steric hindrance is no response 31. 9 with sulfuric acid for removal of protection is acetonide Temsirolimus.
........................................................
SYNTHESIS
Example 11
Rapamycin 42-ester with 2.2-bis-(hydroxymethyl)propionic acid
A solution of the product of Example 10 (2.8 g, 2.65 mmol) in 50 mL THF and
25 mL IN HCl was stirred at room temperature for 4 h. The mixture was diluted with water and extracted three times with EtOAc. The combined organic phases were washed with saturated NaHCO3 solution, saturated NaCl solution, dried over MgSO4, filtered and evaporated to a yellow oily solid. Purification by flash chromatography (3X with EtOAc) afforded the title compound (1.6 g, 59 %).
(-)FAB-MS mlz 1029.6 (M-), 590.4 (southern fragment), 437.3 (northern fragment). !H NMR (400 MHz, d-6 DMSO) δ 4.5 (m, 1 H, C(42)H), 3.45 (s, 4 H), 1.04 (s, 3 H).
*3C NMR (100.6 MHz, d-6 DMSO) δ 174.2, 63.7, 63.6, 49.9, 16.8.
Example 10 Rapamycin 42-ester with 2.2.5-trimethyl.1.3_dioxane-5-carboxyric acid
To a solution of the 2,2-bis(hydroxymethyl)propionic acid isopropylidene ketal (1.041 g, 5.98 mmol) (prepared according to the procedure of Bruice, J. Am. Chem. Soc. 89: 3568 (1967)) and triethylamine (0.83 mL, 5.98 mmol) in 20 mL anhydrous THF at 0 °C under nitrogen was added 2, 4, 6-trichlorobenzoyl chloride (0.93 mL, 5.98 mmol) and the resultant white suspension was stirred 5 h at room temperature. The precipitate was removed by vacuum filtration, rinsing the flask and filter cake with an additional 10 mL dry THF. The filtrate was concentrated by rotary evaporation to a white solid. The residue was dissolved in 20 mL dry benzene, then rapamycin (5.47 g, 5.98 mmol) and DMAP (0.731 g, 5.98 mmol) were added. After stirring overnight at room temperature, the mixture was diluted with EtOAc, washed with H2O and saturated NaCl (aq), dried over MgSO4, filtered and evaporated to a yellow oil. Flash chromatography (5X with 60% EtOAc-hexane) afforded the title compound (2.2 g, 34 %) as a white solid.
(-)FAB-MS mlz 1069.5 (M-), 590.3 (southern fragment), 477.2 (northern fragment). -■H NMR (400 MHz, d-6 DMSO) δ 4.57 (m, 1 H, C(42)H), 4.02 (d, 2 H), 3.60 (d, 2 H), 1.34 (s, 3 H), 1.24 (s, 3 H), 1.06 (s, 3 H). 1 C NMR (100.6 MHz, d-6 DMSO) δ 173.2, 99.0, 65.0, 22.2, 18.1.
..................................................
SYNTHESIS
This scheme
Preparation of 5-Methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [A]
To a suspension of 2,2-bis(hydroxymethyl)propionic acid (131 g, 0.98 mole) in tetrahydrofuran (500 ml) was added a solution of phenylboronic acid (122 g, 1.0 mole) in tetrahydrofuran (500 ml). The mixture was stirred for 3 h and toluene (1.0 L) was added. Water was removed by azeotropic distillation with toluene. Heptanes (500 ml) was added to the precipitated product, heated to reflux and cooled. The mixture was filtered and washed with heptanes (2×300 ml). The solids were dried under vacuum at 70–75° C. until constant weight to give 94% yield. 1H NMR: δ (DMSO-d6) 7.65 (d, 2H, Ar), 7.40 (m, 3H, Ar), 4.35 (d, 2H, CH2), 3.92 (d, 2H, CH2), 1.17 (s, 3H, CH3)
Preparation of Rapamycin 42-ester with 5-methyl-2-phenyl-1,3,2-dioxaborinane-5-carboxylic acid, [B]
As described in U.S. Pat. No. 6,277,983 (2001) a 3 L flask was charged with rapamycin (100 g, 0.104 mole) and dissolved in ethyl acetate (1.50 L). The solution was cooled to 5–10° C. Imidazole (30 g, 0.44 moles, 4.23 eq.) was added and dissolved. Under nitrogen protection, trimethylsilyl chloride (44 g, 0.405 mole, 4.0 eq.) was added over 30–40 min while maintaining the temperature at 0–5° C. during the addition. The mixture was held for a minimum of 0.5 h. The reaction was monitored by TLC (30:70 acetone:heptane eluent). The reaction was complete when all of the rapamycin was consumed.
Two to three drops of the reaction mixture were removed and retained as a 31,42-bis(trimethylsilyl) rapamycin reference standard. 0.5 N Sulfuric acid (300 mL) was added to the 3 L flask over 0.5 h maintaining the temperature 0–5° C. The mixture was stirred vigorously and held for 5 h. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone:heptane eluent). The reaction was complete when essentially no 31,42-bis-(trimethylsilyl) rapamycin was present. The layers were separated and the lower aqueous layer was back extracted with ethyl acetate (500 mL). The combined organic layers were washed with saturated brine (500 mL) and saturated sodium bicarbonate (2×200 mL) until pH 8 was obtained. The organic layer was washed with water (2×500 mL) and brine (500 ml) until pH 6 to 7 was obtained. The solution was dried over magnesium sulfate (100 g) for 30 min, filtered into a 2 L flask and concentrated to a volume of 135 ml. Ethyl acetate (500 ml) was added and concentrated to a volume of 135 ml. The water chase was repeated once more with ethyl acetate (500 ml). Methylene chloride (300 ml) was added and the solution held until needed in the next step.
A 3 L flask equipped with mechanical stirrer was charged with compound [A] (75 g, 0.341 mole) in methylene chloride (400 mL). Diisopropylethylamine (66.1 g, 0.51 mole) was added dropwise over 20 mins and rinsed with methylene chloride (25 mL). 2,4,6-Trichlorobenzoyl chloride (80 g, 0.328 mole) was added and rinsed with methylene chloride (25 mL). The mixture was held at 0–5° C. for 4 h, and cooled to −10±5° C.
The solution of 31-trimethylsilyl rapamycin was added to the 3 L flask containing the mixed anhydride, and rinsed with methylene chloride (25 mL). A solution of dimethylamino pyridine (48.5 g, 0.397 mole) in methylene chloride (150 mL) was prepared, added over 1.5 h, maintaining the temperature <−8° C., and rinsed with methylene chloride (25 mL). The mixture was held for 12 h at −11 to −5° C. The reaction mixture was quenched with 1 N sulfuric acid (600 ml) keeping the temperature <10° C. The mixture was stirred and held for 30 mins. The pH of the upper aqueous layer was ≦2. The layers were separated, and the lower organic layers washed with brine (450 ml), saturated sodium bicarbonate (500 mL) until pH ≧8. The organic layer was washed with water (450 ml) until pH 6–7 was obtained. The solution was concentrated, acetone (250 ml) added and concentrated. This was repeated with another portion of acetone (250 ml) and concentrated.
The solution was diluted with acetone. 0.5 N Sulfuric acid (500 ml) was added dropwise over 30 mins keeping the pot temperature 0–5° C. The mixture was held for a minimum of 5 h, during which time, the product precipitated out of solution. Aqueous sodium bicarbonate (30 g in 375 ml water) was added dropwise over 30 minutes keeping the pot temperature 0 to 5° C.; the mixture was held for a minimum of 30 minutes. Acetic acid (25 ml) was added until pH was 5–6 keeping the pot temperature <10° C. The mixture was warmed to room temperature and held for 16 h. The solid product was filtered and washed with water (2×100 ml) followed by 1:1 acetone:water (2×100 ml). The cake was purified in acetone (375 ml) to give 65 g (58% overall from rapamycin) of product [B]. LC/MS: using an electrospray interface in the positive ion mode afforded the molecular ion [M+Na]=1138.5 atomic mass units (amu).
Preparation of Rapamycin 42-ester with 2,2-bis(hydroxymethyl)-propionic acid, [C]
Compound [B] (200 g, 0.179 mole), was dissolved in tetrahydrofuran (600 ml), 2-methyl-2,4-pentanediol (42.3 g, 0.358 mole, 2.0 eq.) was added and the mixture stirred for a minimum of 3 h. The reaction mixture was concentrated to a foam. Diethyl ether (1.0 L) was added and the mixture stirred for 2 h. Heptanes (1.0 L) was added dropwise over 1 h and the mixture stirred for 2 h. The mixture was filtered and the solid product washed with heptanes (500 ml). The solids were re-dissolved in acetone (400 ml), re-treated with 2-methyl-2,4-pentanediol (21.1 g, 0.179 mole, 1 eq.) in acetone (200 ml), clarified through a 0.2 micron cartridge filter, and rinsed with acetone (200 ml). The solution was concentrated to a foam, diethyl ether (1.0 L), pre-filtered through a 0.2 micron cartridge filter, was added and the mixture stirred for 2 h. The mixture was co-precipitated by adding pre-filtered heptanes (1.0 L). The precipitated solids were filtered and washed with ether:heptane (2×500 ml). The solids were dried (55 to 60° C., 10 mm Hg, minimum 24 h) to give 159 g (86%) of product [C]. LC/MS: using APCl in the positive ion mode afforded the molecular ion [M+NH4]=1047.0 amu. The 1H NMR of the product (CCl-779) was identical to the product described in example 11 of U.S. Pat. No. 5,362,718 (1994).
.......................................
Synthesis
Example 1 - Synthesis of Proline CCI-779
This example describes a method for the synthesis of the proline analog of CCI- 779, which is illustrated in the scheme provided above.
A.
Preparation of 31, 42-Bis (trimethylsilyl) proline rapamycin (Compound B)
A 3 -neck 50 mL flask was charged with proline rapamycin (compound A in the scheme) (1.47 g, 1.63 mmol), imidazole (0.45 g, 6.6 mmol, 4 eq.) and ethyl acetate (22.5 mL). The magnetically stirred mixture became cloudy. The mixture was cooled to 0-5°C. Under nitrogen protection, trimethylsilyl chloride (0.62 g, 5.7 mmol, 3.5 eq.) was added over 0.5 h via syringe while maintaining the temperature at 0-5°C during the addition. The syringe was rinsed with 2.5 ml ethyl acetate and the mixture held for 0.75 hours (0.75 h), whereupon a white precipitate was formed. The reaction was monitored by thin layer chromatography (TLC) (30:70 acetone :heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The mixture was shaken and allowed to settle. The upper organic layer was spotted against the starting material (proline rapamycin). The reaction was complete when no more starting material was present.
B.
Preparation of 31 -trimethylsilyl proline rapamycin, Compound E
When the above reaction was complete, 2-3 drops of the reaction mixture was removed and retained for the following step as the 31,42-bis(trimethylsilyl) proline rapamycin reference standard. To the 50 ml flask was added 0.5 N sulfuric acid (4.5 mL) over 0.5 h maintaining the temperature at 0-5 °C. The mixture became less cloudy. The mixture was held for 2.5 h and was monitored by thin layer chromatography (TLC, 30:70 acetone:heptane eluent). The TLC sample was prepared by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 10 drops ethyl acetate. The reaction aliquot was shaken and allowed to settle. The upper organic layer was spotted against the 31 ,42-bis(trimethylsilyl) proline rapamycin reference. The reaction was complete when essentially no 31,42-bis(trimethylsilyl) proline rapamycin was present. Ethyl acetate (5 mL) was added and the layers separated. The lower aqueous layer is extracted with ethyl acetate (7.5 mL). The combined organic layers were washed with brine (7.5 mL), by washing with saturated sodium bicarbonate (6 mL) followed by washing water (3 x 7.5 mL), in that order. The pH of the last water wash was 6-7. The organic layer was washed again with brine (7.5 mL) and dried over sodium sulfate (4 g) for 20 min. The mixture was filtered into a 250 mL flask and concentrated to dryness.
The solid was dried at room temperature under high vacuum (10 mmHg or less) for 20 h.
Weight = 1.51 g of an off-white foam.
C.
Preparation of Intermediate, Compound F:
A 3 -neck 100 mL flask equipped with mechanical stirrer was charged with
2,2,5-trimethyl[l,3-dioxane]-5-carboxylic acid, Compound C (0.63 g, 3.6 mmol) in methylene chloride (7.5 mL). Dusopropylethylamine (0.77 g, 5.9 mmol) was added, followed by a rinse with methylene chloride (1 mL). 2,4,6-Trichlorobenzoyl chloride (0.85 g, 3.5 mmol) was added, followed by a rinse with methylene chloride (1.5 mL).
The mixture was held at room temperature for 4.5 h. The solution was cooled to -12 ±
2°C. 31 -Trimethylsilyl proline rapamycin, compound E, (1.51 g) in methylene chloride (8 mL) was dissolved and added to the 100 mL flask. Methylene chloride (2 mL) was added as a rinse. A solution of dimethylamino pyridine (DMAP) (0.77 g, 6.8 mmol) in methylene chloride (3 mL) was prepared and added to the 100 mL flask over
2.5 h maintaining the temperature -12 ± 2 °C. Methylene chloride (1 mL) was added as a rinse. The mixture was held for 16 h and was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL water and 0.2 mL ethyl acetate. The HPLC sample was prepared by withdrawing 2 drops of the upper organic layer, blowdrying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04
Wavelength : λ = 280 nm Flow rate : 1 mL / min
Time : 60 min
Retention times : Compound E ~14.0-14.5 min Compound F -33.4-33.8 min
The reaction was complete when < 0.5% of starting material was present. The reaction mixture was quenched with water (6 mL). Methylene chloride (10 mL) was added and the layers separated. The aqueous layer was extracted with methylene chloride (10 mL). The combined organic layers were washed with 0.5 N sulfuric acid (12 mL), brine (10 mL), saturated sodium bicarbonate (6 mL), and water (3 x 10 mL) in that order. The pH of the last water wash was 6-7. The clear yellow solution was concentrated to a foam. The solid was dried at room temperature under high vacuum (10 mmHg or less) for 24 h. Weight = 1.88 g of a yellow foam.
D.
Preparation of crude proline CCI-779
A 1-neck 50 mL flask equipped with mechanical stirrer was charged with Compound F in THF (18.8 mL, 10 vols) and then cooled to 0 - 5 °C (or about -2.5°C). 2 N sulfuric acid (9.4 mL, 5 vols) was added over 2.5 h. After complete addition, the mixture was warmed to 2.5 °C and then held for 45 h. The reaction was monitored by HPLC by quenching 3-4 drops of reaction mixture into 0.25 mL saturated sodium bicarbonate and 0.25 mL ethyl acetate. The HPLC sample was prepared by withdrawing 5 drops of the upper organic layer, blow drying the sample under nitrogen in an HPLC vial and redissolving using the mobile phase.
HPLC column : CSC Hypersil ODS / BDS 5 μm.
Mobile phase : 68.5 % dioxane:water + 0.01M KH2P04 Wavelength : λ= 280 nm Flow rate : 1 mL / min Time : 60 min Retention times Compound F ~33.4-33.8 min Desilylated Compound F ~10.5-11.5 min (intermediate) Proline CCI-779 -5.0-5.5 min The desilylated intermediate of compound F was formed first. The reaction was complete when < 0.5% of the silylated analog remained. Ethyl acetate (27 mL) and brine (7.5 mL) was added and the layers separated. The aqueous layer was extracted with ethyl acetate (10 mL). The combined organic layers were washed with brine (10 mL), saturated sodium bicarbonate (7.5 mL), and water (3 x 7.5 mL) in that order. The pH of the last water wash was 6-7. The mixture was dried over sodium sulfate (5 g) for 30 min, filtered into a 250 L flask and concentrated to dryness. Weight = 1.58 g of a yellow foam.
E.
Chromatographic purification of crude proline CCI-779
A silica gel column (31.6 g, 60 A, 200-400 mesh) (22 cm length x 2.5 cm diameter) was prepared and conditioned with 15:85 acetone:HPLC grade hexane (1 L). The yellow crude proline CCI-779 (1.58 g) in acetone (1.58 mL) was prepared and chromatographed. The column was eluted with the remaining 15:85 acetone :hexane mixture followed by 25:75 acetone:hexane (4 L). The positive fractions were combined and concentrated to dryness. The resulting foam was dried at 35 °C, high vacuum (i.e., 10 mmHg or less) for 24 h. Weight = 1.12 g of a light yellow foam.
F.
Ether treatment of proline CCI-779
A 1 -neck 50 mL flask was charged with proline CCI-779 ( 1.12 g) and dissolved in ether (1.5 mL). The mixture was held for 2 h. The ether was stripped to give a foam. The foam was dried at 35 °C, under high vacuum (10 mmHg or less) for 12 h then at room temperature overnight (12 h). Weight = 1.09 g.
*H NMR (500 and 600 MHz, DMSO-d6) δ 5.45 (H-l), 6.12 (H-2), 6.27 (H-3), 6.41 (H-4), 6.20 (H-5), 3.66 (H-7), 1.14 and 1.86 (H-8), 4.02 (H-9), 1.19 and 1.81 (H-10), 1.52 (H-11), 2.03 (H-12), 3.23 and 3.54 (H-18), 1.76 (H-19), 2.20 and 1.89 (H-21), 4.22 (H-22), 4.87 (H-25), 2.28 and 2.70 (H-26), 3.22 (H-28), 5.11 (H-29), 4.04 (H-31), 4.17 (H-32), 2.25 (H-34), 0.985 and 1.38 (H-35), 2.22 (H-36), 1.76 (H-37), 0.961 and 1.11 (H-38), 1.31 (H-39), 0.726 and 1.90 (H- 40), 3.14 (H-41), 4.46 (H-42), 1.22 and 1.81 (H-43), 0.888 and 1.60 (H-44), 1.60 (H-45), 3.05 (H-46, OCH3), 0.697 (H-47), 6.48 (H-48), 0.821 (H-49), 1.76 (H-50), approx. 5.1- 5.3 (H-51), 3.17 (H-52, OCH3), 0.755 (H-53), 0.966 (H-54), 0.805 (H-55), 3.29 (H-56, OCH3), 3.46 (H-59), 1.01 (H-60), approx. 4.3-4.7 (0-61)
13C NMR (75 MHz, DMSO- d6) δ 139.12 (C-1), 130.53 (C-2), 132.49 (C-3), 127.08 (C-4), 127.21 (C-5), 137.12 (C-6), 81.93 (C-7), 40.40 (C-8), 65.83 (C-9), 29.45 (C-10), 25.87 (C-l l), 34.21 (C-12), 99.25 (C-13), 198.17 (C-15), 165.55 (C-16), 47.01 (C-18), 24.04 (C-19), 28.93 (C-21), 58.50 (C-22), 170.44 (C-23), 73.24 (C-25), 39.96 (C-26), 207.67 (C-27), 44.51 (C-28), 123.92 (C-29), 136.56 (C-30), 75.84 (C-31), 84.86 (C-32), 209.49 (C-33), 40.76 (C-34), 39.20 (C-35), 35.05 (C-36), 32.73 (C-37), 38.42 (C-38), 32.06 (C-39), 36.01 (C-40), 80.12 (C- 41), 75.92 (C-42), 29.25 (C-43), 30.24 (C-44), 10.27 (C-45), 55.48 (C-46, OCH3), 15.46 (C-47), 15.59 (C-49), 14.41 (C-50), 56.56 (C-52, OCH3), 12.67 (C-53), 21.50 (C-54), 14.89 (C-55), 57.27 (C-56, OCH3), 174.22 (C-57), 49.90 (C-58), 63.59 and 63.98 (C-59), 16.82 (C-60). MS [M+NH ] 1033.5, [ESI(+), M+Na+] 1038.7.
Example 3 - Synthesis of CCI-779:
A. Synthesis of CCI-779 via intermediate A Method 1 : A mixture of rapamycin (6 g), vinyl ester I (2 g), lipase PS-C "Amano" II (6 g) in anhydrous TBME (36 mL) was heated at 45 °C under Ar2 for 2 days. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, the oily residue was added to heptane while stirring. The batch was then cooled to -15 °C for 2 h, collect the solid on the Buchner funnel and washed with cold heptane, A was obtained as off-white solid, crude yield : 98%.MS (El): 1070 Above crude A (6g), dissolved in n-PrOH (24 mL) cooled to 0 °C with an ice-water bath, to this solution was added aqueous H2S04 (12 mL, 1.2N). The mixture was stirred for 24 h at 0°C and was then added to cold phosphate buffer (300 ml, pH=7.8), collect the solid on a Buchner funnel and washed with DI water and dry under vacuum, silica gel column purification eluting with hexane-acetone furnished CCI-779 as a white solid (5.2 g, 90%). MS (El): 1030 Method 2: A mixture of rapamycin (30.0 g, 32.8 mmol), vinyl ester I (10.0 g, 50 mmol), lipase PS-C "Amano" II (30 g) and molecular sieves (5 A) (10.0 g) in anhydrous TBME (150 mL) was heated at 42-43 °C under Ar2 for 48 hours. THF (100 mL) was added to dissolve the precipitation and the mixture was cooled to room temperature. Enzyme was removed by filtration and washed with THF (200 mL), the filtrate was concentrated to about 60 mL and diluted with THF (320 mL). The solution was then cooled to 0-5 °C, H2S04 (180 mL, 2N) was added dropwise over lh. The mixture was stirred for 48 h at 0-5 °C or until the disappearance of A as monitored by TLC. The mixture was diluted with brine (300 mL) and extracted with EtOAc (three times). The combined organic layer was washed with H20, 5% NaHC03, then brine and dried
(MgS04). Evaporation of solvent gave a light yellowish semi solid which was purified by flash chromatography (hexane/acetone, 2:1) to give CCI-779 as a white solid (30.77 g, 91% for two steps). B. Synthesis of CCI-779 via intermediate B: A mixture of rapamycin (3 g), vinyl ester II (1.2 g), lipase PS-C "Amano" II (5 g) in anhydrous TBME (45 mL) was heated at 45 °C under Ar2 for 60 h. The mixture was cooled to room temperature and enzyme was removed by filtration, the filtrate was concentrated, MeOH (20 mL) was added to the residue and concentrated to dryness. Silica gel column purification of crude eluting with hexane-acetone furnished CCI-779 as a white solid (2.3 g), and recovered rapamycin (0.81 g). The yield is 93% based on the recovered rapamycin.
proline analog of CCI-779 (proline-rapamycin42-ester with 2,2-bis(hydroxymethyl)propionic acid or proline-CCI-779) and methods of synthesizing same. Proline-CCI-779 is an active drug substance useful in oncology and other associated indications (immunosuppression, anti-inflammatory, anti-proliferation and anti-tumor). In one aspect, the synthesis of proline-CCI-779 is accomplished through bis- silylation of proline rapamycin, mono-de-protecting 31 ,42-bis-trimethylsilyl proline rapamycin, and acylating the mono-silyl proline rapamycin followed by hydrolysis. In another aspect, the invention provides a two-step enzymatic process involving a regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to give CCI-779.
Example 4 - Synthesis of Proline-CCI-779 The enzymatic procedure of the invention can also be applied to the synthesis of proline CCI-779 from proline-rapamycin under essentially the same conditions as described in Example 2, procedure A for the synthesis of CCI-779 from rapamycin.
proline-rapamycin proline-CCI-779
......................
more info added for readers
synthesis of CCI-779 or Proline CCI-779 (Temsirolimus) which is useful as an antineoplastic agent having the structure
It is stated to be effective in multiple applications, including inhibition of tumor growth, the treatment for multiple sclerosis and rheumatoid arthritis.
2. The Prior Arts

U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.
U.S. Pat. No. 7,202,256 disclosed methods for the synthesis of CCI-779 (Temsirolimus), providing two-step enzymatic process involving regiospecific acylation of rapamycin, using a microbial lipase and an activated ester derivative of 2,2-bis(hydroxymethyl)propionic acid in an organic solvent, followed by deprotection to obtain the CCI-779 (as shown in scheme 1). A number of drawbacks of the synthesis route depicted in scheme 1 are high-priced PdCl2 and poisonous trimethylboroxine.
A selective synthesis of 42-monoacylated product was previously conducted by reacting rapamycin 31,42-bis-silyl ether, and then the 42-sily ether protection group is selectively removed to provide rapamycin-OH-31-sily ether (U.S. Pat. No. 5,563,145). In addition, a regioselective process for the preparation of CCI-779 is also described in U.S. Pat. No. 6,277,983 (Scheme2). First, rapamycin (compound 4b) is treated with excess chlorotrimethylsilane to form rapamycin31,42-bis-trimethylsilyl ether (compound 5), and then 42-trimethylsilyl ether protection group is selectively removed in mild acid to provide rapamycin 42-OH-31-trimethylsilyl ether (compound 6). This free 42-OH was then acylated with 2,4,6-trichlorobenzyl mixed anhydride of 2,2,5-trimethyl[1,3-dioxane]-5-carboxylic acid (compound 7) at −15° C. for 16 h to give rapamycin 31-trimethylsilyl ether 42-ester (compound 8). Following treatment with mild acid for a certain period, CCI-779 can be isolated. 2,4,6-trichlorobenzyl chloride is irritant, moisture sensitive and costly.
Further, as below-depicted in Scheme 3, U.S. Pat. No. 7,153,957 disclose another method for the CCI-779. It can be prepared by the acylation of 31-silyl ether of rapamycin with the anhydride derived from the 2-phenylboronate acid to give rapamycin 31-silyl ether, 42-boronate. Thereafter, it is hydrolyzed under mild acid condition to form rapamycin 42-ester boronate. After being treated with a suitable diol, CCI-779 was obtained (Scheme 3). Mixed anhydride is not satisfactory for commercial scale synthesis because it can be kept stable only for 48 hr at −5˜0° C., not durable for longer time.
synthesis ofTemsirolimus in a more economic way.
..............
PAPERS
CCI-779
Drugs Fut 2002, 27(1): 7
Drugs Fut 2002, 27(1): 7
Organic Letters, 2005 , vol. 7, 18 pg. 3945 - 3948 seenmr
PATENTS
| United States | 5362718 | APPROVED 1994-04-18 | EXPIRY 2014-04-18 |
| Canada | 2429020 | 2009-05-26 | 2021-11-13 |
| Canada | 2187024 | 2004-08-10 | 2015-04-14 |
| US8198280 | 6-13-2012 | N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF |
| US2011281891 | 11-18-2011 | N-HYDROXYAMIDE DERIVATIVES AND USE THEREOF |
| US7998964 | 8-17-2011 | N-Hydroxyamide Derivatives and Use Thereof |
| US7973039 | 7-6-2011 | Sulfonyl Amino Cyclic Derivatives and Use Thereof |
| US7838522 | 11-24-2010 | Benzothiazole Formulations and Use Thereof |
| US2010292231 | 11-19-2010 | Indazole Compounds for Treating Inflammatory Disorders, Demyelinating Disorders and Cancers |
| US2010249415 | 9-31-2010 | Process for preparation of temsirolimus |
| US2010098691 | 4-23-2010 | COMBINATION OF BENZIMIDAZOLE ANTI-CANCER AGENT AND A SECOND ANTI-CANCER AGENT |
| US7605257 | 10-21-2009 | Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants |
| US2009149511 | 6-12-2009 | Administration of an Inhibitor of HDAC and an mTOR Inhibitor |
| US2007128731 | 6-8-2007 | Methods for preparing crystalline rapamycin and for measuring crystallinity of rapamycin compounds using differential scanning calorimetry |
| US7202256 | 4-11-2007 | Proline CCI-779, production of and uses therefor, and two-step enzymatic synthesis of proline CCI-779 and CCI-779 |
| US2007004767 | 1-5-2007 | Methods for treating neurofibromatosis 1 |
| US7074804 | 7-12-2006 | CCI-779 Isomer C |
| US5362718 | 18 Apr 1994 | 8 Nov 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US6197967 | 13 Dec 1999 | 6 Mar 2001 | Clariant Gmbh | Process for the preparation of paraoxadiazolyphenylboronic acids |
| US6277983 | 27 Sep 2000 | 21 Aug 2001 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| WO1995028406A1 | 14 Apr 1995 | 26 Oct 1995 | American Home Prod | Rapamycin hydroxyesters, process for their preparation and pharmaceutical compositions containing them |
| US7553843 | 6 Dec 2006 | 30 Jun 2009 | Wyeth | Process for the preparation of purified crystalline CCI-779 |
| US7605258 | 16 Oct 2007 | 20 Oct 2009 | Wyeth | Processes for the synthesis of individual isomers of mono-peg CCI-779 |
| US7622578 | 6 Dec 2006 | 24 Nov 2009 | Wyeth | Scalable process for the preparation of a rapamycin 42-ester from a rapamycin 42-ester boronate |
| US7625726 | 29 Sep 2008 | 1 Dec 2009 | Wyeth | Process for preparing rapamycin 42-esters and FK-506 32-esters with dicarboxylic acid, precursors for rapamycin conjugates and antibodies |
| US7875612 | 24 Apr 2002 | 25 Jan 2011 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US7910594 | 13 May 2003 | 22 Mar 2011 | Endocyte, Inc. | Vitamin-mitomycin conjugates |
| US8026276 | 25 Jul 2003 | 27 Sep 2011 | Wyeth Llc | Parenteral CCI-779 formulations containing cosolvents, an antioxidant, and a surfactant |
| US8044200 | 14 Mar 2006 | 25 Oct 2011 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| US8105568 | 10 Jul 2009 | 31 Jan 2012 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US8288557 | 22 Jul 2005 | 16 Oct 2012 | Endocyte, Inc. | Bivalent linkers and conjugates thereof |
| US8299116 | 10 Aug 2011 | 30 Oct 2012 | Wyeth Llc | CCI-779 concentrate formulations |
| US8455539 | 15 Oct 2012 | 4 Jun 2013 | Wyeth Llc | CCI-779 concentrate formulations |
| US8465724 | 18 Aug 2006 | 18 Jun 2013 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US8470822 | 7 May 2010 | 25 Jun 2013 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| US8524893 | 28 Jan 2011 | 3 Sep 2013 | Fresenius Kabi Oncology Limited | Process for the preparation of temsirolimus and its intermediates |
| WO2011092564A2 | 20 Jan 2011 | 4 Aug 2011 | Fresenius Kabi Oncology Ltd | Process for the preparation of temsirolimus and its intermediates |
amcrasto@gmail.com
.................................................................
3 EVEROLIMUS

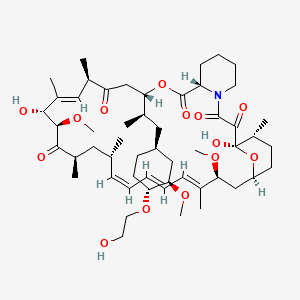 EVEROLIMUS
EVEROLIMUS









3 EVEROLIMUS

Afinitor (everolimus)
40-O-(2-hydroxyethyl)-rapamycin
Company: Novartis
Approval Status: Approved July 2012
Treatment Area: hormone receptor-positive, HER2-negative breast cancer
Approval Status: Approved July 2012
Treatment Area: hormone receptor-positive, HER2-negative breast cancer
Everolimus is a derivative of Rapamycin (sirolimus), and works similarly to Rapamycin as an mTOR (mammalian target of rapamycin) inhibitor. It is currently used as an immunosuppressant to prevent rejection of organ transplants. In a similar fashion to other mTOR inhibitors Everolimus' effect is solely on the mTORC1 protein and not on the mTORC2 protein.
Also known as: Afinitor, Certican, Zortress, SDZ-RAD, RAD001, Everolimus [USAN], 42-O-(2-Hydroxyethyl)rapamycin, RAD 001
Molecular Formula: C53H83NO14 Molecular Weight: 958.22442
159351-69-6 CAS NO
BRANDS
| Afinitor | Novartis |
| Certican | Novartis |
| VOTUBIA | Novartis |
| Zortress | Novartis |
Afinitor (everolimus), an inhibitor of mTOR (mammalian target of rapamycin), is an antineoplastic agent.
Afinitor is specifically approved for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole.
Afinitor is supplied as a tablet for oral administration. The recommended dose of Afinitor for breast cancer is 10 mg, to be taken once daily, at the same time every day, either consistently with food or consistently without food.
FDA Approval
The FDA approval of Afinitor for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer was based on a randomized, double-blind, multicenter study in 724 postmenopausal women with estrogen receptor-positive, HER 2/neu-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole.
The FDA approval of Afinitor for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer was based on a randomized, double-blind, multicenter study in 724 postmenopausal women with estrogen receptor-positive, HER 2/neu-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole.
Everolimus is indicated for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer (advanced HR+ BC) in combination with exemestane, after failure of treatment with letrozole or anastrozole. Indicated for the treatment of adult patients with progressive neuroendocrine tumors of pancreatic origin (PNET) with unresectable, locally advanced or metastatic disease. Indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib. Indicated for the treatment of adult patients with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. Indicated in pediatric and adult patients with tuberous sclerosis complex (TSC) for the treatment of subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected.
Everolimus (RAD-001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors for use in a number of cancers.
It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
EVEROLIMUS
AFINITOR (everolimus), an inhibitor of mTOR, is an antineoplastic agent.
The chemical name of everolimus is (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18- dihydroxy-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-aza-tricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20pentaone.
The molecular formula is C53H83NO14 and the molecular weight is 958.2. The structural formula is:
 |
AFINITOR Tablets are supplied for oral administration and contain 2.5 mg, 5 mg, 7.5 mg, or 10 mg of everolimus. The tablets also contain anhydrous lactose, butylated hydroxytoluene, crospovidone, hypromellose, lactose monohydrate, and magnesium stearate as inactive ingredients.
AFINITOR DISPERZ (everolimus tablets for oral suspension) is supplied for oral administration and contains 2 mg, 3 mg, or 5 mg of everolimus. The tablets for oral suspension also contain butylated hydroxytoluene, colloidal silicon dioxide, crospovidone, hypromellose, lactose monohydrate, magnesium stearate, mannitol, and microcrystalline cellulose as inactive ingredients.
- R.N Formica Jra, K.M Lorberb, A.L Friedmanb, M.J Biaa, F Lakkisa, J.D Smitha, M.I Lorber (March 2004). “The evolving experience using everolimus in clinical transplantation”. Elsevier 36 (2): S495–S499.
- “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 2009-03-30. Retrieved April 6, 2009.
- “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 2010-04-22. Retrieved April 26, 2010.
- “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 Nov 2010.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- “US FDA approves Novartis drug Afinitor for breast cancer”. 20 Jul 2012.
PATENTS
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 6440990 | 1993-09-24 | 2013-09-24 |
| Canada | 2145383 | 2004-11-16 | 2013-09-24 |
| Canada | 2225960 | 2004-05-11 | 2016-07-12 |
| United States | 7297703 | 1999-12-06 | 2019-12-06 |
10-28-2011
|
METHODS OF TREATMENT
| |
1-21-2011
|
ANTI-IGF1R
| |
1-14-2011
|
HISTONE H2AX (HH2AX) BIOMARKER FOR FTI SENSITIVITY
| |
3-24-2010
|
Thermal treatment of a drug eluting implantable medical device
| |
1-13-2010
|
Therapeutic phosphonate compounds
| |
10-21-2009
|
Processes for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants
| |
10-16-2009
|
Heparin Prodrugs and Drug Delivery Stents Formed Therefrom
| |
9-11-2009
|
PHOSPHONATE COMPOUNDS HAVING IMMUNO-MODULATORY ACTIVITY
| |
12-31-2008
|
Phosphonate compounds having immuno-modulatory activity
| |
10-8-2008
|
Anti-inflammatory phosphonate compounds
|
6-27-2008
|
Genes Involved in Neurodegenerative Conditions
| |
10-24-2007
|
Fluid treatment of a polymeric coating on an implantable medical device
| |
7-11-2007
|
Oxepane isomer of 42-O-(2-hydroxy)ethyl-rapamycin
| |
2-9-2007
|
40-O-(2-hydroxy)ethyl-rapamycin coated stent
| |
1-5-2007
|
Methods for treating neurofibromatosis 1
| |
9-8-2006
|
Anti-inflammatory phosphonate compounds
|
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| WO2007135397A1 * | May 18, 2007 | Nov 29, 2007 | Christoph Beckmann | 36 -des (3 -methoxy-4 -hydroxycyclohexyl) 36 - (3 -hydroxycycloheptyl) derivatives of rapamycin for the treatment of cancer and other disorders |
| EP0663916A1 | Sep 24, 1993 | Jul 26, 1995 | Novartis AG | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US5665772 | Sep 24, 1993 | Sep 9, 1997 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20030125800 | Apr 24, 2002 | Jul 3, 2003 | Shulze John E. | Drug-delivery endovascular stent and method for treating restenosis |
...........................................
Rapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433'- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
..............
SYNTHESIS
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — --> RAP-O-CH2-CH2-O-P — - > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
............................
SYNTHESIS
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
.......................
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
 - natural product derived anticancer drugs")
..........................................
4 RIDAFOROLIMUS
 RIDAFOROLIMUS
RIDAFOROLIMUS
 Ridaforolimus
Ridaforolimus









Phase 3 , breast cancer, Ridaforolimus (MK-8669; AP23573; formerly Deforolimus) Merck, license,Ariad Pharmaceuticals

Ridaforolimus
572924-54-0
(1R,2R,4S)-4-[(2R)-2-[(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28Z,30S,32S,35R)-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraen-12-yl]propyl]-2-methoxycyclohexyl dimethylphosphinate
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
42-(dimethylphosphinate) Rapamycin
Deforolimus, MK-8669, AP-23573, S1022_Selleck, AP23573, AP23573, MK-8669, Ridaforolimus, Deforolimus, 572924-54-0, MK 8669
- AP 23573
- AP23573
- Deforolimus
- MK 8669
- MK-8669
- MK8669
- Ridaforolimus
- Taltorvic
- UNII-48Z35KB15K
Molecular Formula: C53H84NO14P Molecular Weight: 990.206122
An mTOR inhibitor for the treatment of cancer.
Ridaforolimus (MK-8669; AP23573; formerly Deforolimus)
Merck, under exclusive worldwide license agreement with Ariad Pharmaceuticals
Method of Action: Oral inhibitor of mammalian target of rapamycin inhibitor (mTOR)
Indications/Phase of Trial: Maintenance therapy for metastatic soft-tissue sarcoma and bone sarcomas after at least four chemotherapy cycles (under review after receiving Complete Response letter from FDA in June; NME)
Ridaforolimus is an investigational small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN, known to be important to malignancy.
TARGET- mTOR
Ridaforolimus (also known as AP23573 and MK-8669; formerly known as Deforolimus[1]) is an investigational targeted and small-molecule inhibitor of the protein mTOR, a protein that acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival, integrating signals from proteins, such as PI3K, AKT and PTEN known to be important to malignancy. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism, and angiogenesis.
It has had promising results in a clinical trial for advanced soft tissue and bone sarcoma.
| Commercial arrangements |
Ridaforolimus is being co-developed by Merck and ARIAD Pharmaceuticals. On May 5, 2010, Ariad Pharmaceuticals and Merck & Company announced a clinical development and marketing agreement. With this agreement, Ariad received $125 million in upfront payments from Merck and $53 million in milestone payments. Future payments are triggered upon acceptance of the NDA by the FDA with another payment when the drug receives marketing approval. There are similar milestones for acceptance and approval in both Europe and Japan. Other milestone payments are tied to revenue goals for the drug.[2] ARIAD has opted to co-promote ridaforolimus in the U.S. Merck plans to submit a New Drug Application (NDA) for ridaforolimus to the U.S. Food and Drug Administration (FDA) and a marketing application in the European Union in 2011.[3]
Clinical trials
Phase III SUCCEED
On June 6, 2011, Ariad and Merck announced detailed results from the largest randomized study ever in the soft tissue and bone sarcoma population, the Phase III SUCCEED clinical trial. SUCCEED evaluated oral ridaforolimus, in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. In this patient population, ridaforolimus improved progression-free survival (PFS) compared to placebo, the primary endpoint of the study. The complete study results were presented by Sant P. Chawla, M.D., director, Sarcoma Oncology Center, Santa Monica, CA, during the 2011 American Society of Clinical Oncology (ASCO) annual meeting.
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind study of oral ridaforolimus administered at 40 mg/day (five of seven days per week) in patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy. Oral ridaforolimus was granted a Special Protocol Assessment (SPA) by the FDA for the SUCCEED trial.
Based on 552 progression-free survival (PFS) events in 711 patients, (ridaforolimus (N=347), placebo (N=364) determined by an independent radiological review committee, the study achieved its primary endpoint of improvement in PFS, with a statistically significant (p=0.0001) 28 percent reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo (hazard ratio=0.72).
Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group. Furthermore, based on the full analysis of PFS determined by investigator assessment, there was a statistically significant (p<0.0001) 31 percent reduction by ridaforolimus in the risk of progression or death compared to placebo (hazard ratio=0.69). In the investigator assessment analysis, median PFS was 22.4 weeks for those treated with ridaforolimus compared to 14.7 weeks in the placebo group [4
EU WITHDRAWAL IN NOV 2012
Merck, known as MSD outside the U.S. and Canada, announced today that it has formally notified the European Medicines Agency (EMA) of Merck's decision to withdraw the Marketing Authorisation Application (MAA) for ridaforolimus.
The application for Marketing Authorisation for ridaforolimus was accepted by the EMA in August 2011. At the time of the withdrawal it was under review by the Agency’s Committee for Medicinal Products for Human Use (CHMP). In its letter to the EMA, Merck said that the withdrawal of ridaforolimus was based on the provisional view of the CHMP that the data available to date and provided in the Marketing Authorisation Application were not sufficient to permit licensure of ridaforolimus in the European Union for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor.
Although the application for these uses was withdrawn, Merck is studying ridaforolimus in combination with other drugs in other tumor types. The withdrawal of the European application of ridaforolimus for the maintenance treatment of patients with soft tissue sarcoma or primary malignant bone tumor does not change Merck’s commitment to the ongoing clinical trials with ridaforolimus.
Ridaforolimus
Description
42-(dimethylphosphinate) Rapamycin (Ridaforolimus) represented by the following formula I:
2. Description of RelatedArt
The mammalian target of Rapamycin (mTOR) is known as a mechanistic target of Rapamycin (H), which is found in the studies of Rapamycin. On the other hand, 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) is a derivative of Rapamycin (II), which is also a kind of mTOR inhibitor. Ridaforolimus (I) can inhibit cell division and possibly lead to tumor cell death. Hence, there are many studies related to solid tumor treatments and blood cancer treatments using Ridaforolimus (I). In addition, in 2011, Merck also applied a certification of this compound against soft tissue and bone cancer.
U.S. Pat. No. 7,091,213 discloses a process for preparing 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I), and the process thereof is shown in the following Scheme I.
In this process, a solution of Rapamycin (II) in dichloromethane (DCM) was respectively added with 2,6-di-tert-butyl-4-methylpyridine or 3,5-lutidine as a base, and followed by the addition of a solution of dimethylphosphinic chloride (DMP-Cl) to perform a phosphorylation reaction at 0° C., under a stream of N2(g). The crude product was purified by flash chromatography (eluted with MeOH/DCM/EtOAc/hexane=1:10:3:3) to provide 42-(dimethyl- phosphinate) Rapamycin (Ridaforolimus) (I), which is a phosphorylated compound at 42-hydroxyl position of Rapamycin (II). In addition, this patent also disclosed a side product of 31,42-bis(dimethyl phosphinate) Rapamycin (III), which is a phosphorylated compound at both 31- hydroxyl position and 42- hydroxyl position of Rapamycin (II).
.......................
SYNTHESIS
Some additional transformations of potential interest to the practitioner are shown below, including the preparation of reagents for generating the described C-43 phosphorus-containing rapalogs:
Preparation of Diakyl/diaryl Chlorophoshates
Preparation of Alkyl Halide Phosphonates
Illustrative routes for using the foregoing sorts of reagents to prepare certain rapalogs of this invention are shown below.
The synthesis of compounds of this invention often involves preparation of an activated form of the desired moiety “J”, such as a phosphoryl chloride as shown above (e.g. (R)(RO)P—Cl or RR′P(═O)—Cl, etc), and reaction of that reagent with rapamycin (or the appropriate rapalog) under conditions yielding the desired product, which may then be recovered from residual reactants and any undesired side products. Protecting groups may be chosen, added and removed as appropriate using conventional methods and materials.
Purification of Compounds of the Invention
A variety of materials and methods for purifying rapamycin and various rapalogs have been reported in the scientific and patent literatures and may be adapted to purification of the rapalogs disclosed herein. Flash chromatography using a BIOTAGE prepacked cartridge system has been particularly effective. A typical protocol is disclosed in the Examples which follow.
Physicochemical Characterization of Compounds of the Invention
The identity, purity and chemical/physical properties of the rapalogs may be determined or confirmed using known methods and materials, including HPLC, mass spectral analysis, X ray crystallography and NMR spectroscopy. High resolution 1D 1H and 31P NMR spectra acquired using a typical relaxation delay of 3 seconds have proved useful, as has reverse phase HPLC analysis (analytical column, 3 micron particle size, 120 ansgstrom pore size, thermostatted to 50° C. with a mobile phase of 50% acetonitrile, 5% methanol and 45% water (all % s by volume), for example, in an isocratic elution system, with elution of product and impurity peaks followed by UV detection at 280 nanometers). Normal phase HPLC may also be used, especially to evaluate the level of residual rapamycin or rapalog by-products. The presence of residual solvent, heavy metals, moisture and bioburden may be assessed using conventional methods.
Example 9
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
Dimethyl-phosphinic Acid C-43 Rapamycin Ester
To a cooled (0° C.) solution of rapamycin (0.1 g, 0.109 mmol) in 1.8 mL of dichloromethane was added 0.168 g (0.82 mmol) of 2,6-di-t-butyl-4-methyl pyridine, under a stream of N2, followed immediately by a solution of dimethylphosphinic chloride (0.062 g, 0.547 mmol) in 0.2 mL of dichloromethane. The slightly yellow reaction solution was stirred at 0° C., under an atmosphere of N2, for 3.5 h (reaction monitored by TLC). The cold (0° C.) reaction solution was diluted with ˜20 mL EtOAc then transferred to a separatory funnel containing EtOAc (150 mL) and saturated NaHCO3 (100 mL). Upon removing the aqueous layer, the organic layer was washed successively with ice cold 1N HCl (1×100 mL), saturated NaHCO3 (1×100 mL), and brine (1×100 mL), then dried over MgSO4 and concentrated. The crude product was purified by silica gel flash chromatography (eluted with 1:10:3:3 MeOH/DCM/EtOAc/hexane) to provide 0.092 g of a white solid:
1H NMR (300 MHz, CDCl3) d 4.18 (m, 1H), 4.10 (m, 1H), 3.05 (m, 1H), 1.51 (m, 6H);
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
31P NMR (121 MHz, CDCl3) d 53.6; 1013 m/z (M+Na).
Example 9
Alternative Synthesis
Rapamycin and dichloromethane are charged into a nitrogen-purged reaction flask. The stirred solution is cooled to approximately 0° C. (an external temperature of −5±5° C. is maintained throughout the reaction). A solution of dimethylphosphinic chloride (2.0 molar equivalents) in dichloromethane is then added over a period of approximately 8–13 minutes.
This is followed immediately by the addition of a solution of 3,5-lutidine (2.2 molar equivalents) in dichloromethane over a period of approximately 15–20 minutes. Throughout both additions, the internal temperature of the reaction sssstays below 0° C. The cooled reaction solution is stirred for 1 hour and then transferred, while still cold, to an extractor containing saturated aqueous NaHCO3 and methyl-t-butyl ether (MTBE), ethyl acetate or diethyl ether. In-process samples are removed at 30 and 60 minute time points.
Samples are prepared in a similar fashion to that described for the reaction workup. Reaction progress is monitored by TLC (1:10:3:3 MeOH/DCM/EtOAc/hexanes) and reverse-phase HPLC analyses. The isolated organic layer is successively washed with ice cold 1N HCl, saturated aqueous NaHCO3 (2×), saturated aqueous NaCl, and dried over sodium sulfate. Upon filtration and solvent removal, the residue undergoes solvent exchange with acetone followed by concentration in vacuo to provide crude product, which may be analyzed for purity by normal- and reversed-phase HPLC.
.........................
SYNTHESIS
The process of the present invention is shown in the following Scheme II.
EXAMPLE 7
Preparation of 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I)
42-(dimethylphosphinate)-31-triethylsilylether Rapamycin (VI-b) (2.312 g, available from 1.945 mmole of Rapamycin -28-triethylsilylether) and tetrahydrofuran (60 mL) was placed into a flask, and the resulting solution was cooled to 0˜−5° C. Next, a sulthric acid solution (2 N, 6 mL) was slowly added into the resulting solution dropwise. When the 42-(dimethylphosphinate)-31-triethylsilylether Rapamycin (VI-b) was less than 2%, ethyl acetate (1000 mL) was added into the resulting solution. Then, the organic layer was successively washed with a NaCl saturated solution (300 mL), a NaHCO3saturated solution (200 mL) and a NaCl saturated solution (200 mL), dried over anhydrous sodium sulfate and concentrated to obtain a crude product of 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) (2.341 g). The crude product was then purified by Licrhoprep RP-18 silica gel chromatography (eluted with acetonitrile: 0.02 M ammonium formate solution=6:4, wherein the pH of the ammonium formate solution was adjusted to 4.0 with formic acid), extracted with ethyl acetate, concentrated and dried to obtain a white foam solid 42-(dimethylphosphinate) Rapamycin (Ridaforolimus) (I) (1.840 g, purity=99.48%). The yield thereof was 95.55% based on 2.0 g of 31-triethylsilyl ether Rapamycin.
1H-NMR(400 MHz, CDCl3)d 4.18(m, 1H), 4.10(m, 1H), 3.05(m, 1H),1.51(m, 6H); 31P-NMR(161 MHz, CDCl3)d 53.33; 1012.6 m/z [M+Na]+.
- “ARIAD Reports First Quarter 2009 Development Progress and Financial Results- Ridaforolimus New USAN Name to Replace Deforolimus”. ARIAD Pharmaceuticals. 2009. Retrieved 2009-05-07.
- “ARIAD – News release”. Phx.corporate-ir.net. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-03-17. Retrieved 2012-10-07.
- “ARIAD – News release”. Phx.corporate-ir.net. 2011-06-06. Retrieved 2012-10-07.
| US8216571 | 7-11-2012 | FULLY HUMAN ANTI-VEGF ANTIBODIES AND METHODS OF USING |
| US2011262525 | 10-28-2011 | METHODS OF TREATMENT |
| US2011014117 | 1-21-2011 | ANTI-IGF1R |
| US2007004767 | 1-5-2007 | Methods for treating neurofibromatosis 1 |
| US2004073024 | 4-16-2004 | Phosphorus-containing compounds and uses thereof |
Share this:
.............................................
5 ZOTAROLIMUS
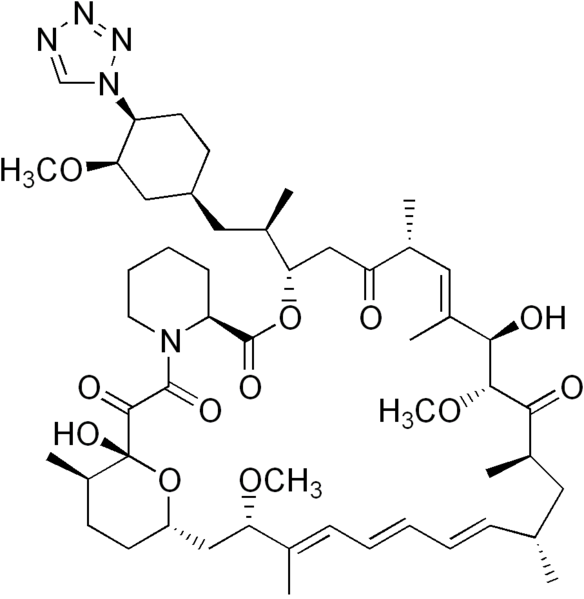


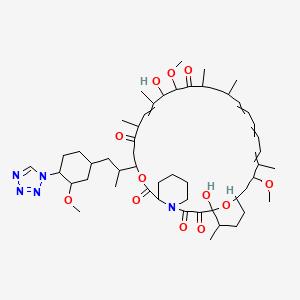












The FDA has approved the zotarolimus-eluting stent (Medtronic).
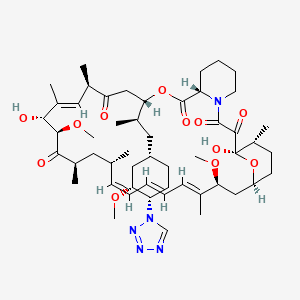
8 TACROLIMUS
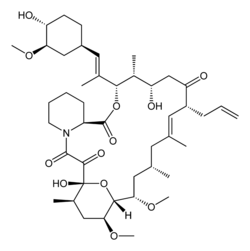
 CTK8E6891, 109581-93-3 MONOHYDRATE TACROLIMUS
CTK8E6891, 109581-93-3 MONOHYDRATE TACROLIMUS




 Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from 'Tsukuba macrolide immunosuppressant'.
Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from 'Tsukuba macrolide immunosuppressant'.




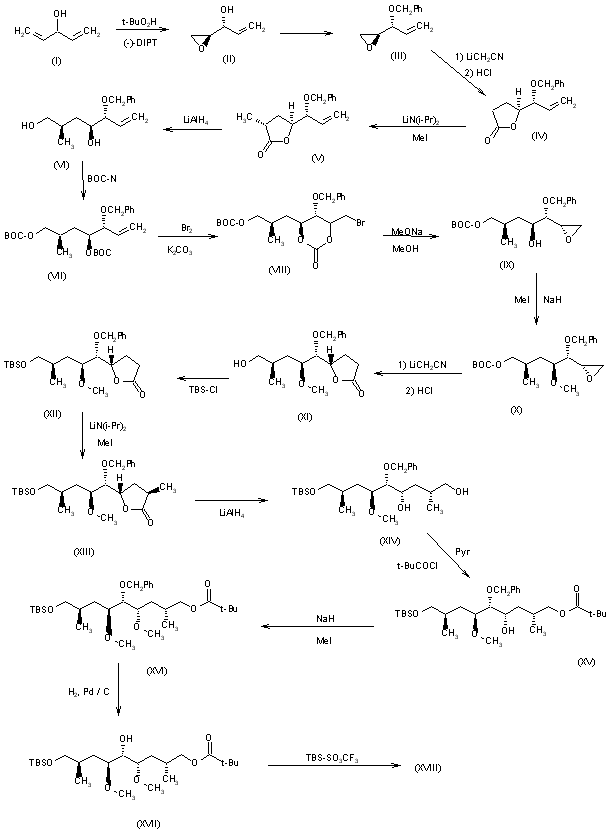

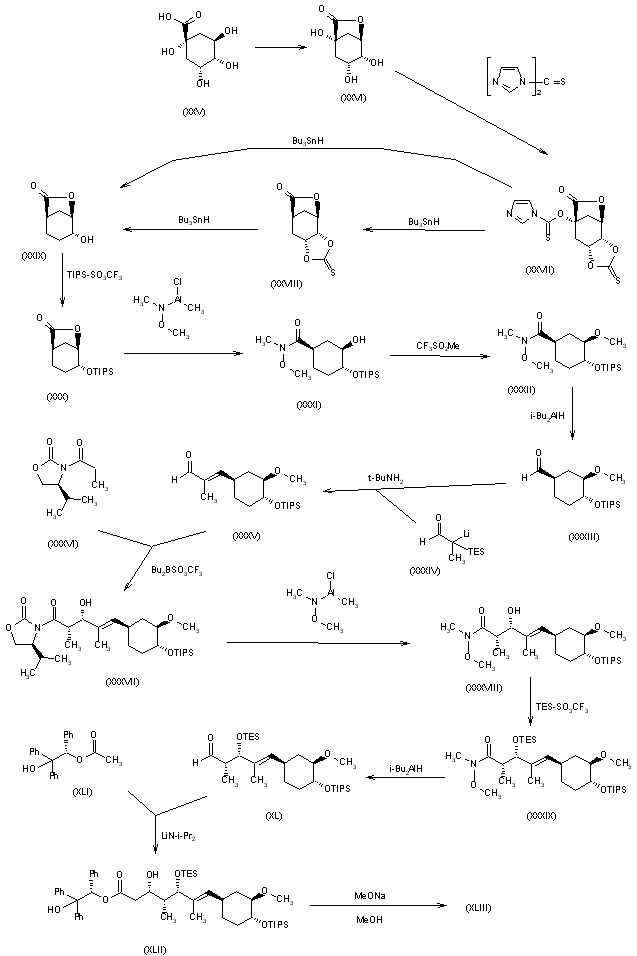

Zotarolimus
221877-54-9 CAS
A 179578; ABT 578; Resolute; 42-(1-Tetrazolyl)rapamycin; (42S)-42-Deoxy-42-(1H-tetrazol-1-yl)rapamycin
| Molecular Formula: C52H79N5O12 |
| Molecular Weight: 966.21 |
A tetrazole-containing Rapamycin analog as immunomodulator and useful in the treatment of restenosis and immune and autoimmune diseases.
(3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,27-dihydroxy-10,21-dimethoxy-3-{(1R)-2-[(1S,3R,4S)-3-methoxy-4-(1H-tetrazol-1-yl)cyclohexyl]-1-methylethyl)-6,8,12,14,20,26-hexamethyl-4,9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-heptadecahydro-3H-23,27-epoxypyrido[2,1-c][1,4]oxazacyclohentriacontine-1,5,11,28,29(6H,31H)-pentone, cas no 221877-54-9
zotarolimus in U.S. Patent Nos. 6,015,815 and 6,329,386 , and PCT Application No. WO 1999/015530
Zotarolimus (INN, codenamed ABT-578) is an immunosuppressant. It is a semi-synthetic derivative of rapamycin. It was designed for use in stents with phosphorylcholine as a carrier. Coronary stents reduce early complications and improve late clinical outcomes in patients needing interventional cardiology.[1] The first human coronary stent implantation was first performed in 1986 by Puel et al.[1][2] However, there are complications associated with stent use, development of thrombosis which impedes the efficiency of coronary stents, haemorrhagic and restenosis complications are problems associated with stents.[1]
These complications have prompted the development of drug-eluting stents. Stents are bound by a membrane consisting of polymers which not only slowly release zotarolimus and its derivatives into the surrounding tissues but also do not invoke an inflammatory response by the body.
Medtronic are using zotarolimus as the anti-proliferative agent in the polymer coating of their Endeavor and Resolute products.[3]
The inherent growth inhibitory properties of many anti-cancer agents make these drugs ideal candidates for the prevention of restenosis. However, these same properties are often associated with cytotoxicity at doses which block cell proliferation. Therefore, the unique cytostatic nature of the immunosuppressant rapamycin was the basis for the development of zotarolimus by Johnson and Johnson. Rapamycin was originally approved for the prevention of renal transplant rejection in 1999. More recently, Abbott Laboratories developed a compound from the same class, zotarolimus (formerly ABT-578), as the first cytostatic agent to be used solely for delivery from drug-eluting stents to prevent restenosis.[4]
Drug-eluting stents
Drug-eluting stents have revolutionized the field of interventional cardiology and have provided a significant innovation for preventing coronary artery restenosis. Polymer coatings that deliver anti-proliferative drugs to the vessel wall are key components of these revolutionary medical devices. The development of stents which elute the potent anti-proliferative agent, zotarolimus, from a synthetic phosphorylcholine-based polymer known for its biocompatible profile. Zotarolimus is the first drug developed specifically for local delivery from stents for the prevention of restenosis and has been tested extensively to support this indication. Clinical experience with the PC polymer is also extensive, since more than 120,000 patients have been implanted to date with stents containing this non-thrombogenic coating.[4]
Structure and properties
Zotarolimus is a analog made by substituting a tetrazole ring in place of the native hydroxyl group at position 42 in rapamycin that is isolated and purified as a natural product from fermentation. This site of modification was found to be the most tolerant position to introduce novel structural changes without impairing biologic activity. The compound is extremely lipophilic, with a very high octanol:water partition coefficient, and therefore has limited water solubility. These properties are highly advantageous for designing a drug-loaded stent containing zotarolimus in order to obtain a slow sustained release of drug from the stent directly into the wall of coronary vessels. The poor water solubility prevents rapid release into the circulation, since elution of drug from the stent will be partly dissolution rate-limited. The slow rate of release and subsequent diffusion of the molecule facilitates the maintenance of therapeutic drug levels eluting from the stent. In addition, its lipophilic character favors crossing cell membranes to inhibit neointimal proliferation of target tissue. The octanol:water partition coefficients of a number of compounds, recently obtained in a comparative study, indicate that zotarolimus is the most lipophilic of all DES drugs [4]
Stents are used to treat serious decreases in vessel or duct diameter due to a variety of diseases and conditions, especially atherosclerotic diseases, and are often used after angioplasty. While frequently used in arteries, stents are also used in other structures, including veins, bile ducts, esophagus, trachea, large bronchi, ureters, and urethras. Stents are the innovation of the English dentist Charles Stent (1845-1901).
While effective in treating deleterious lumen narrowing, vascular stents in an instance of medical irony, also risk re-creating the condition that they were used to treat. Stents can incur the development of thick endothelial tissue inside the lumen—the neointima. While the degree of development varies, the neointima can grow to occlude the vessel lumen, a type of restenosis.
Previous Syntheses of Zotarolimus
Mollison presented several methods to generate zotarolimus from sirolimus (Mollison, 2000). For example, C-40 hydroxyl of sirolimus is activated with the formation of triflate, and the triflate is then purified by column chromatography. During triflate purification, some of the activated intermediate reverts to sirolimus and its epimer, epi-sirolimus, due to presence of the water during chromatography. The purified triflate is then reacted in a second step with tetrazole to produce the 40-epi-tetrazole derivative of sirolimus, that is, zotarolimus. The crude product is then purified by column chromatography. However, even with this purification, the end product could contain sirolimus and epi-sirolimus impurities.
ISOMERS
ABT-578 [40-epi-(1-tetrazolyl)-rapamycin], known better today as zotarolimus, is a semi-synthetic macrolide triene antibiotic derived from rapamycin. Zotarolimus' structure is shown in Formula D.
...........................
zotarolimus having one of the following structures:
A representative procedure is shown in Scheme 1.
As shown in Scheme 1, conversion of the C-42 hydroxyl of rapamycin to a trifluoromethanesulfonate or fluorosulfonate leaving group provided A. Displacement of the leaving group with tetrazole in the presence of a hindered, non-nucleophilic base, such as 2,6-lutidine, or, preferably, diisopropylethyl amine provided epimers B and C, which were separated and purified by flash column chromatography.
Synthetic Methods
The foregoing may be better understood by reference to the following examples which illustrate the methods by which the compounds of the invention may be prepared and are not intended to limit the scope of the invention as defined in the appended claims.
Example 1 42-Epi-(tetrazolyl)-rapamycin (less polar isomer) Example 1AA solution of rapamycin (100 mg, 0.11 mmol) in dichloromethane (0.6 mL) at −78° C. under a nitrogen atmosphere was treated sequentially with 2,6-lutidine (53 uL, 0.46 mmol, 4.3 eq.) and trifluoromethanesulfonic anhydride (37 uL, 0.22 mmol), and stirred thereafter for 15 minutes, warmed to room temperature and eluted through a pad of silica gel (6 mL) with diethyl ether. Fractions containing the triflate were pooled and concentrated to provide the designated compound as an amber foam.
Example 1B 42-Epi-(tetrazolyl)-rapamycin (less polar isomer)A solution of Example 1A in isopropyl acetate (0.3 mL) was treated sequentially with diisopropylethylamine (87 L, 0.5 mmol) and 1H-tetrazole (35 mg, 0.5 mmol), and thereafter stirred for 18 hours. This mixture was partitioned between water (10 mL) and ether (10 mL). The organics were washed with brine (10 mL) and dried (Na2SO4). Concentration of the organics provided a sticky yellow solid which was purified by chromatography on silica gel (3.5 g, 70-230 mesh) eluting with hexane (10 mL), hexane:ether (4:1(10 mL), 3:1(10 mL), 2:1(10 mL), 1:1(10 mL)), ether (30 mL), hexane:acetone (1:1(30 mL)). One of the isomers was collected in the ether fractions.
MS (ESI) m/e 966 (M)−;
Example 2 42-Epi-(tetrazolyl)-rapamycin (more polar isomer) Example 2A 42-Epi-(tetrazolyl)-rapamycin (more polar isomer)Collection of the slower moving band from the chromatography column using the hexane:acetone (1:1) mobile phase in Example 1B provided the designated compound.
MS (ESI) m/e 966 (M)−.
..........................................................
sirolimus (commercially available or produced as described ((Paiva et al., 1991; Sehgal et al., 1975; Vezina et al., 1975) is dissolved in DCM:toluene (such as 1:2) 100. The reaction mixture is concentrated to dryness 105, and the azeo-drying process 105 is repeated 1-5 times more, more preferably 2-4 times, most preferably twice, preferably with DCM:toluene. The resulting foamy solid is dissolved in IPAc 110, and then 2,6-Lutidine is added 115. The solution is cooled to −30° C. 115. Triflic anhydride is then slowly added to the solution 115. After stirring the reaction mixture, the solution is filtered under nitrogen. The recovered salts 120 are washed with IPAc 125.
To the salts is added 1-H-tetrazole and DIEA 130. The reaction mixture is stirred at room temperature (e.g., 22-25° C.) 135and then concentrated. The crude reaction mixture is purified, using for example, a silica gel column and using, e.g., 1:1 THF:heptane to elute 140. The fractions are monitored for the N-1 isomer (which elutes more slowly than the N-2 isomer), pooled and concentrated, forming an oil. The oil is dissolved in minimum DCM and the solution loaded on a silica gel column packed in, for example, 65:35 heptane:acetone 145. The column is eluted with, for example, 65:35 heptane:acetone, the fractions monitored for the pure product, pooled and concentrated 150.
The purified product is then dissolved in t-BME, and then n-heptane is slowly added to form a precipitate while vigorously stirring the solution 150. The precipitated solids are stirred at 5-10° C., filtered, washed again with heptane, and dried on the funnel with nitrogen. The product is dissolved in acetone and treated with BHT 155. The solution is concentrated, dissolved in acetone, and then concentrated to dryness. The product is then dried under vacuum at 47° C. 160.
EXAMPLES
Example 1 Dichloromethane-Toluene Isopropylacetate One-Pot Process with Filtration (1)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane, toluene and isopropylacetate; the preparation was then purified, concentrated, and dried. The purified product was then characterized by its 1H, 13C NMR resonances from COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 25 ml) and toluene (50 ml). The reaction mixture was concentrated to dryness. This azeo-drying process was repeated twice with DCM/toluene. The foamy solid was dissolved in isopropylacetate (IPAc, 65 ml), and 2,6-Lutidine (3.2 ml) was added. The solution was cooled to −30° C. acetonitrile-dry ice bath, and triflic anhydride (2.8 ml) was added slowly in 10 minutes. The reaction mixture was stirred for 30 minutes, and then filtered under nitrogen atmosphere. The salts were washed with IPAc (10 ml). 1-H-tetrazole (2.3 g), followed by diisopropylethylamine (DIEA, 7.4 ml) were added. The reaction mixture was stirred for 6 hours at room temperature, and then concentrated. The crude reaction mixture was purified on a silica gel column (350 g) eluting with 1:1 THF/heptane. The fractions containing product that eluted later (predominantly N-1 isomer) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column packed in 65:35 heptane:acetone. The column was eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was then dissolved in t-butylmethyl ether (t-BME, 13.5 g), and n-heptane (53 g) was added slowly with vigorous stirring. The precipitated solids were stirred at 5-10° C. for 2 hours, filtered, washed with heptane and dried on the funnel with nitrogen to give 3.2 g wet product. The solids (1.0 g) were dissolved in acetone (10 ml) and treated with 2,6-di-tert-butyl-4-ethylphenol (DEP, 0.2%). The solution was concentrated, dissolved in acetone (10 ml) and concentrated to dryness. The product was dried under vacuum for 18 hours at 47° C., yielding 0.83 g of zotarolimus. The product was characterized by its 1H, 13C NMR resonances from its COSY, ROESY, TOCSY, HSQC, and HMBC spectra.
1H-NMR (DMSO-d6, position in bracket): ppm 0.73 (Me, 43); 0.81 (Me, 49); 0.84 (Me, 46); 0.89 (Me, 48); 0.98 (Me, 45); 1.41, 1.05 (CH2, 24); 1.18, 1.10 (CH2, 36); 1.52 (CH, 37); 1.53 (CH2, 12 & 42); 1.59, 1.30 (CH2, 5); 1.41, 1.67 (CH2, 4); 1.11, 1.73 (CH2, 38); 1.21, 1.83 (CH2, 15); 1.21, 1.83 (CH2, 13); 1.62 (Me, 44); 1.73 (Me, 47); 1.76 (CH, 35); 1.60, 2.09 (CH2, 3); 1.93, 2.21 (CH2, 41); 2.05 (CH, 11); 2.22 (CH, 23); 2.47 (CH, 25); 2.40, 2.77 (CH2, 33); 3.06 (OCH3, 50); 3.16 (OCH3, 51); 3.22, 3.44 (CH2, 6); 3.29 (OCH2, 52); 3.29 (CH, 31); 3.60 (CH, 39), 3.62 (CH, 16); 3.89 (CH, 27); 4.01 (CH, 14); 4.02 (CH, 28); 4.95 (CH, 2); 5.02 (CH, 34); 5.10 (═CH, 30); 5.17 (CH, 40); 5.24 (OH, 28); 5.46 (═CH, 22); 6.09 (═CH, 18); 6.15 (═CH, 21); 6.21 (═CH, 20); 6.42 (═CH, 19); 6.42 (OH, 10), 9.30 (CH, 53).
13C NMR (DMSO-d6, position in bracket): ppm 10.4 (Me, 44); 13.1 (Me, 47); 13.6 (Me, 46); 14.5 (Me, 49); 15.5 (Me, 43 & 48); 20.3 (CH2, 4); 21.6 (Me, 45); 24.4 (CH2, 4); 26.2 (CH2, 12); 26.4 (CH2, 3); 26.8 (CH2, 41); 27.2 (CH2, 42); 29.6 (CH2, 13); 31.6 (CH2, 38), 31.7 (CH, 37); 32.9 (CH, 35); 34.8 (CH, 11); 35.2 (CH, 23); 38.2 (CH2, 36); 39.1 (CH, 25); 39.4 (CH2, 33); 39.6 (CH2, 24), 40.0 (CH2, 15); 43.4 (CH2, 6); 45.2 (CH, 31); 50.6 (CH, 2); 55.4 (OCH3, 50); 55.8 (OCH3, 52); 57.0 (OCH3, 52); 55.9 (CH, 40); 66.2 (CH, 14); 73.4 (CH, 34); 75.6 (CH, 28); 77.4 (CH, 39); 82.3 (CH, 16); 85.7 (CH, 27); 99.0 (CH, 10); 125.3 (═CH, 30); 127.0 (═CH, 18 & 19); 130.4 (═CH, 21); 132.2 (═CH, 20); 137.2 (═CMe, 29); 137.7 (═CMe, 17); 139.2 (═CH, 22); 144.6 (CH, 53); 167.0 (C═O, 8); 169.1 (C═O, 1); 199.0 (C═O, 9); 207.5 (C═O, 32); 210.7 (C═O, 26).
Example 2 Dichloromethane-Isopropylacetate One-Pot Process (2)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane and isopropylacetate. The compound was then purified, concentrated, and dried.
Rapamycin (10 g) was dissolved in dichloromethane (DCM, 100 g). 2,6-Lutidine (2.92 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (4.62 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then warmed to 10° C. within 15 minutes. The reaction solution was then concentrated. The residue was dissolved in IPAc (55 g). 1-H-tetrazole (2.68 g), followed by diisopropylethylamine (DIEA, 7.08 g) were then added. The reaction mixture was stirred for 6 hours at room temperature and then concentrated. The crude reaction mixture was purified on a silica gel column (360 g), eluting with 1:1 THF:heptane. The fractions containing product that eluted later (principally N-1) were collected and concentrated. The concentrated oil was dissolved in minimum DCM and loaded on a silica gel column (180 g) that was packed in 65:35 heptane:acetone. The column was then eluted with 65:35 heptane:acetone, and fractions containing pure product were concentrated.
The purified product was dissolved in t-butylmethyl ether (t-BME, 23 g) and added slowly to n-heptane (80 g) with vigorous stirring. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with heptane and dried on the funnel with nitrogen. BHT (0.015 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone two times (20 g), and concentrated each time to dryness. The product was then dried under vacuum for 18 h at not more than 50° C. to give 2.9 g of zotarolimus.
Example 3 Dichloromethane One Pot Process (3)
In this example, zotarolimus was prepared from rapamycin in a one-pot process using dichloromethane. The compound was then purified, concentrated, and dried as described in Example 2.
Rapamycin (7.5 g) was dissolved in DCM (30 g). 2,6-Lutidine (1.76 g) was added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (2.89 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then assayed for the presence of rapamycin to determine consumption in the reaction. 1-H-tetrazole (1.44 g), followed by DIEA (5.29 g) was added. The reaction mixture was stirred for 6 hours at room temperature, and then directly loaded on a silica gel (270 g) column prepared in 1:1 THF:n-heptane (v/v). The crude reaction mixture was purified with 1:1 THF:n-heptane. The fractions containing product that elute later were collected and concentrated. The concentrated solids were dissolved in minimum DCM and loaded on a silica gel column (135 g) packed in 70:30 n-heptane:acetone. The column was eluted with 70:30 n-heptane:acetone, and fractions containing pure product, as identified by thin-layer chromatography (TLC), were concentrated.
The purified product was dissolved in t-BME (9 g), and added slowly to n-heptane (36 g) with vigorous stirring at 10±10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.006 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g each) and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 2.5 g of zotarolimus.
The above process, when carried out with rapamycin presence of 2,6-di-tert-butylpyridine or 2,4,6-collidine (2,3,5-trimethylpyridine) as a non-nucleophilic in step 1a gave zotarolimus of acceptable purity, but a lower yield.
Example 4 High-Pressure Liquid Chromatography HPLC Purification of Zotarolimus Prepared by the One-Pot Synthesis Method
In this example, zotarolimus was made from rapamycin using a one-pot synthesis method of the invention (using DCM), and then subjected to an additional round of purification using HPLC.
Rapamycin (3.75 g) was dissolved in dichloromethane (DCM, 15 g). 2,6-Lutidine (0.88 g) was then added. The solution was cooled to −30° C. in acetonitrile-dry ice bath, and triflic anhydride (1.45 g) was added slowly in 10 minutes. The reaction mixture was stirred for 20 minutes, and then 1-H-tetrazole (0.72 g), followed by DIEA (2.65 g) was added. The reaction mixture was stirred for 6 hours at 25° C., and then directly loaded on a silica gel (115 g) column prepared in 70:30 n-heptane:acetone. The crude reaction mixture was purified with 70:30 n-heptane:acetone. The fractions containing product were collected, and concentrated.
The concentrated solids were dissolved in acetonitrile-water and loaded on a C-18 TechniKrom column (5 cm×25 cm), and eluted with 64:36 acetonitrile-water containing 0.1% BHT. Fractions were analyzed by reverse phase (RP)—HPLC, and product fractions pooled and concentrated to remove acetonitrile. The product was extracted with ethyl acetate or isopropyl acetate, dried (sodium sulfate) and concentrated.
The purified product was dissolved in t-BME (4.5 g), and added slowly to n-heptane (18 g) with vigorous stirring at −10° C. The precipitated solids were stirred at 5-10° C. for not longer than 1 hour, filtered, washed with n-heptane and dried on the funnel with nitrogen. BHT (0.005 g) was added to the solids. The solids were dissolved in acetone (20 g), passed through a filter, and concentrated. The residue was treated with acetone twice (20 g), and concentrated each time to dryness. The product was dried under vacuum for not longer than 18 hours at not more than 50° C. to give 1.2 g of high quality zotarolimus.
- Braunwald E, Zipes D, Libby P, ed. (2001). Heart diseases: a textbook of cardiovascular disease (6th ed.). Philadelphia: Saunders Elsevier.
- Sigwart, U; Puel, J; Mirkovitch, V; Joffre, F; Kappenberger, L (1987). "Intravascular stents to prevent occlusion and restenosis after transluminal angioplasty". The New England journal of medicine 316 (12): 701–6. doi:10.1056/NEJM198703193161201. PMID 2950322.
- "Medtronic Receives FDA Approval for Endeavor Zotarolimus-Eluting Coronary Stent System".
- Burke, Sandra E.; Kuntz, Richard E.; Schwartz, Lewis B. (2006). "Zotarolimus (ABT-578) eluting stents". Advanced Drug Delivery Reviews 58 (3): 437–46.doi:10.1016/j.addr.2006.01.021. PMID 16581153.
- Heitman, J; Movva, NR; Hall, MN (1991). "Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast". Science 253 (5022): 905–9. PMID 1715094.
The FDA has approved the zotarolimus-eluting stent (Medtronic).
| US8129127 | 3-7-2012 | ASSAY FOR IMMUNOSUPPRESSANT DRUGS |
| US8129521 | 3-7-2012 | ONE POT SYNTHESIS OF TETRAZOLE DERIVATIVES OF RAPAMYCIN |
| US2011171662 | 7-15-2011 | NON-DENATURING LYSIS REAGENT |
| US2011091997 | 4-22-2011 | IMMUNOSUPPRESSANT DRUG EXTRACTION REAGENT FOR IMMUNOASSAYS |
| US7914999 | 3-30-2011 | NON-DENATURING LYSIS REAGENT |
| US7820812 | 10-27-2010 | METHODS OF MANUFACTURING CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS |
| US7812032 | 10-13-2010 | CRYSTALLINE FORMS OF RAPAMYCIN ANALOGS |
| US7700614 | 4-21-2010 | One pot synthesis of tetrazole derivatives of rapamycin |
| US2009258054 | 10-16-2009 | Heparin Prodrugs and Drug Delivery Stents Formed Therefrom |
| US2009047323 | 2-20-2009 | Medical Devices Containing Rapamycin Analogs |
| US2009047324 | 2-20-2009 | Medical Devices Containing Rapamycin Analogs |
| US7455853 | 11-26-2008 | Medical devices containing rapamycin analogs |
| US2008287675 | 11-21-2008 | CASCADE SYSTEM |
| US2008213278 | 9-5-2008 | Method Of Treating Disorders Using Compositions Comprising Zotarolimus And Paclitaxel |
| US2008175884 | 7-25-2008 | Medical Devices Containing Rapamycin Analogs |
| US7399480 | 7-16-2008 | Methods of administering tetrazole-containing rapamycin analogs with other therapeutic substances using medical devices |
| US2008153790 | 6-27-2008 | Medical Devices Containing Rapamycin Analogs |
| US2006198867 | 9-8-2006 | COMPOSITIONS AND METHODS OF ADMINISTERING RAPAMYCIN ANALOGS USING MEDICAL DEVICES FOR LONG-TERM EFFICACY |
| WO2001087372A1 * | Apr 25, 2001 | Nov 22, 2001 | Cordis Corp | Drug combinations useful for prevention of restenosis |
| EP1826211A1 * | Feb 20, 2007 | Aug 29, 2007 | Cordis Corporation | Isomers and 42-Epimers of rapamycin alkyl ether analogs, methods of making and using the same |
| US5151413 * | Nov 6, 1991 | Sep 29, 1992 | American Home Products Corporation | Rapamycin acetals as immunosuppressant and antifungal agents |
| US5362718 * | Apr 18, 1994 | Nov 8, 1994 | American Home Products Corporation | Rapamycin hydroxyesters |
| US7193078 | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| US7220755 | Nov 12, 2003 | May 22, 2007 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US7279571 | Dec 1, 2005 | Oct 9, 2007 | Teva Gyógyszergyár Zártkörüen Müködö Részvénytársaság | Methods of preparing pimecrolimus |
| US7812155 | Nov 26, 2007 | Oct 12, 2010 | Terumo Kabushiki Kaisha | Process for preparing an o-alkylated rapamycin derivative and o-alkylated rapamycin derivative |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
| US20050101624 * | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20090209572 | Nov 19, 2008 | Aug 20, 2009 | Biotica Technology Limited | 36-Des(3-Methoxy-4-Hydroxycyclohexyl) 36-(3-Hydroxycycloheptyl) Derivatives of Rapamycin for the Treatment of Cancer and Other Disorders |
| US20100204466 | Feb 23, 2010 | Aug 12, 2010 | Abbott Laboratories | One pot synthesis of tetrazole derivatives of rapamycin |
| US20100249415 | Mar 29, 2010 | Sep 30, 2010 | Kwang-Chung Lee | Process for preparation of temsirolimus |
READ
ANONYMOUS: "Randomised comparison of zotarolimus eluting and sirolimus-eluting stents in patients with coronary artery disease (ENDEAVOUR III)" JOURNAL OF AMERICAN COLLEGE OF CARDIOLOGY, vol. 46, no. 11, 6 December 2005 (2005-12-06), pages CS5-CS6, XP009089338
...............................
6. BIOLIMUS

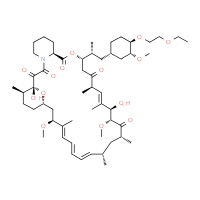











Umirolimus, Biolimus
Biosensors (Originator)
40 -O-[(2′-ethoxy) ethyl]rapamycin
(1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-12-{(2R)-1-[(1S,3R,4R)-4-(2-Ethoxyethoxy)-3-methoxycyclohexyl]-2-propanyl}-1,18-dihydroxy-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36 -dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentone
Umirolimus [INN], Umirolimus [USAN:INN], UNII-U36PGF65JH, TRM-986, 42-O-(2-ethoxyethyl) rapamycin, cas no 851536-75-9
Molecular Formula: C55H87NO14 Molecular Weight: 986.27758
Umirolimus (INN/USAN), is a macrocyclic lactone, a highly lipophilic derivative of sirolimus, an immunosuppressant. This drug is proprietary toBiosensors International, which uses it in its own drug-eluting stents, and licenses it to partners such as Terumo.
Biosensors had been developing a Biolimus A9(R)-eluting coronary stent for the treatment of arterial restenosis. No recent development has been reported. The product candidate was developed with BioMatrix(R), the company's low-profile, rapid-exchange delivery system. Specifically engineered for use on stents, Biolimus A9(R), a new rapamycin derivative, readily attaches to and enters smooth muscle cell membranes and binds to immunophilins inside the cell, causing cell cycle arrest at G0. Animal and in vitro studies suggest potency and safety profiles comparable to sirolimus.
Umirolimus inhibits T cell and smooth muscle cell proliferation, and was designed for use in drug eluting stents. This analog has a chemical modification at position 40 of the rapamycin ring. It has potent immunosuppressive properties that are similar to those of sirolimus, but the drug is more rapidly absorbed by the vessel wall, readily attaches and enters smooth muscle cell membranes causing cell cycle arrest at G0, and is comparable to sirolimus in terms of potency.
The key biologic event associated with the restenotic process is clearly the proliferation of smooth muscle cells in response to the expansion of a foreign body against the vessel wall. This proliferative response is initiated by the early expression of growth factors such as PDGF isoforms, bFGF, thrombin, which bind to cellular receptors.
However, the key to understanding the mechanism by which compounds like umirolimus inhibit cell proliferation is based on events which occur downstream of this growth factor binding. The signal transduction events which culminate in cell cycle arrest in the G1 phase are initiated as a result of ligand binding to an immunophilin known as FK binding protein-12. The FK designation was based on early studies conducted with tacrolimus, formerly known as FK-506, which binds this cytoplasmic protein with high affinity.
Subsequent investigations showed that rapamycin also binds to this intracellular target, forming an FKBP12–rapamycin complex which is not in itself inhibitory, but does have the capacity to block an integral protein kinase known as target of rapamycin (TOR). TOR was first discovered in yeast J.N. Heitman, N.R. Movva and M.N. Hall, Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast, Science 253 (1991), pp. 905–909. View Record in Scopus | Cited By in Scopus (434)and later identified in eukaryotic cells, where it was designated as mTOR, the mammalian target of rapamycin. The importance of mTOR is based on its ability to phosphorylate a number of key proteins, including those associated with protein synthesis (p70s6kinase) and initiation of translation (4E-BP1).
Of particular significance is the role that mTOR plays in the regulation of p27kip1, an inhibitor of cyclin-dependent kinases such as cdk2. The binding of agents like rapamycin and umirolimus to mTOR is thought to block mTOR's crucial role in these cellular events, resulting in arrest of the cell cycle, and ultimately, cell proliferation.
Introduction
It is known that Biolimus A9, a rapamycin derivative, is an immunosuppressant, and is also proven to have anti-tumor and anti-fungal effect.
Several prior arts had disclosed the improvements of the product yield of rapamycin derivatives. U.S. Pat. No. 7,193,078 to Isozaki et al. disclosed a process for producing Biolimus A9, giving an example to obtain a yield of 46% by reacting rapamycin with 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate) in an organic solvent.
However, the Isozaki's prior art still has the following drawbacks:
- 1. Even one example ever showed a 46% yield of Biolimus A9, it however just revealed a small-scale laboratory experiment with only one gram (1.09 mmol) of rapamycin and 1.95g (8.78 mmol) of 2-ethoxyethyl triflate. After amplifying or expanding the process to be larger scale, the yield will be remarkably reduced to thereby decrease the commercial or industrial value of this prior art (Note: The low yield after simulated process amplification will be hereinafter discussed in Examples 3, 4 of this application).
- 2. Even the reactant of 2-ethoxyethyl triflate is a compound with high activity, it is unstable and will be decomposed such as after being stored for one week at room temperature. Also, the triflate is not UV-absorbable and is therefore unsuitable for process tracking when proceeding the reaction. Such poor properties will affect the material storage, production scheduling and process tracking for commercially making the Biolimus A9.
sirolimus 42 - ether derivatives are a class of sirolimus derivative, is a new generation macrolide immunosuppressant and anticancer drugs. The compounds discovered by the Swiss company Sandoz, mainly applicable to organ transplant recipient's immune suppression and cancer. The synthesis of such substances currently on the patent literature have W09409010, CN102127092A and CN102268015A.
Patent Document W09409010 on Synthesis of this type of structure are used sirolimus protected materials in acidic or neutral reaction conditions, and then removing the protective group to obtain the target product. Such as 42-0 - (4 - hydroxymethyl) benzyl - sirolimus, the first synthesis of the formula V, and then removal of silyl ether protecting groups have the formula VI.
This synthetic method has several drawbacks: 1, the reaction reagent relatively difficult to obtain; 2, the intermediate prepared in the reaction yield is low;
3, sirolimus, structural part to participate in a two-step reaction, reduction reaction yield, costs.
CN102127092A mention a synthetic sirolimus 42 - ether derivative everolimus one way. This synthetic route similar to the W09409010 (route of reaction formula 1), but with silane reagents and reaction conditions are different.First reaction toluene as solvent, 50 ° C _60 ° C between the reaction and after-treatment of the intermediate after the first column chromatography, yield 32%.The second step in tetrahydrofuran as a solvent, the reaction overnight at 0 ° C, after treatment by a column chromatography to give the product, the yield was 66%, with a total yield of 21.1%.
Reaction Scheme I:
This method has the defects include: 1, the reaction reagent relatively difficult to obtain. 2, the structure part of sirolimus to attend two-step reaction, reduction reaction yield, costs. 3, the use of highly toxic solvents, are not suitable for practical application. 4, the reaction temperature is relatively harsh, difficult to control.
CN102268015A discloses a method for synthesizing everolimus. The first step to sirolimus or sirolimus derivatives as raw materials in -20 ° C was added dropwise trifluoromethanesulfonic anhydride and incubated for 3 hours, was isolated intermediates 02, the yield was 87.4% or 95.32%. The second step of the intermediate 02 with ethylene glycol mono-protected in 50 ° C reaction intermediate 03 was isolated in a yield of 79.0% or 76.78%. The third step of dilute hydrochloric acid was added dropwise at room temperature intermediate 03 was deprotected product everolimus. The total yield was 48.4% or 52.5%. See Reaction Scheme 2 synthetic route.
Reaction Scheme 2:
The method of the defects as follows: 1, to protection and deprotection of ethylene glycol. 2, the reaction steps excessive structural part to participate in the sirolimus-step reaction. 3, the reaction yield improved, but also greatly increased operating costs.
....................
.
Examples 5,42-0-(2_ ethoxy) ethyl - Synthesis of sirolimus
[0044] In the IOOml three-necked flask was added 2g of sirolimus, 2. 78g of 4_ dimethylaminopyridine, 5. 34g of chlorine acid glycol ester and 20ml of acetonitrile, 35 ° C The reaction was stirred 36 hours ended. The reaction solution was poured into an equal volume of saturated sodium bicarbonate solution and extracted with 5% potassium bisulfate solution was washed twice with a saturated sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated through the column. Silica gel column chromatography (EA: PE = I :20-2: 1), obtained by rotary evaporation 42-0 - (2 - ethoxy) ethyl - sirolimus I. 57g (yield: 73.3 %). HPLC analysis showed that: a purity of 88.2%.
............
A process for making Biolimus A9 represented by formula (1),
comprising reacting sirolimus of formula (2),
with
2-ethoxyethyl pentafluorobenzene sulfonate
under catalyzing by an organic base,
and in the presence of an organic solvent,
to undergo a nucleophilic substitution reaction to obtain Biolimus A9 of formula (1).
2. A process according to claim 1, wherein said 2-ethoxyethyl pentafluorobenzene sulfonate as used in the reaction is 1˜20 moles per mole of said sirolimus of formula (2).
the Biolimus A9 of the present invention will be presented as below-mentioned:
Biolimus A9
Biolimus A9
Reaction Parameters
Quantity of Alkylbenzene Sulfonate: 1˜20 equivalents, preferably being 5˜10 equivalents, per equivalent of sirolimus.
Reaction Temperature: 40˜80° C., preferably being 55˜65° C. Reaction Time: 12˜72 hours, preferably being 16˜30 hours.
After the reaction is completed, the rough product is collected, washed, dried and purified to obtain the Biolimus A9 of the present invention with high yield of 45%.
Since the product Biolimus A9 is a polyene macrolide, which is easily oxidized and decomposed during the storage or material handling.
Accordingly, a proper antioxidant may be homogeneously mixed with the Biolimus A9 to enhance the stability when stored or handled.
The proper antioxidants may be selected from: Butylatd hydroxytoluene (BHT), DL-α-tocopherol, propyl gallate, ascorbyl palmitate, 3-tert-butyl-4-hydroxyanisole, 2-tert-butyl-4-hydroxyanisole, and fumaric acid.
The Butylated hydroxytoluene (BHT) is the most preferable antioxidant adapted for use in the present invention.
The process for making Biolimus A9 in accordance with the present invention will be described in detail in view of the following examples:
EXAMPLE 1
A. Synthesis of 2-ethoxyethyl pentafluorobenzene sulfonate
In a reaction flask, 25 grams (93.8 mmol) of pentafluorobenzene sulfonyl chloride (or pentafluorobenzene sulfochloride) and 86 ml of tetrahydrofuran were added and nitrogen gas was filled into the flask.
The flask is then cooled to 0° C. and is dripped therein with 2-ethoxyethanol (8.5g, 94.5 mmol) and triethyl amine (15 g, 148.5 mmol). After dripping, the reaction solution is stirred for 30 minutes, and then filtered, concentrated and the residue is separated from the solution and further purified by silica gel column chromatography to obtain a colorless oily product of 2-ethoxyethyl pentafluorobenzene sulfonate (26.6 g, 83.1 mmol) having a yield of 88.6%.
B. Synthesis of Biolimus A9
In a reaction flask, 1 g (1.1 mmol) of sirolimus, 7.8 g (60.3 mmol) of ethyl di-isopropyl amine, 3.5 ml of methylene chloride and 2.8 g (8.7 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate as previously obtained were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 23 hours. It is cooled, and further added therein with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (100 ml) and saturated saline (100 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(0.49 g, 0.5 mmol) with a yield of 45.4%.
EXAMPLE 2 Process Amplification of Example 1B
In a reaction flask, 10 g (10.9 mmol) of sirolimus, 78 g (603.5 mmol) of ethyl di-isopropyl amine, 35 ml of methylene chloride and 28 g (87.4 mmol) of 2-ethoxyethyl pentrafluorobenzene sulfonate were added therein.
The reaction mixture in the flask was heated to 60° C. and agitated for 24 fours. It is cooled, and further added therein with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml ) under agitation.
Then, it is settled for separating the organic and aqueous layers. The organic layer is collected, and washed with pure water (500 ml) and saturated saline (400 ml). The washed organic liquid is then dried and concentrated. The residue is then separated from the liquid and further purified by silica gel column chromatography to obtain white solid product of Biolimus A9(4.8 g, 4.9 mmol) with a yield of 44.5%.
This example is a process amplification of the previous Example 1, Step B, by amplifying or expanding the quantity of each reactant for about 10 times of that of the Example 1 (of small scale).
By the way, the production yield (44.5%) of this Example is still as high as that of the previous Example 1 of small scale. It indicates that the reproducibility of high yield can still be obtained in accordance with the present invention even after process amplification, proving that the present invention is suitable for commercialization or mass production. The product may be further purified to obtain a high-purity final product of Biolimus A9 such as by middle-performance liquid chromatography or the like. The Biolimus A9 thus obtained is identified by the X-ray powder diffractogramm as shown in the single drawing FIGURE as attached herewith.
EXAMPLE 3 Comparative Example for Simulating the Process of the Prior Art of U.S. Pat. No. 7,193,078
A. Synthesis of 2-ethoxyethyl trifluoromethane sulfonate
In a reaction flask, 2-ethoxyethanol (10 g, 111 mmol), methylene chloride (177 ml) and 2,6-dimethyl pyridine (23.8 g, 222.3 mmol) were added into the flask, which is filled therein with nitrogen gas. It is cooled to 0° C. and added dropwise with trifluoromethane sulfonic acid anhydride (37.6 g, 133.4 mmol). After completing the dripping of said sulfonic acid anhydride, the reaction mixture is agitated for one hour and a saturated aqueous solution of ammonium chloride (20 ml) is added and further agitated for 10 minutes.
It is then settled for separating the layers. The organic layer is collected, and is respectively washed with aqueous solution of hydrochloric acid (1N, 100 ml), pure water (100 ml), saturated aqueous solution of sodium bicarbonate (100 ml) and saturated saline (100 ml). The washed organic layer is dried, concentrated and the residue is then separated and further purified with silica gel column chromatography to obtain the oily product of 22.5 g (101.3 mmol) of 2-ethoxyethyl trifluoromethane sulfonate (or 2-ethoxyethyl triflate), with a yield of 91.3%.
B. Synthesis of Biolimus A9
In a reaction flask, sirolimus (1 g, 1.1 mmol), ethyl di-isopropyl amine (7.8 g, 60.3 mmol), methylene chloride (3.5 ml) and 2-ethoxyethyl triflate (2.0 g, 8.8 mmol) as previously made in Example 3A were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (100 ml) and aqueous solution of hydrochloric acid (1N, 100 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (100 ml), saturated saline (80 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (0.48 g, 0.49 mmol), with a yield of 44.5%.
EXAMPLE 4 Comparative Example for Simulative Process Amplification of Example 3B
In a reaction flask, sirolimus (10 g, 10.9 mmol), ethyl di-isopropyl amine (78 g, 603.5 mmol), methylene chloride (35 ml) and 2-ethoxyethyl triflate (20 g, 88 mmol), each having a quantity about 10 times of that used in Example 3B, were added into the flask, which is filled with nitrogen gas. The reaction mixture is heated to 60° C. and is agitated for one hour and twenty minutes. Then, it is cooled, added with ethyl acetate (500 ml) and aqueous solution of hydrochloric acid (1N, 500 ml) and is further agitated. After agitation, it is settled for separating the layers. The organic layer is collected and respectively washed with pure water (500 ml), saturated saline (400 ml). The washed organic layer is dried and concentrated. The residue is then separated and purified by silica gel column chromatography to obtain white product of Biolimus A9 (2.9 g, 2.9 mmol), having a yield of 26.8% only.
Comparatively, via this process amplification, the yield of Biolimus A9 of the prior art is remarkably reduced in comparison with its small-scale production (Example 3B). Therefore, the prior art of U.S. Pat. No. 7,193,078 may be considered as a process especially suitable for small-scale production, such as a laboratory experiment, rather than a large-scale commercial or industrial production, which is thus inferior to this application, when compared with this application which has shown the high yields both in small-scale process (Example 1) and large-scale process (Example 2).
Accordingly, this application is more suitable for commercialization for mass production.
Moreover, the essential reactant of 2-ethoxyethyl triflate of the prior art (U.S. Pat. No. 7,193,078), even having high activity, is unstable because it will be decomposed into unknown compounds after one-week storage (by NMR spectrographic detection) as accompanied with physical change from its original colorless transparent liquid to a black viscous oily product, to thereby be inferior to this application because the 2-ethoxyethyl pentafluoro benzene sulfonate (which is obviously different from the 2-ethoxyethyl triflate as used in the prior art) of this application is still stable after one-week storage as aforementioned.
Furthermore, the 2-ethoxyethyl pentafluorobenzene sulfonate of this application may absorb ultra-violet rays to have a better tractability during the process proceeding than that of the 2-ethoxyethyl triflate (which is not UV-absorbable) of the prior art. So, this application is also beneficial for better production scheduling, reliable process tracking and efficient production management than the prior art.
So, this application is more suitable for commercial production even when considering the stability of product storage and improvement of process monitoring, control and management.
EXAMPLE 5
The Biolimus A9, as obtained from Example 2, is respectively added with anti-oxidant, namely Butylated Hydroxytoluene (or BHT), for 0.1%, 0.2, 0.5%, and 1% (w/w) based on 100% (wt) of Biolimus to enhance its stability at 40° C. by revealing a high yield of more than 99.4% even after six-week storage. Comparatively, a control test is provided by adding 0% of anti-oxidant (BHT) into Biolimus A9, resulting in a reduction of yield to be 69.7% after six-week storage. The yield data of different amounts of anti-oxidant as added into Biolimus A9 with respect to time lapse of weeks are summarized in Table 1 as below-mentioned.
...............................
The O-(2-ethoxyethyl)-rapamycin can be produced by reaction between rapamycin and 2-ethoxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride.
An example of the O-alkylrapamycin derivative (with R=hydroxyalkyl) is O-(2-hydroxyethyl)-rapamycin represented by the general formula 3 below.
The O-(2-hydroxyethyl)-rapamycin can be produced by reaction between rapamycin and t-butyldimethylsilyloxyethyl triflate in the presence of N,N-diisopropylethylamine in methylene chloride, followed by deprotecting of t-butyldimethylsilyl group.
EXAMPLES
The invention will be described with reference to the following examples, which demonstrate the efficient production of O-alkylrapamycin derivatives by the process of the present invention.
Example 1
- (1) Synthesis of 2-ethoxyethyl Triflate
In a round bottom flask containing a stirring bar was placed 9.0 g (100 mmol) of ethoxyethanol. The atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. The flask was given 160 mL of methylene chloride and 23.3 mL (120 mmol) of 2,6-lutidine. The flask cooled with ice was given dropwise 20.2 mL (120 mmol) of trifluoromethanesulfonic acid anhydride over 20 minutes. After stirring for 1 hour, the reaction liquid was mixed with 20 mL of saturated solution of ammonium chloride. The resulting mixture was washed sequentially with 1N hydrochloric acid (100 mL), deionized water (100 mL), saturated solution of sodium hydrogen carbonate (100 mL), and saturated aqueous solution of sodium chloride (100 mL). The organic layer was separated and dried with anhydrous sodium sulfate. With the sodium sulfate filtered off, the solution was concentrated under reduced pressure. The residue underwent silica gel chromatography. Thus there was obtained 15.03 g (67.6% yields) of 2-ethoxyethyl triflate from the fraction in eluate of 20% ethyl acetate-hexane.
- (2) Synthesis of 40-O-[(2′-ethoxy)ethyl]rapamycin
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the previously synthesized 2-ethoxyethyl triflate with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated by using a rotary evaporator. The concentrated solution was purified using a column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1 v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 494 mg (0.501 mmol) of the desired product (46% yields).
Example 2
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of chloroform for dissolution. To the flask was further added 10 mL (57.5 mmol) of N,N-diisopropylethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 451 mg (0.458 mmol) of the desired product (42% yields).
Example 3
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of methylene chloride for dissolution. To the flask was further added 8 mL (57.4 mmol) of triethylamine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica-gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 344 mg (0.349 mmol) of the desired product (32% yields).
Example 4
In 2 mL of methanol was dissolved 500 mg of the 40 -O-[(2′-ethoxy)ethyl]rapamycin which had been obtained in Example 1. The resulting solution was added dropwise to 20 mL of deionized water with stirring. The solids which had precipitated out were filtered off and washed with a small amount of water and finally dried under reduced pressure at 40° C. for more than 10 hours. Thus there was obtained 483 mg of white powder.
This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40 -O-[(2′-ethoxy) ethyl]rapamycin represented by the general formula 4.
Comparative Example
A sample of 40-O-[(2′-ethoxy)ethyl]rapamycin was synthesized by the process disclosed in WO94/09010 official gazette so as to evaluate yields.
In a round bottom flask containing a stirring bar was placed 1.0 g (1.09 mmol) of rapamycin. With the flask connected to a condenser, the atmosphere in the flask was replaced with nitrogen by using a nitrogen bubbler. To the flask was added 3.5 mL of toluene for dissolution. To the flask was further added 467 mg (4.36 mmol) of 2,6-lutidine and 1.95 g (8.78 mmol) of the 2-ethoxyethyl triflate previously synthesized in Example 1 with vigorous stirring. With the flask kept at 60° C. in an oil bath, the content was stirred for 1 hour and 20 minutes. The resulting mixture was diluted with 100 mL of ethyl acetate and washed sequentially with 100 mL of 1N hydrochloric acid, 100 mL of deionized water, and 80 mL of saturated aqueous solution of sodium chloride. The ethyl acetate phase was separated and then stirred with 5 g of anhydrous sodium sulfate for 20 minutes. With the sodium sulfate filtered off, the solution was concentrated using a rotary evaporator. The concentrated solution was purified using column chromatograph, with a silica gel bed measuring 4 cm in diameter and 26 cm high. Elution was accomplished by flowing sequentially 300 mL of ethyl acetate/n-hexane (1:1, v/v), 1000 mL of ethyl acetate/n-hexane (3:2, v/v), and 300 mL of ethyl acetate/n-hexane (7:3, v/v). The desired fraction was collected and concentrated, and the concentrate was vacuum dried in a desiccator. Thus there was obtained 247 mg (0.251 mmol) of the desired product (23% yields).
NMR
This product gave an NMR chart as shown in FIG. 1. This NMR chart indicates the structure of 40-O-[(2′-ethoxy)ethyl]rapamycin represented by the general formula 4.
| US20050101624 | Nov 12, 2003 | May 12, 2005 | Betts Ronald E. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| US20050131008 | Nov 12, 2004 | Jun 16, 2005 | Sun Biomedical, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| WO1994009010A1 | Sep 24, 1993 | Apr 28, 1994 | Sandoz Ag | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US7193078 * | Mar 1, 2005 | Mar 20, 2007 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| WO2012017449A1 | Aug 2, 2011 | Feb 9, 2012 | Meril Life Sciences Pvt. Ltd | Process for preparation of novel 42-0-(heteroalkoxyalkyl) rapamycin compounds with anti-proliferative properties" |
| US7872122 | May 8, 2009 | Jan 18, 2011 | Chunghwa Chemical Synthesis & Biotech Co., Ltd. | Process for making Biolimus A9 |
............................
7 PIMECROLIMUS







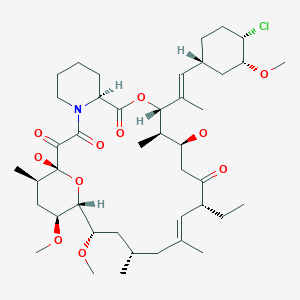






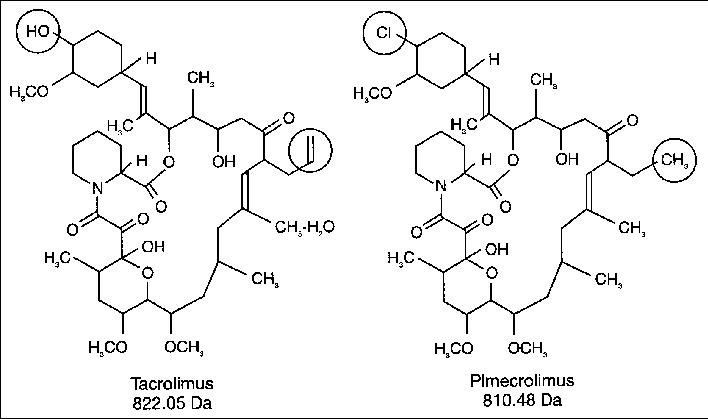
Pimecrolimus
137071-32-0 cas
(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)- 3-{(E)-2-[(1R,3R,4S)-4-Chloro-3-methoxycyclohexyl]- 1-methylvinyl}-8-ethyl-5,6,8,11,12,13,14,15,16,17,
18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy- 14,16-dimethoxy-4,10,12, 18-tetramethyl-15,19-epoxy- 3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1, 7,20,21(4H,23H)-tetrone
The systematic name of pimecrolimus is (lR,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(lE)-2- {(1 R,3R,4S)-4-chloro-3-methoxycyclohexyl} - 1 -methylvinyl] - 17-ethyl- 1,14- dihydroxy-23,25-dimethoxy-13,19,21,27-tetramethyl-ll,28-dioxa-4-aza- tricyclo[22.3.1.04'9]octacos-18-ene-2,3,10,16-tetraone.
Pimecrolimus is the 32 epichloro derivative of ascomycin.
Elidel, NCGC00167506-01, DSSTox_CID_26674, DSSTox_RID_81811, DSSTox_GSID_46674, 137071-32-0, Tox21_112504
Molecular Formula: C43H68ClNO11 Molecular Weight: 810.45312
| US2008085858 | 4-11-2008 | Pharmaceutical Composition |
| Canada | 2200966 | 2006-12-19 | expiry 2015-10-26 |
| United States | 6423722 | 1998-12-26 | 2018-12-26 |
PATENT AND EXPIRY DATE
| 5912238 | Jun 15, 2016 | |
| 5912238*PED | Dec 15, 2016 | |
| 6352998 | Oct 26, 2015 | |
| 6352998*PED | Apr 26, 2016 | |
| 6423722 | Jun 26, 2018 | |
| 6423722*PED | Dec 26, 2018 |
Viktor Gyollai, Csaba Szabo, “Methods of preparing pimecrolimus.” U.S. Patent US20060142564, issued June 29, 2006.

NDA..021302, 13 DEC 2001... VALEANT BERMUDA..ELIDEL1% TOPICAL CREAM
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is currently available as a topical cream, once marketed by Novartis, (however Galderma will be promoting the molecule in Canada in early 2007) under the trade name Elidel.
Pimecrolimus is an immunomodulating agent used in the treatment of atopic dermatitis (eczema). It is available as a topical cream, once marketed by Novartis (however, Galderma has been promoting the compound in Canada since early 2007) under the trade name Elidel.
Pimecrolimus is an ascomycin macrolactam derivative. It has been shown in vitro that pimecrolimus binds to macrophilin-12(also referred to as FKBP-12) and inhibits calcineurin. Thus pimecrolimus inhibits T-cell activation by inhibiting the synthesis and release of cytokines from T-cells. Pimecrolimus also prevents the release of inflammatory cytokines and mediators from mast cells.
Pimecrolimus is a chemical that is used to treat atopic dermatitis (eczema). Atopic dermatitis is a skin condition characterized by redness, itching, scaling and inflammation of the skin. The cause of atopic dermatitis is not known; however, scientists believe that it may be due to activation of the immune system by various environmental or emotional triggers. Scientists do not know exactly how pimecrolimus reduces the manifestations of atopic dermatitis, but pimecrolimus reduces the action of T-cells and mast cells which are part of the immune system and contribute to responses of the immune system. Pimecrolimus prevents the activation of T-cells by blocking the effects of chemicals (cytokines) released by the body that stimulate T-cells. Pimecrolimus also reduces the ability of mast cells to release chemicals that promote inflammation.
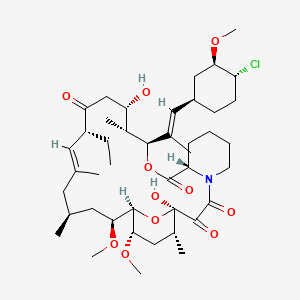
Pimecrolimus, like tacrolimus, belongs to the ascomycin class of macrolactam immunosuppressives, acting by the inhibition of T-cell activation by the calcineurin pathway and inhibition of the release of numerous inflammatory cytokines, thereby preventing the cascade of immune and inflammatory signals.[1] Pimecrolimus has a similar mode of action to that of tacrolimus but is more selective, with no effect on dendritic (Langerhans) cells.[2] It has lower permeation through the skin than topical steroids or topical tacrolimus[3] although they have not been compared with each other for their permeation ability through mucosa. In addition, in contrast with topical steroids, pimecrolimus does not produce skin atrophy.[4] It has been proven to be effective in various inflammatory skin diseases, e.g., seborrheic dermatitis,[5] cutaneous lupus erythematosus,[6]oral lichen planus,[7] vitiligo,[8] and psoriasis.[9][10] Tacrolimus and pimecrolimus are both calcineurin inhibitors and function as immunosuppressants.[11]
Ascomycin macrolactams belong to a new group of immunosuppressive, immunomodulatory and anti-inflammatory agents and include, e.g., ascomycin (FK520), tacrolimus (FK506) and pimecrolimus (ASM 981). The main biological effect of ascomycin macrolactams appears to be the inhibition of the synthesis of both Th1 and Th2-type cytokines in target cells.
As used herein, the term “ascomycin macrolactam” means ascomycin, a derivative of ascomycin, such as, e.g., tacrolimus and pimecrolimus, or a prodrug or metabolite of ascomycin or a derivative thereof.
Ascomycin, also called immunomycin, is a structurally complex macrolide produced by Streptomyces hygroscopicus. Ascomycin acts by binding to immunophilins, especially macrophilin-12. It appears that ascomycin inhibits the production of Th1 (interferon- and IL-2) and Th2 (IL-4 and IL-10) cytokines. Additionally, ascomycin preferentially inhibits the activation of mast cells, an important cellular component of the atopic response. Ascomycin produces a more selective immunomodulatory effect in that it inhibits the elicitation phase of allergic contact dermatitis but does not impair the primary immune response when administered systemically. The chemical structure of ascomycin is depicted below.
Tacrolimus (FK506) is a synthetic derivatives of ascomycin. As a calcineurin inhibitor, it works through the FK-binding protein and inhibits the dephosphorylation of nuclear factor of activated T cells (NFAT), thereby preventing the transport of the cytoplasmic component of NFAT to the cell nucleus. This leads to transcriptional inhibition of proinflammatory cytokine genes such as, e.g., interleukin 2, which are dependent on the nuclear factor of activated NFAT. The chemical structure of tacrolimus is depicted below.
Pimecrolimus, an ascomycin derivative, is a calcineurin inhibitor that binds with high affinity to the cytosolic receptor macrophilin-12, inhibiting the calcium-dependent phosphatase calcineurin, an enzyme required for the dephosphorylation of the cytosolic form of the nuclear factor of the activated T cell (NF-AT). It thus targets T cell activation and proliferation by blocking the release of both TH1 and TH2 cytokines such as IF-g, IL-2, -4, -5, and -10.3 It also prevents the production of TNF-a and the release of proinflammatory mediators such as histamine, hexosaminidase, and tryptase from activated mast cells.3 It does not have general antiproliferative activity on keratinocytes, endothelial cells, and fibroblasts, and in contrast to corticosteroids, it does not affect the differentiation, maturation, functions, and viability of human dendritic cells. The chemical structure of pimecrolimus is depicted below.
Pimecrolimus is an anti-inflammatory compound derived from the macrolactam natural product ascomycin, produced by certain strains of Streptomyces.
In January 2006, the United States Food and Drug Administration (FDA) announced that Elidel packaging would be required to carry a black box warning regarding the potential increased risk of lymph node or skin cancer, as for the similar drug tacrolimus. Whereas current practice by UKdermatologists is not to consider this a significant real concern and they are increasingly recommending the use of such new drugs.[12]
Importantly, although the FDA has approved updated black-box warning for tacrolimus and pimecrolimus, the recent report of the American Academy of Dermatology Association Task Force finds that there is no causal proof that topical immunomodulators cause lymphoma or nonmelanoma skin cancer, and systemic immunosuppression after short-term or intermittent long-term topical application seems an unlikely mechanism.[13] Another recent review of evidence concluded that postmarketing surveillance shows no evidence for this systemic immunosuppression or increased risk for any malignancy.[14] However, there are still some strong debates and controversies regarding the exact indications of immunomodulators and their duration of use in the absence of active controlled trials.[15] Dermatologists' and Allergists' professional societies, the American Academy of Dermatology[1], and the American Academy of Allergy, Asthma, and Immunology, have protested the inclusion of the black box warning. The AAAAI states "None of the information provided for the cases of lymphoma associated with the use of topical pimecrolimus or tacrolimus in AD indicate or suggest a causal relationship."[2].
Pimecrolimus binds with high affinity to macrophilin-12 (FKBP-12) and inhibits the calcium-dependent phosphatase, calcineurin. As a consequence, it inhibits T cell activation by blocking the transcription of early cytokines. In particular, pimecrolimus inhibits at nanomolar concentrations Interleukin-2 and interferon gamma (Th1-type) and Interleukin-4 and Interleukin-10 (Th2-type) cytokine synthesis in human T cells. Also, pimecrolimus prevents the release of inflammatory cytokines and mediators from mast cells in vitro after stimulation by antigen/lgE.
ELIDEL® (pimecrolimus) Cream 1% contains the compound pimecrolimus, the immunosuppressant 33-epi-chloro-derivative of the macrolactam ascomycin.
Chemically, pimecrolimus is (1R,9S,12S,13R,14S,17R,18E,21S,23S,24R,25S,27R)-12-[(1E)-2{(1R,3R,4S)-4-chloro-3-methoxycyclohexyl}-1-methylvinyl]-17-ethyl-1,14-dihydroxy-23,25 dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-aza-tricyclo[22.3.1.0 4,9]octacos-18-ene2,3,10,16-tetraone.
The compound has the empirical formula C43H68CINO11 and the molecular weight of 810.47. The structural formula is
 |
Pimecrolimus is a white to off-white fine crystalline powder. It is soluble in methanol and ethanol and insoluble in water.
Each gram of ELIDEL Cream 1% contains 10 mg of pimecrolimus in a whitish cream base of benzyl alcohol, cetyl alcohol, citric acid, mono- and di-glycerides, oleyl alcohol, propylene glycol, sodium cetostearyl sulphate, sodium hydroxide, stearyl alcohol, triglycerides, and water.
The second representative of the immunosuppressive macrolides for topical application - after tacrolimus (Protopic ®) - has 21 October in the trade. Pimecrolimus is approved for short-term and intermittent long-term treatment for patients aged two years who suffer from mild to moderate atopic dermatitis.
Pimecrolimus is a lipophilic derivative of macrolactam Ascomycin. The macrolides inhibit the production and release of pro-inflammatory cytokines by blocking the phosphatase calcineurin.The anti-inflammatory effect unfolds the drug in the skin. Since he is only minimally absorbed to not measurable, it hardly affects the local or systemic immune response. Therefore, the authorization neither restricts nor a maximum daily dose treatable area or duration of therapy.The cream can also be applied on the face, head and neck, and in skin folds, but not simultaneously with other anti-inflammatory topical agents such as glucocorticoids.
In studies in phases II and III patients aged three months and treated a maximum of one year.In two six-week trials involving 186 infants and young children as well as 403 children and adolescents, the verum symptoms and itching decreased significantly better than the cream base. Already in the first week of itching in 44 percent of children and 70 percent of the infants improved significantly. In adults, pimecrolimus was less effective than 0.1 percent betamethasone 17-valerate.
In the long-term treatment the verum significantly reduced the incidence of flares, revealed two studies with 713 and 251 patients. About a half and one year each about twice as many of the small patients were free of acute disease exacerbations than with the cream base (example: 61 versus 34 per cent of children, 70 versus 33 percent of infants older than six months). Moreover, the use of topical corticosteroids decreased significantly.
In a study of 192 adults with moderate to severe eczema half suffered six months no relapses more (24 percent with placebo). In the long-term therapy pimecrolimus was less effective than 0.1 percent triamcinolone acetonide cream and 1 percent hydrocortisone cream in adults.
The new topicum is-apart from burning and irritation at the application site - relatively well tolerated. It is neither kontaktsensibilisierend still phototoxic or sensitizing and does not cause skin atrophy. As in atopic Ekzen but usually a long-term therapy is necessary studies can reveal long-term adverse effects of the immunosuppressant on the skin only beyond one year.Also available from direct comparative studies between tacrolimus and pimecrolimus. They could help to delineate the importance of the two immunosuppressants.
Pimecrolimus (registry number 137071-32-0; Figure 1) is a macro lide having anti-inflammatory, antiproliferative and immunosuppressive properties. This substance is present as an active ingredient in the Elidel ® drug recently approved in Europe and in the USA for topical treatment of inflammatory conditions of the skin such as atopic dermatitis.
Figure 1: structural formula of pimecrolimus
19th Ed., vol. π, pg. 1627, spray-drying consists of bringing together a highly dispersed liquid and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. Spray-drying however is often limited to aqueous solutions unless special expensive safety measures are taken. Also, in spite of the short contact time, certain undesirable physical and chemical characteristics of the emerging solids are in particular cases unavoidable. The turbulence present in a spray-drier as a result of the moving air may alter the product in an undesirable manner. Modifications to the spray-drying technique are disclosed in WO 03/063821 and WO 03/063822. [00012] European Patent EP 427 680 Bl discloses a method of synthesizing amorphous pimecrolimus (Example 66a). The method yields amorphous pimecrolimus as a colorless foamy resin.
U.S. Patent No. US 6,423,722 discloses crystalline forms of pimecrolimus, such as form A, form B, etc. US 722 also contend that by performing example 66a from the European Patent EP 427 680 Bl, amorphous pimecrolimus is obtained.
The preparation of pimecrolimus was described for the first time in the patent application EP427680 on behalf of Sandoz. Used as raw material in such document is ascomycin (compound identified by registry number 11011-38-4), a natural product obtained through fermentation from Streptomyces strains (such as for example Streptomyces hygroscopicus var ascomyceticus, or Streptomyces hygroscopicus tsukubaensis N°9993). Pimecrolimus is obtained from the ascomycin through a sequence of four steps of synthesis (scheme 1)
Scheme 1 : synthesis process described in EP427680
From a structural point of view, pimecrolimus is the 33-epi-chloro derivative of ascomycin. As described in EP427680, the simultaneous presence - in the structure of ascomycin - of two secondary hydroxyl groups in position 24 and in position 33, requires the protection of the hydroxyl in position 24 before substituting the second hydroxyl in position 33 with an atom of chlorine.
In order to obtain the monoprotection of the hydroxyl in position 24 of ascomycin, such synthesis process provides for the preparation of 24,33-disilyl derivative and the subsequent selective removal of the silyl ester in position 33.
The high ratio between the silylating agent and the substrate and the non-complete selectivity of the subsequent step of deprotection requires carrying out two chromatographic purifications on the column of silica gel (Baumann K., Bacher M., Damont A., Hogenauer K., Steck A. Tetrahedron, (2003), 59, 1075-1087). The general yields of such synthesis process are not indicated in literature; an experiment by the applicant revealed that such yields amount to about 16% molar starting from ascomycin.
Other synthesis processes were recently proposed as alternatives to the synthesis of EP427680.
In particular, the International patent application WO2006040111 on behalf of Novartis provides for the direct substitution of the hydroxyl in position 33 of ascomycin with an atom of chlorine and a second alternative, described in the international patent application WO2006060614 on behalf of Teva, uses - as a synthetic intermediate - a sulfonate derivative in position 33 of ascomycin. Both the proposed synthetic alternatives are not entirely satisfactory in that in WO2006040111 the proposed halogenating agents (chlorophosphorane and N- chlorosuccinimide) are not capable, according to the same authors, of regioselectively substituting the hydroxyl function in position 33, while in WO2006060614 the quality characteristics of the obtained product are, even after chromatographic purification and/or crystallisation, low for a product to be used for pharmaceutical purposes (i.e. purity of 96% as described in the experimental part).
Generally, purified enzymatic systems may be used for the organic synthesis of polyfunctional molecules (Wang Y-F, Wong C-H. J Org Chem (1988) 53, 3127- 3129; Santaniello E., Ferraboschi P., Grisenti P., Manzocchi A. Chem. Rev. (1992), 92(5), 1071-140; Ferraboschi P., Casati S., De Grandi S., Grisenti P., Santaniello E. Biocatalysis (1994), 10(1-4), 279-88); WO2006024582). WO2007103348 and WO2005105811 describe the acylation of rapamycin in position 42 in the presence of lipase from Candida antartica.
.........................
Scheme 2: synthesis of pimecrolimus for enzymatic transesterification of ascomycin.
Scheme 3. Synthesis of pimecrolimus for enzyme-catalyzed alcoholysis from 33,24- diacetate of ascomycin
Example 1
Preparation of the 33-acetyl derivative of ascomvcin (compound I of scheme II)
Lipase from Candida antarctica (CAL B, Novozym 435) [0.140 g (2 U/mg)
FLUKA] was added to a solution of ascomycin (100 mg; 0.126 mmol) in toluene (8 ml) and vinyl acetate (4.5 eq; 0.473 g). The reaction is kept under stirring at the temperature of 30° C for 80 hrs then the enzyme is taken away for filtration and the filtrate is concentrated at low pressure to obtain 105 mg of 33-acetyl ascomycin.
A sample of such intermediate was purified for analytical purposes by chromatography on silica gel (n-hexane/acetone = 8/2 v/v as eluents) and thus crystallised by acetone/water.
The following analysis were carried out on such sample: 1H-NMR (500MHz) δ:
2.10 (CH3CO), 3.92 and 4.70 (24CH and 33CH); IR (cm-1): 3484.245, 2935.287,
1735.331, 1649.741, 1450.039,
1372.278; DSC: endotherm at 134.25° C; [α]D=-74,0° (c=0.5 CHCl3).
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H7iNO13: C 64.80%; H, 8.58%; N, 1.68%;
O, 24.94%
Elementary analysis found: C 64.78%; H, 8.54%; N, 1.59%; O, 24.89%
Preparation of the 24-tgrt-butyldimethylsilylether-33 -acetyl derivative of ascomvcin (intermediate 24-silyl-33-Oac; compound II of scheme 2)
2,6-lutidine (0.29Og; 2.7 mmolels) and tert-butyldimethylsilyl triflate (0.238g; 0.9 mmoles) are added to a solution of 33-acetyl derivative of ascomycin (150 mg;
0.18 mmoles) in dichloromethane (5ml). The reaction is left under stirring at ambient temperature for 30 minutes. After this period the reaction mixture is washed with a solution saturated with sodium bicarbonate (5 ml) and organic phase obtained is washed in sequence with HCl 0.1N (5 ml 3 times) and with a solution at 30% of NaCl (5ml). The organic phase is anhydrified on sodium sulphate, filtered and concentrated to residue under vacuum to obtain 128 mg of product.
Spectrum of MS (ESI +): m/z: 970.5 (M+23; 100.0%)
1H-NMR (500 MHz) δ: 0.05 and 0.06 ((CHs)2Si), 0.90 ((CH3)3C-Si), 2.10
(CH3CO), 4.70 (33CH)
IR (cm-'): 3462.948, 2934.450, 1739.236, 1649.937
Elementary analysis calculated for C51H85NOi3Si: C 64.59%; H, 9.03%; N, 1.48%; O, 21.93%
Elementary analysis found: C 64.50%; H, 9.05%; N, 1.41%; O, 21.88%
DSC= endoderma a 236,43° C. [α]D=-81,4° (c=0.5 CHCl3).
Preparation of 24-tert-butyldimethylsilylether of ascomycin (intermediate 24- silyl-33-OH; compound III of scheme 2) n-octan-1-ol (0.035g; 0.265 mmoles) and CAL B (Novozym 435) [0.100 g (2
U/mg) FLUKA] are added to a solution of 24-tert-butyldimethylsilylether-33- acetyl derivative of ascomycin (50 mg; 0.053 mmoles) in tert-butylmethylether (4 ml). The reaction is kept under stirring at the temperature of 40° C for 120 hours.
After this period the reaction mixture is filtered and the filtrate is evaporated to residue under vacuum to obtain a reaction raw product which is purified by chromatography on silica gel: 44 mg of product (0.048 mmoles) are recovered through elution with petroleum ether/acetone 7/3.
The chemical/physical properties of the obtained product match those of a reference sample obtained according to patent EP427680.
Preparation of 24-tert-butyldimethylsilylether-33-epi-chloro ascomycin
(intermediate 24-silyl-33-chloro; compound IV of scheme 2)
A solution of 24-silyl FR520, i.e. 24-silyl ascomycin (165 g; 0.18 moles) in anhydrous toluene (1.4 litres) and pyridine (50 ml) is added to a suspension of dichlorotriphenylphosphorane (99.95g) in anhydrous toluene (1.1 litres), under stirring at ambient temperature (20-25 °C) in inert atmosphere.
After adding, the reaction mixture is heated at the temperature of 60° C for 1 hour.
After this period the temperature of the reaction mixture is taken to 25° C and thus the organic phase is washed in sequence with water (1 time with 1 L) and with an aqueous solution of NaCl at 10% (4 times with 1 L each time), then it is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain about 250 g of a moist solid of toluene. Such residue product is retaken with n- hexane (500 ml) and then evaporated to dryness (in order to remove the toluene present). The residue product is diluted in n-hexane (500 ml) under stirring at ambient temperature for about 45 minutes and then the undissolved solid taken away for filtration on buckner (it is the sub-product of dichlorophosphorane).
The filtrate is concentrated at low pressure to obtain 148.6 g of a solid which is subsequently purified by chromatography on silica gel (elution with n- heptane/acetone = 9/1) to obtain 123 g (0.13 moles) of product.
The chemical/physical properties of the obtained product match those described in literature (EP427680).
Preparation of the pimecrolimus from 24-fert-butyldimethylsilylether-33-epi- chloro ascomycin
The intermediate 24-silyl-33 chloro (123g; 0.13 Moles; compound IV of scheme
2) is dissolved under stirring at ambient temperature in a dichloromethane/methanol mixture=l/l=v/v (1.1 litres) then p-toluenesulfonic acid monohydrate (10.11 g) is added.
The reaction is kept under stirring at the temperature of 20-25° C for 72 hours, thus a solution of water (600 ml) and sodium bicarbonate (4.46 g) is added to the reaction mixture. The reaction mixture is kept under stirring at ambient temperature for 10 minutes, the organic phase is then prepared and washed with an aqueous solution at 10% of sodium chloride (600 ml).
The organic phase is anhydrified on sodium sulphate, filtered and concentrated under vacuum to obtain 119 g of raw pimecrolimus. Such raw product is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 66 g (81.5 mmoles) of purified pimecrolimus.
The chemical/physical data obtained matches the data indicated in literature.
Example 2
Preparation of ascomvcin 24.33-diacetate (intermediate 24, 33-diacetate; compound V of scheme 3)
DMAP (4.5 eq; 0.136 g) and acetic anhydride (4.5 eq; 0.114 g) are added to a solution of ascomycin (200 mg; 0.25 mmoles) in pyridine (2.5 ml), under stirring at the temperature of 0° C.
The reaction is kept under stirring for 1.5 hours at the temperature of 0° C then it is diluted with water and it is extracted with ethyl acetate (3 times with 5 ml). The organic extracts are washed with HCl 0.5 N (5 times with 10 ml), anhydrified on
Na2SO4 concentrated under vacuum.
The residue product was purified by chromatography on silica gel (n- hexane/acetone 8/2 v/v as eluent) to obtain ascomycin 24,32-diacetate (210 mg;
0.24 mmoles).
We carried out the following analysis on such purified sample:
1H-NMR (500 MHz) δ: 2.02 and 2.06 (2 CH3CO), 5.20 and 4.70 (24CH and
33CH);
IR (Cm-1): 3462.749, 2935.824, 1734.403, 1650.739, 1449.091, 1371.079.
DSC: endothermic peak at 234.10° C ; [α]D=- 100.0° (C=0.5 CHCl3).
Spectrum of MS (ESI+): m/z: 898.4 (100.0%; m+23).
Elementary analysis calculated for C47H73NO14: C 64.44%; H 8.40%; N 1.60%; O
25.57%
Elementary analysis found: C 64.55%; H 8.44%; N 1.61%; O 25.40%
Preparation of the 24-acetyl ascomycin (intermediate 24-acetate-33-OH; compound VI of scheme 3)
Lipase from Candida antartica (CAL B Novozym 435) [1.1 g (2 U/mg) FLUKA] is added to a solution of ascomycin 33,24-diacetate (500 mg; 0.57 mmol) in
TBDME (25 ml) and n-octan-1-ol (4.5 eq; 0.371 g). The reaction is kept under stirring at 30° C for 100 hours, then the enzyme is taken away for filtration and the obtained filtrate is concentrated under low pressure to obtain 425 mg (0.51 mmoles) of product.
A sample was purified for analytical purposes by chromatography on silica gel (n- hexane/acetone = 7:3 v/v as eluents) and thus crystallised by acetone/water.
We carried out the following analysis on such purified sample: 1H-NMR
(500MHz) δ: 2.05 (CH3CO); IR (an 1): 3491.528, 2935.860, 1744.728, 1710.227,
1652.310, 1448.662, 1371.335. DSC: endothermic peak at 134.68° C; [α]D=-
102.7° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 856.4 (M+23; 100.0%)
Elementary analysis calculated for C45H71NO13: C 64.80%; H, 8.58%; N, 1.68%;
0, 24.94%
Elementary analysis found: C 64.71%; H, 8.49%; N, 1.60%; O, 24.97%
Preparation of the 24-acetyl-33epi-chloro ascomycin (intermediate 24-Acetate-33- chloro; compound VII of scheme 3) Supported triphenylphosphine (0.335 g; 1.1 mmoles) is added to a solution of 24- acetyl ascomycin (400 mg; 0.48 mmoles) in carbon tetrachloride (5 ml). The reaction mixture is kept under reflux for 3 hours then it is cooled at ambient temperature. The obtained suspension is filtered and the filtrate is concentrated to residue under vacuum to obtain 0.45g of reaction raw product which is purified by chromatography on silica gel: 163mg (0.19 mmoles) of product are obtained by elution with petroleum ether/acetone = 90/10.
1H-NMR δ: 2.08 (CH3CO); 4.60 (33CH); IR (Cm"1)= 3464.941, 2934.360,
1738.993, 1650.366, 1450.424, 1371.557; DSC: endothermic peak at 231.67° C
[α]D=-75.2° (c=0.5 CHCl3)
Spectrum of MS (ESI +): m/z: 874.3 (M+23; 100.0%)
Elementary analysis calculated for C45H70ClNO12: C 63.40%; H, 8.28%; Cl,
4.16%; N, 1.64%; O, 22.52%
Elementary analysis found: C 63.31%; H, 8.30%; Cl, 4.05%; N, 1.58%; O,
22.42%.
Preparation of pimecrolimus from 24-acetyl-33-epi-chloro ascomycin
A solution of 24-acetyl-33-epi-chloro ascomycin (200 mg; 0.23 mmoles; compound VII) in methanol (2 ml) and HCl 3N (1 ml) is stirred at ambient temperature for 40 hours. After this period, the reaction is neutralised with an aqueous bicarbonate solution, the methanol evaporated under vacuum. The mixture is extracted with dichloromethane (3 times with 5 ml), anhydrified on sodium sulphate, filtered and concentrated to residue to obtain a residue product which is purified by chromatography on silica gel (n-hexane/acetone as eluents) and thus crystallised by ethyl acetate, cyclohexane/water to obtain 78 mg of purified pimecrolimus (0.096 mmoles).
The chemical/physical characteristics of the obtained product matches the data indicated in literature for pimecrolimus.
Example 4 (comparative*)
Verification of the method of synthesis of pimecrolimus described in EP427680 Imidazole (508 mg) and tert-Butyldimethylsilylchloride (1.125 g) are added in portions to a solution of 2g (2.53 mmoles) of ascomycin in anhydrous N,N- dimethylformamide (40 ml). The reaction mixture is kept under stirring at ambient temperature for 4.5 days. The reaction is thus processed diluting it with ethyl acetate (200 ml) and processing it using water (5 x 100 ml). The organic phase is separated, anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy raw product which is subsequently purified by chromatography on silica gel (1:30 p/p): 2.1 g (2.05 mmoles; yields 81% molars) of ascomycin 24,33 disilyl intermediate are obtained by elution with n- hexane/ethyl acetate 3/1. The chemical/physical data of such intermediate matches that indicated in EP427680.
2.1 g (2.05 mmoles) of ascomycin 24,33 disilyl intermediate are dissolved in a solution under stirring at the temperature of 0°C composed of acetonitrile (42 ml) and aqueous HF 40% (23.1 ml). The reaction mixture is kept under stirring at the temperature of 0°C for 2 hours then it is diluted with dichloromethane (30 ml). Then the reaction is washed in sequence with a saturated aqueous solution using sodium bicarbonate (30 ml) and water (30 ml). The separated organic phase is anhydrified on sodium sulphate, filtered and evaporated to residue under vacuum to obtain a foamy residue which is subsequently purified by chromatography on silica gel (1:30 p/p): 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate are obtained by elution with dichloromethane/methanol 9/1. The chemical/physical data of such intermediate matches that obtained on the compound III scheme 2 and matches the data of literature indicated in EP427680. A mixture of 839 mg (0.92 mmoles; yields 45% molars) of ascomycin 24 monosilyl intermediate, triphenylphosphine (337 mg) in carbon tetrachloride (36.4 ml) is heated under stirring under reflux for 15 hours. After this period the reaction mixture is evaporated to residue under vacuum to obtain a solid product purified by chromatography on silica gel (1:30 p/p): 535 mg (0.57 mmoles; yields 63% molars) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are obtained by elution with n-hexane/ethyl acetate 2/1. The chemical/physical data of such intermediate matches those we obtained on compound IV scheme 2 and matches the data of literature indicated in EP427680.
535 mg (0.57 mmoles) of ascomycin 24 monosilyl intermediate, 33-chloro derivative are dissolved under stirring at ambient temperature in acetonitrile (16.4 ml) and aqueous HF 40% (0.44 ml). The reaction mixture is kept under stirring at ambient temperature for 45' and then it is diluted with ethyl acetate (100 ml). The organic phase is thus washed in sequence with an aqueous solution of sodium bicarbonate (70 ml) with water (2 x 70 ml) and thus it is anhydrified on sodium sulphate, filtered and evaporated under vacuum to obtain a solid which is subsequently purified by chromatography on silica gel (1 :30 p/p): 323 mg (0.399 mmoles; yields 70% molars) of pimecrolimus is obtained by elution with n- hexane/ethyl acetate 2/3. The chemical/physical characteristics of the obtained product matches the data indicated in literature regarding pimecrolimus; the overall yield of the process is 16%.
.............................
POLYMORPHS.......WO2006060615A1
Example 7: Preparation of amorphous pimecrolimus by precipitation [00094] 19,5 g purified pimecrolimus (colorless resin) was dissolved in 217 ml acetone at 4O0C and concentrated. Residue: 38,76 g. The residue was diluted with 6 ml distilled water with stirring. Finally 1 ml acetone was added. This solution was added slowly to 2 L chilled distilled water that was stirred efficiently. After the addition had been completed, the suspension was stirred 20 min at O0C. Then the solid was filtered and dried at 450C in vacuum oven overnight. Product: 15,65 g yellowish solid. Amorphous (XRD, DSC).
Example 8: Preparation of amorphous pimecrolimus by grinding
[00095] Procedure of grinding: 200 mg of Pimecrolimus sample was ground gently in an agate mortar using a pestle for half a minute. ,
References
- Allen BR, Lakhanpaul M, Morris A, Lateo S, Davies T, Scott G, Cardno M, Ebelin ME, Burtin P, Stephenson TJ (2003). "Systemic exposure, tolerability, and efficacy of pimecrolimus cream 1% in atopic dermatitis patients". Arch Dis Child 88 (11): 969–973. doi:10.1136/adc.88.11.969.PMC 1719352. PMID 14612358.
- Meingassner JG, Kowalsky E, Schwendinger H, Elbe-Bürger A, Stütz A (2003). "Pimecrolimus does not affect Langerhans cells in murine epidermis". Br J Dermatol 149 (4): 853–857.doi:10.1046/j.1365-2133.2003.05559.x. PMID 14616380.
- Billich A, Aschauer H, Aszódi A, Stuetz A (2004). "Percutaneous absorption of drugs used in atopic eczema: pimecrolimus permeates less through skin than corticosteroids and tacrolimus". Int J Pharm 269 (1): 29–35. doi:10.1016/j.ijpharm.2003.07.013.PMID 14698574.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). "Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial". Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Firooz A, Solhpour A, Gorouhi F, Daneshpazhooh M, Balighi K, Farsinejad K, Rashighi-Firoozabadi M, Dowlati Y (2006). "Pimecrolimus cream, 1%, vs hydrocortisone acetate cream, 1%, in the treatment of facial seborrheic dermatitis: a randomized, investigator-blind, clinical trial". Archives of Dermatology 142 (8): 1066–1067. doi:10.1001/archderm.142.8.1066.PMID 16924062.
- Kreuter A, Gambichler T, Breuckmann F, Pawlak FM, Stücker M, Bader A, Altmeyer P, Freitag M (2004). "Pimecrolimus 1% cream for cutaneous lupus erythematosus". J Am Acad Dermatol 51(3): 407–410. doi:10.1016/j.jaad.2004.01.044. PMID 15337984.
- Gorouhi F, Solhpour A, Beitollahi JM, Afshar S, Davari P, Hashemi P, Nassiri Kashani M, Firooz A (2007). "Randomized trial of pimecrolimus cream versus triamcinolone acetonide paste in the treatment of oral lichen planus". J Am Acad Dermatol 57 (5): 806–813.doi:10.1016/j.jaad.2007.06.022. PMID 17658663.
- Boone B, Ongenae K, Van Geel N, Vernijns S, De Keyser S, Naeyaert JM (2007). "Topical pimecrolimus in the treatment of vitiligo". Eur J Dermatol 17 (1): 55–61. doi:10.1111/j.1610-0387.2006.06124.x. PMID 17081269.
- Kreuter A, Sommer A, Hyun J, Bräutigam M, Brockmeyer NH, Altmeyer P, Gambichler T (2006). "1% pimecrolimus, 0.005% calcipotriol, and 0.1% betamethasone in the treatment of intertriginous psoriasis: a double-blind, randomized controlled study". Arch Dermatol 142 (9): 1138–1143. doi:10.1001/archderm.142.9.1138. PMID 16983001.
- Jacobi A, Braeutigam M, Mahler V, Schultz E, Hertl M (2008). "Pimecrolimus 1% cream in the treatment of facial psoriasis: a 16-week open-label study". Dermatology 216 (2): 133–136.doi:10.1159/000111510. PMID 18216475.
- Scheinfeld N (2004). "The use of topical tacrolimus and pimecrolimus to treat psoriasis: a review". Dermatol. Online J. 10 (1): 3. PMID 15347485.
- N H Cox and Catherine H Smith (December 2002). "Advice to dermatologists re topical tacrolimus" (DOC). Therapy Guidelines Committee. British Association of Dermatologists.
- Berger TG, Duvic M, Van Voorhees AS, VanBeek MJ, Frieden IJ; American Academy of Dermatology Association Task Force (2006). "The use of topical calcineurin inhibitors in dermatology: safety concerns Report of the American Academy of Dermatology Association Task Force". J Am Acad Dermatol 54 (5): 818–823. doi:10.1016/j.jaad.2006.01.054.PMID 16635663.
- Spergel JM, Leung DY (2006). "Safety of topical calcineurin inhibitors in atopic dermatitis: evaluation of the evidence". Curr Allergy Asthma Rep 6 (4): 270–274. doi:10.1007/s11882-006-0059-7. PMID 16822378.
- Stern RS (2006). "Topical calcineurin inhibitors labeling: putting the "box" in perspective".Archives of Dermatology 142 (9): 1233–1235. doi:10.1001/archderm.142.9.1233.PMID 16983018.
| WO2005105811A1 | Apr 12, 2005 | Nov 10, 2005 | Ping Cai | Regiospecific synthesis of rapamycin 42-ester derivatives |
| WO2006024582A1 | Jul 26, 2005 | Mar 9, 2006 | Poli Ind Chimica Spa | A method for the preparation of mycophenolate mofetil by enzimatic transesterification |
| WO2006040111A2 | Oct 10, 2005 | Apr 20, 2006 | Novartis Ag | Heteroatoms-containing tricyclic compounds |
| WO2006060614A1 | Dec 1, 2005 | Jun 8, 2006 | Teva Gyogyszergyar Zartkoeruen | Methods for preparing pimecrolimus |
| WO2007103348A2 | Mar 5, 2007 | Sep 13, 2007 | Wyeth Corp | Process for preparing water-soluble polyethylene glycol conjugates of macrolide immunosuppressants |
| EP0427680A1 | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
- Elidel official homepage
- FDA News
- NPS RADAR
- Article about American Academy of Dermatology speaking out against black box warning
- Report of the Calcineurin Task Force of the ACAAI and AAAAI
| WO2005117837A1 * | Jun 1, 2005 | Dec 15, 2005 | Lorant Gyuricza | Process for preparation of amorphous form of a drug |
| EP0427680A1 * | Nov 7, 1990 | May 15, 1991 | Sandoz Ltd. | Heteroatoms-containing tricyclic compounds |
| EP0480623A1 * | Oct 2, 1991 | Apr 15, 1992 | Merck & Co., Inc. | New halomacrolides and derivatives having immunosuppressive activity |
| US6423722 * | Oct 17, 2000 | Jul 23, 2002 | Novartis Ag | Crystalline macrolides and process for their preparation |
....................................
8 TACROLIMUS
Tacrolimus, Fujimycin
104987-11-3 CAS, 804.0182, C44H69NO12
- Astagraf XL
- FK 506
- FR 900506
- FR900506
- LCP-Tacro
- Prograf
- Protopic
- Tacrolimus
- Tacrolimus hydrate
- Tsukubaenolide hydrate
- UNII-WM0HAQ4WNM
3S-[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5, 19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c] [1,4] oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone, monohydrate
17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetraone
Astellas Pharma (Originator), LAUNCHED 1993
Tacrolimus (also FK-506 or Fujimycin) is an immunosuppressive drug whose main use is after organ transplant to reduce the activity of the patient’s immune system and so the risk of organ rejection. It is also used in a topical preparation in the treatment of severe atopic dermatitis, severe refractory uveitis after bone marrow transplants, and the skin condition vitiligo. It was discovered in 1984 from the fermentation broth of a Japanese soil sample that contained the bacteria Streptomyces tsukubaensis. Tacrolimus is chemically known as a macrolide. It reduces peptidyl-prolyl isomerase activity by binding to the immunophilin FKBP-12 (FK506 binding protein) creating a new complex. This FKBP12-FK506 complex interacts with and inhibits calcineurin thus inhibiting both T-lymphocyte signal transduction and IL-2 transcription.
PATENT
| Canada | 2037408 | 2002-12-17 | EXPIRY 2011-03-01 |
| Canada | 1338491 | 1996-07-30 | 2013-07-30 |
| United States | 5665727 | 1994-09-09 | 2014-09-09 |
| United States | 5260301 | 1994-02-28 | 2011-02-28 |
Pan Sup Chang, Hoon Cho, “Water soluble polymer-tacrolimus conjugated compounds and process for preparing the same.” U.S. Patent US5922729, issued April, 1997.
Tacrolimus is a naturally-occurring macrolide isolated from the fermentation broth of Streptomyces tsukubaensis that was originally discovered by Fujisawa (now Astellas Pharma) in 1984. Tacrolimus possesses immunosuppressive properties and suppresses IL-2 production from helper T-cells, resulting in inhibition of the activation and proliferation of cytotoxic T-cells. In the cell, tacrolimus binds to an immunophilin called FKBP-12 and forms a tacro-immunophilin complex that, in turn, binds to calcineurin and prevents the dephosphorylation of cytoplasmic NF-AT thus disallowing it from reaching the nucleus, thereby strongly inhibiting IL-2 gene transcription. As a result, T-cell activation and proliferation is inhibited.
In 1993, Prograf(R) (tacrolimus capsules and injection) received clearance from the Japanese Ministry of Health and Welfare and was introduced in Japan the same year for the treatment of kidney and liver transplant rejection. Based on two large phase III comparative clinical trials, the product received clearance from the FDA in April 1994, and was made available two months later for commercial use in the U.S. The product is available extensively for transplant rejection. Prograf(R) was also launched in Japan for the treatment of myasthenia gravis and for the treatment of heart transplant rejection; the latter indication was approved in the U.S. in 2006 and launched in 2007. In 2008, Astellas Pharma preregistered the compound in Japan for the oral treatment of all cases of myasthenia gravis. The same year, Senju launched the product in Japan for the treatment of vernal and perennial allergic conjunctivitis in patients unresponsive to anti-allergic drugs. In 2009, the product was approved and commercialized in Japan for the treatment of ulcerative colitis. In 1999, Astellas Pharma launched Protopic(R) (tacrolimus ointment) in Japan for the treatment of atopic dermatitis and in 2001, Protopic(R) was commercialized in the U.S. and Europe. In April 2005, tacrolimus (capsules) was commercialized again by Astellas Pharma in Japan for the treatment of rheumatoid arthritis (RA) in patients who respond insufficiently to current therapies. The following year, Senju received approval in Japan for the use of tacrolimus for the treatment of vernal conjunctivitis and perennial allergic conjunctivitis. A once-daily capsule was approved in the E.U. in 2006. The compound was launched in 2007 in Japan for lupus nephritis. In 2009, the product was approved in US for the prophylaxis of organ rejection in allogeneic kidney transplantation in combination with mycophenolate mofetil and, in the E.U., for the prophylaxis of transplant rejection in adult and pediatric, kidney, liver or heart allograft recipients. In 2011, the compound was launched in Japan for the prophylaxis of organ rejection in patients receiving allogeneic small bowel transplants. In 2013, the indication for interstitial pneumonia associated with polymyositis/dermatomyositis was approved in Japan and an extended release formulation was approved in the U.S. for the prophylaxis of organ rejection in adult patients receiving kidney transplants. This extended release formulation was launched in the U.S. in August 2013. Veloxis Pharmaceuticals (formerly LifeCycle Pharma) is developing a once-daily tablet formulation of tacrolimus (Envarsus®) with improved bioavailability and reduced variability compared with the modified-release version of the compound. Envarsus® has been pre-registered in E.U. and the U.S. for the prevention of transplant rejection in kidney transplant patients. The company is also evaluating the compound in phase II trials for the treatment of autoimmune hepatitis.
In terms of clinical development, the National Cancer Institute (NCI) is developing tacrolimus in phase III for the treatment of graft-versus-host disease (GVHD). Phase III trials are also underway at Astellas Pharma for the treatment of psoriasis, ulcerative colitis and chronic focal encephalitis (Rasmussen's encephalitis), while early clinical trials are ongoing for asthma. In 2009, Astellas Pharma withdrew an NDA seeking approval in the U.S. based on potential clinical challenges that would result from FDA requirements to conduct additional clinical studies. Kyoto University had been conducting phase II clinical studies for the treatment of Crohn's disease; however, no recent development has been reported for this research.
In 2003, Sucampo Pharmaceuticals obtained a license from Astellas Pharma to develop and market tacrolimus for ophthalmic indications in the U.S. and Europe, however, in June 2005, the company voluntarily discontinued its tacrolimus eye drops development program due to FDA safety concerns. In 2005, Senju and Astellas Pharma established an agreement to codevelop an eye drop formulation of tacrolimus in Japan. Also, Astellas Pharma granted Senju exclusive manufacturing and marketing rights of the compound. In 2003, Astellas Pharma and GlaxoSmithKline signed an agreement for the copromotion of Protopic(R) in the U.S for atopic dermatitis. An additional agreement for the copromotion of Protopic(R) in South America for the same indication was signed in 2004 between Astellas Pharma and Roche. Tacrolimus was designated orphan drug status in Japan in 1993 and in 2005 for the suppression of organ rejection in allogenic kidney transplantation and for the treatment of vernal conjunctivitis, respectively, in patients unresponsive to anti-allergic drugs. In the E.U., the latter indication was assigned orphan drug designation in 2004. The product was withdrawn from the community register of designated orphan medicinal products in the E.U. in April 2010 on request of the sponsor. In 1998 and 2005, the FDA assigned orphan drug designation for the prophylaxis of GVHD and for the prophylaxis of organ rejection in patients receiving heart transplants. Finally, in 2008, orphan designation was received in Japan for the treatment of myasthenia gravis. In 2012, an additional orphan drug designation was assigned in the U.S. for the treatment of hemorrhagic cystitis. This designation was granted in Japan in 2012 for the treatment of interstitial pneumonia accompanied with polymyositis/dermatomyositis complex. In 2012, orphan drug designation was assigned in Japan for the treatment of interstitial pneumonia accompanied with polymyositis/dermatomyositis complex. In 2012, the product was licensed by Veloxis Pharmaceuticals to Chiesi on an exclusive basis for the commercialization and distribution in Europe, Turkey and CIS countries for the prevention of rejection in kidney transplant recipients. In 2013, an additional orphan drug designation was assigned in the U.S. for the prophylaxis of organ rejection in patients receiving allogeneic kidney transplant.
Tacrolimus, also known as FK-506 or FR-900506, has the chemical tricyclic structure shown below:
corresponding to C44H69NO-|2- Tacrolimus appears in the form of white crystals or crystalline powder. It is practically insoluble in water, freely soluble in ethanol and very soluble in methanol and chloroform. The preparation of tacrolimus is described in EP-A-0 184 162 and analogues of tacrolimus are disclosed e.g. in EP-A-0444659 and US 6,387,918
Tacrolimus is an immunosuppressive agent produced by Streptomyces tsukubaensis No. 9993 and is the compound of formula (I) wherein R.sub.1 and R.sub.2 are both hydrogen. Tacrolimus, which is also called FK-506, has first discovered by Tanaka, Kuroda and their colleague in Japan see, J. Am. Chem. Soc., 1987, 109, 5031 and U.S. Pat. No. 4,894,366 issued on Jan. 16, 1990!.
July 19, 2013 /PRNewswire/ -- Astellas Pharma US, Inc. ("A.stellas"), a U.S. subsidiary of Tokyo-based Astellas Pharma Inc., announced today that the U.S. Food and Drug Administration (FDA) has approved Astagraf XL (tacrolimus extended-release capsules) for the prophylaxis of organ rejection in patients receiving a kidney transplant with mycophenolate mofetil (MMF) and corticosteroids, with or without basiliximab induction.
"Each transplant recipient is different and requires a personalized treatment approach. The approval of Astagraf XL marks an important milestone in post-transplant care as it provides physicians with a new treatment option for kidney t recipients," said Sef Kurstjens, M.D., PhD., chief medical officer, Astellas Pharma, Inc. "Astellas is pleased to continue our more than 20-year commitment to the field of transplant immunology."
PROTOPIC (tacrolimus) Ointment contains tacrolimus, a macrolide immunosuppressant produced by Streptomyces tsukubaensis. It is for topical dermatologic use only. Chemically, tacrolimus is designated as [3S[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*]]5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethenyl]-14,16-dimethoxy-4,10, 12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c][1,4] oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone,monohydrate. It has the following structural formula:
 |
Tacrolimus has an empirical formula of C44H69NO12•H2O and a formula weight of 822.03. Each gram of PROTOPIC Ointment contains (w/w) either 0.03% or 0.1% of tacrolimus in a base of mineral oil, paraffin, propylene carbonate, white petrolatum and white wax.
FK-506 (also Tacrolimus or fujimycin) is a potent calcineurin (protein phosphatase 2B) inhibitor that requires FK 506-binding protein 12 (FKBP12) for activity (IC50 = 3 nM). FK-506 inhibits secretion of IL-1, IL-2 (IC50 = 1 nM), IL-3, IL-4, IL-6 (IC50 = 35 nM), GM-CSF, TNFα (IC50 = 10 nM), IFNγ and Myc from activated T-cells in vitro. FK-506 exhibits potent immunosuppressive, neuroprotective and anticonvulsant activity in vivo. The physiological effects of FK-506 also include regulation of nitric oxide neurotoxicity, neurotransmitter release, and regulation of Ca2+ release via the ryanodine and inositol-(1,4,5)-trisphosphate (IP3) receptors. Furthermore, it has become clear that, predominantly as a result of CaN inhibition, FK506 alters multiple biochemical processes in a variety of cells besides lymphocytes. FK506 and ascomycin inhibit signaling pathways in astrocytes and change the pattern of cytokine and neurotrophin gene expression.
Tacrolimus (also FK-506 or fujimycin, trade names Prograf, Advagraf, Protopic) is an immunosuppressive drug that is mainly used after allogeneic organ transplant to reduce the activity of the patient's immune system and so lower the risk of organ rejection. It is also used in a topical preparation in the treatment of atopic dermatitis (eczema), severe refractory uveitis after bone marrow transplants, exacerbations of minimal change disease, and the skin condition vitiligo.
It is a 23-membered macrolide lactone discovered in 1984 from the fermentation broth of a Japanese soil sample that contained the bacteria Streptomyces tsukubaensis. It reduces interleukin-2 (IL-2) production by T-cells.
 Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from 'Tsukuba macrolide immunosuppressant'.
Tacrolimus was discovered in 1984; it was among the first macrolide immunosuppressants discovered, preceded by the discovery of rapamycin (sirolimus) on Rapa Nui (Easter Island) in 1975.It is produced by a type of soil bacterium, Streptomyces tsukubaensis. The name tacrolimus is derived from 'Tsukuba macrolide immunosuppressant'.
Tacrolimus 0.1%
| Indication | For use after allogenic organ transplant to reduce the activity of the patient's immune system and so the risk of organ rejection. It was first approved by the FDA in 1994 for use in liver transplantation, this has been extended to include kidney, heart, small bowel, pancreas, lung, trachea, skin, cornea, and limb transplants. It has also been used in a topical preparation in the treatment of severe atopic dermatitis. |
|---|---|
| Pharmacodynamics | Tacrolimus is a macrolide antibiotic. It acts by reducing peptidyl-prolyl isomerase activity by binding to the immunophilin FKBP-12 (FK506 binding protein) creating a new complex. This inhibits both T-lymphocyte signal transduction and IL-2 transcription. Although this activity is similar to cyclosporine studies have shown that the incidence of acute rejection is reduced by tacrolimus use over cyclosporine. Tacrolimus has also been shown to be effective in the topical treatment of eczema, particularly atopic eczema. It suppresses inflammation in a similar way to steroids, but is not as powerful. An important dermatological advantage of tacrolimus is that it can be used directly on the face; topical steroids cannot be used on the face, as they thin the skin dramatically there. On other parts of the body, topical steroid are generally a better treatment. |
| Mechanism of action | The mechanism of action of tacrolimus in atopic dermatitis is not known. While the following have been observed, the clinical significance of these observations in atopic dermatitis is not known. It has been demonstrated that tacrolimus inhibits T-lymphocyte activation by first binding to an intracellular protein, FKBP-12. A complex of tacrolimus-FKBP-12, calcium, calmodulin, and calcineurin is then formed and the phosphatase activity of calcineurin is inhibited. This prevents the dephosphorylation and translocation of nuclear factor of activated T-cells (NF-AT), a nuclear component thought to initiate gene transcription for the formation of lymphokines. Tacrolimus also inhibits the transcription for genes which encode IL-3, IL-4, IL-5, GM-CSF, and TNF-, all of which are involved in the early stages of T-cell activation. Additionally, tacrolimus has been shown to inhibit the release of pre-formed mediators from skin mast cells and basophils, and to downregulate the expression of FceRI on Langerhans cells. |
Tacrolimus was first approved by the Food and Drug Administration (FDA) in 1994 for use in liver transplantation; this has been extended to include kidney, heart, small bowel, pancreas, lung, trachea, skin, cornea, bone marrow, and limb transplants.
The branded version of the drug is owned by Astellas Pharma, and is sold under the trade names Prograf given twice daily, Advagraf, a sustained release formulation allowing once daily dosing, and Protopic (Eczemus in Pakistan by Brookes Pharma), the topical formulation. Advagraf is available in 0.5, 1, 3 and 5 mg capsules, the ointment is concentrations of 0.1% and 0.03%.
A second once-daily formulation of tacrolimus is in Phase 3 clinical trials in the U.S. and Europe. This formulation also has a smoother pharmacokinetic profile that reduces the peak-to-trough range in blood levels compared to twice-daily tacrolimus.Data from the first Phase 3 trial in stable kidney transplant patients showed that this once-daily formulation was non-inferior in efficacy and safety compared to twice-daily tacrolimus. A second Phase 3 trial in de novo patients is ongoing.
Tacrolimus, which is also referred to as FK-506 (Fermentek catalogue number 506), is a 23-membered macrolide lactone and belongs to the group of polyketides. Tacrolimus was first isolated in the 1980's from the fermentation broth of the soil bacteria Streptomyces tsukubaensis. The antibiotic macrolide compound tacrolimus was e.g. reported in 1984 by Kino et al. (J. Antibiotics 40, 1249-1255, 1984). Later on tacrolimus was prepared as a microbial natural product by using different microorganisms, i.e. soil bacteria such as Streptomyces sp. MA6858 (US 5,116,756) ATCC 55098, Streptomyces tsukubaensis NRRL 18488 (EP-B 0 356 399 and US 5,200,41 1 ), Streptomyces clavuligerus CKD 1119 (KR-B 100485877) or Streptomyces glaucescens MTCC 5115 (US 2007191415).
The product tacrolimus exhibits immunosuppressive activities which are due to its effect to reduce the activity of the enzyme peptidyl-propyl isomerase and to the binding to the protein immunophilin FKBP12 (FK506 binding protein). Tacrolimus and the structurally similar polyketides ascomycin and rapamycin require initial binding to the highly conserved protein cyclophilin FKBP12 in order to be physiologically active. The rapamycin/FKBP12 complex binds to mTOR (mammalian target of rapamycin), a serine- threonine kinase that appears to act as a central controller for sensing the cellular environment and regulating translation initiation (see e.g. Easton J. B. and Houghton P.J., 2004, Expert Opin Ther Targets; 8(6):551-64). However, the tacrolimus/FKBP12 complex was found to bind to a different cellular target and inhibits the phosphatase activity of calcineurin, in analogy to cyclosporine (see Allison A.C., 2000, Immunopharmacology; 47(2-3):63-83).
Tacrolimus is often used for immunosuppression following e.g. organ transplantation. Furthermore, tacrolimus and its derivatives have been shown to be effective in treating a number of diseases such as asthma, inflammatory diseases and hyperproliferative skin disease. Tacrolimus and other immunosuppressant such as rapamycin, cyclosporine, or a combination thereof are also useful in the treatment of various auto-immmune diseases. For many years calcineurin inhibitors (e.g. cyclosporine and tacrolimus) have been the mainstay of immunosuppressive therapy. These two compounds are potent suppressors of cellular immune response and have significantly improved the outcome of organ transplants during the past two decades (see Allison A.C., 2000, Immunopharmacology; 47(2-3):63-83). Gene clusters encoding the biosynthetic pathways of a great number of medically important drugs of microbial origin have already been cloned and sequenced, including the gene cluster of macrolides rapamycin, ascomycin and tacrolimus. With respect to cloning of the tacrolimus gene cluster, a partial sequence, mostly encompassing genes encoding polyketide synthase (PKS), was reported in the literature (see Motamedi H. and Shafiee A. 1998, Eur J Biochem; 256(3):528-34). On the other hand, scientists reported cloning of the ascomycin gene cluster in 2000 (see Wu K et al. 2000, Gene; 251(1 ):81- 90, US 6,503,737). Tacrolimus structurally and by the biosynthetic origin resembles ascomycin (FK520) and rapamycin (see Reynolds et al.; Drugs and the Pharmaceutical Sciences, 1997, 82, 497-520. They all can be synthesised by combined polyketide (PKS) and non-ribosomal peptide biosynthetic pathways (NRPS) (see McDaniel R et al. 2005, Chem Rev; 105(2):543-58).
Tacrolimus and ascomycin are structurally similar. As only structural difference, the allyl side chain at carbon 21 of tacrolimus is replaced by an ethyl side chain in ascomycin. The structures of tacrolimus (FK506) and ascomycin (FK520) compounds are shown as formulae (Ia) and (Ib). The structures of ascomycin and tacrolimus already suggest complex biosynthetic pathways which can be divided into four steps considering the biosynthetic mechanism:
1. chain initiation using the unusual shikimate derived starter,
2. chain elongation common to most PKS derived compounds,
3. chain termination and cyclization by incorporation of L-pipecolic acid and
4. post-PKS processing.
During the tacrolimus fermentation process, undesired ascomycin (FK520) product is also produced as an impurity, thus lowering the final yield of tacrolimus and causing significant additional costs to the downstream isolation processes of tacrolimus.
(Ia) FK506, R = -CH2-CH = CH2
(Ib) FK520, R = CH2-CH3
For oral administration, tacrolimus is currently formulated and marketed as soft gelatine capsules comprising the equivalent of 0.5, 1 or 5 mg anhydrous tacrolimus and marketed under the trade name Prograf® and Protropic®. The recommended initial oral dose is from about 0.1 to 0.2 mg/kg/day in patients. The dose aims at a certain trough plasma level from about 5 to about 20 ng/ml. Prograf® is indicated for the prophylaxis of organ rejection in patients receiving allogeneic liver or kidney transplants. There remains a need for novel pharmaceutical compositions and/or dosage forms comprising tacrolimus exhibiting enhanced bioavailability. An increased bioavailability may allow a reduction in the dosage units taken by a patient, e.g. down to a single dose daily, and may also reduce or negate the need for food to be takes simultaneously with the dosage form thereby allowing patients more freedom on when the drug is taken. Furthermore, it is contemplated that fluctuations in the plasma concentration versus time profile may be significantly reduced. Further, enhanced bioavailability may also result in a more reproducible (i.e. less variable compared to that of Prograf®) release profile....
......................
h) Determination of tacrolimus and ascomycin production with HPLC of thiostrepton resistant ccr disrupted mutants derived by secondary homologous recombination using pKC1 139-ccrTs.:
Method for tacrolimus and ascomycin determination: The analysis for determination of tacrolimus or ascomycin production thereof was carried out by isocratic reversed phase HPLC using an appropriate column and running conditions: column Nucleosil-100 C18 (150x4.0 mm, particle size 3 μm), flow 1.5 ml/min, T°C=60°C, mobile phase: 560 ml water, 335 ml acetonitrile, 70 ml MTBE and 0.2 ml 85% H3PO4, detection 210 nm, sample injection 20 μl.
The tacrolimus and ascomycin content in samples quantification was performed by using external standards of tacrolimus and ascomycin, where tacrolimus was eluted at 12.5 min and ascomycin at 11.5 min. Results are expressed as % of ascomycin production compared to tacrolimus production in samples.
............................
A new total synthesis of FK-506 is described: This synthesis has been performed by previous construction of two building fragments (XXIV) and (LI), which later were coupled and cyclized. (Schemes 1-3): 1) (1R*S*,3R,5S,6R,7S,9R)-6-(tert-butyldimethylsilyloxy)-9-(1,3-dithian-2-yl)-5,7-dimethoxy-1-methyldecyl diphenyl phosphine oxide (XXIV). The Sharpless asymetric epoxidation of 1,4-pentadien-3-ol (I) with (-)-diisopropyltartrate and tert-butylhydroperoxide gives the epoxy alcohol (II) with high optical purity, which is benzylated in the usual way to (III). The reaction of (III) with lithioacetonitrile and then HCl yields lactone (IV), which is methylated with lithium diisopropylamide and methyl iodide to lactone (V) as major isomer (separated by chromatography on SiO2). The reduction of (V) with LiAlH4 affords the diol (VI), which is converted into the bis(tert-butyl carbonate) (VII) with 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile (BOC-N). The reaction of (VII) with Br2 and K2CO3 in dichloromethane gives the bromocarbonate (VIII), which by selective saponification of the cyclic carbonate with NaOCH3 in methanol yields the epoxy alcohol (IX). Methylation of (IX) with NaH and methyl iodide affords the methyl ether (X), which is converted into the butyrolactone (XI) with lithioacetonitrile as before. The protection of the OH group of (XI) with TBS-Cl gives the silyl ether (XII), which by trans-selective methylation with lithium diisopropylamide and methyl iodide yields lactone (XIII). The reduction of (XIII) with LiAlH4 affords diol (XIV) as major isomer (separated by column chromatography). The selective esterification of the primary OH group of (XIV) with pivaloyl chloride gives the hydroxy ester (XV), which is methylated with NaH and methyl iodide as usual to the methoxy derivative (XVI). Debenzylation of (XVI) by hydrogenolysis with H2 over Pd/C yields the hydroxy ester (XVII), which is silylated with TBS-SO3CF3 to the fully protected compound (XVIII).
Selective deprotection of (XVIII) with trifluoroacetic acid in THF - water affords the primary alcohol (XIX), which is oxidized with oxalyl chloride and DMSO in dichloromethane to the aldehyde (XX). The protection of the aldehyde group of (XX) with propane-1,3-dithiol and BF3 gives the dithiane derivative (XXI), which is resilylated with TBS-SO3CF3 as before to the dithiane (XXII). The pivaloyl group of (XXII) is eliminated with LiAlH4 in THF yielding the alcohol (XXIII), which is finally treated with benzenesulfonyl chloride and then with ethyl diphenylphosphine oxide and butyllithium in THF to obtain the first building group, the phosphine derivative (XXIV).
2) [2S,3S,5S,6R,7S,8E,9(1'R,3'R,4'R)]-2-Allyl-3-(tert-butyldimethylsilylox y)-6,8-dimethyl-7-(triethylsilyloxy)-5-(triisopropylsilyloxy)-9-[3-meth oxy-4-(triisopropylsilyloxy)cyclohexyl]-8-nonenal (LI). Quinic acid (XXV) is converted into the lactone (XXVI) by known methods. Then this lactone is treated with thiocarbonyldiimidazole in refluxing dichloroethane yielding the bis(thiocarbonyl)lactone (XXVII), which by reaction with tributyltin hydride and AIBN in refluxing xylene is converted into the lactone (XXIX), either directly or through the intermediate thiocarbonyl-lactone (XXVIII). The silylation of (XXIX) with TIPS-SO3CF3 as usual affords the protected lactone (XXX). Opening of the lactone ring with methylchloroaluminum N-methoxy-N-methylamide gives the methoxyamide (XXXI), which is methylated with methyl trifluoromethylsulfonate to the methoxy-N-methoxyamide (XXXII). The reduction of (XXXII) with diisobutylaluminum hydride gives the aldehyde (XXXIII), which is condensed with 2-lithio-2-(triethylsilyl)propanal (XXXIV), yielding unsaturated aldehyde (XXXV). The condensation of (XXXV) with the boron enolate of oxazolidone (XXVI) affords the oxazolidone derivative (XXXVII), which is treated with methylchloroaluminum N-methoxy-N-methylamide to give the methoxyamide (XXXVIII). The silylation of (XXXVIII) with TES-SO3CF3 as usual yields the silylated amide (XXXIX), which is reduced with diisobutylaluminum hydride to the aldehyde (XL). The condensation of (XL) with chiral acetate (XLI) by means of lithium diisopropylamide in THF affords the hydroxy ester (XLII). Transesterification of (XLII) with NaOCH3 and methanol gives methyl ester (XLIII).
..............
Use of the microorganism streptomyces tsukubaensis No. 9993 for the production of the FR-900506 substance of the formula:
- [II]
Synthetic Processes
- :(1)
Process 1
- (Introduction of common Hydroxy-Protective Group)

(2)
Process 2
- (Introduction of common Hydroxy-Protective Group)

(3)
Process 3
- (Formation of Double Bond)

(4)
Process 4
- (Oxidation of Hydroxyethylene Group)

(5)
Process 5
- (Reduction of Allyl Group)
 in which
in which
R¹, R², R³, n and the symbol of a line and dotted line are each as defined above,
R 1 a and R 2 a
and R 2 a are each commonly protected hydroxy, and
are each commonly protected hydroxy, and
R 2 b is a common leaving group.
is a common leaving group.- THE MICROORGANISM
- The microorganism which can be used for the production of the FR-900506, FR-900520 and/or FR-900525 substances is FR-900506 FR-900520 and/or FR-900525 substance(s)-producing strain belonging to the genusStreptomyces, among which Streptomyces tsukubaensis No. 9993 has been newly isolated from a soil sample collected at Toyosato-cho, Tsukuba-gun, Ibaraki Prefecture, Japan.
- A lyophilized sample of the newly isolated Streptomyces tsukubaensis No. 9993 has been deposited with the Fermentation Research Institute, Agency of Industrial Science and Technology (No. 1-3, Higashi 1-chome, Yatabemachi Tsukuba-gun, Ibaraki Prefecture, Japan) under the deposit number of FERM P-7886 (deposited date: October 5th, 1984), and then converted to Budapest Treaty route of the same depository on October 19, 1985 under the new deposit number of FERM BP-927.
- The Streptomyces tsukubaensis No. 9993 has the following morphological, cultural, biological and physiological characteristics.
- This white powder of the FR-900506 substance could be transformed into a form of crystals by recrystallization thereof from acetonitrile, which possess the following physical and chemical properties.
(1) Form and Color:
colorless prisms
(2)Elemental Analysis: C: 64.30 %, H: 8.92 %, N: 1.77 % 64.20 %, 8.86 %, 1.72 %, (3) Melting Point:
127 - 129 °C
(4) Specific Rotation:
[α] 23 D : -84.4° (c = 1.02, CHCl₃)
: -84.4° (c = 1.02, CHCl₃)
(5) ¹³C Nuclear Magnetic Resonance Spectrum: the chart of which being shown in Figure 3,
the chart of which being shown in Figure 3,
(6) ¹H Nuclear Magnetic Resonance Spectrum:
the chart of which being shown in Figure 4. - Other physical and chemical properties, that is, the color reaction, solubility, ultraviolet absorption spectrum, infrared absorption spectrum, thin layer chromatography and property of the substance of the colorless prisms of the FR-900506 substance were the same as those for the white powder of the same under the identical conditions.
- From the above physical and chemical properties and the analysis of the X ray diffraction, the FR-900506 substance could be determined to have the following chemical structure.
 17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetraone........................
17-Allyl-1,14-dihydroxy-12-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylvinyl]-23,25-dimethoxy-13,19,21,27-tetramethyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetraone........................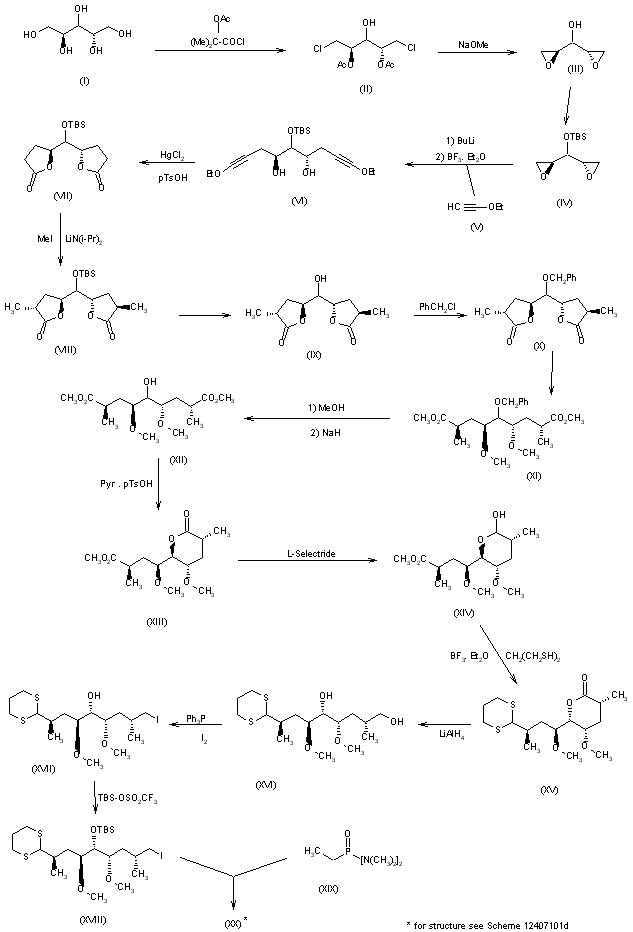 The total synthesis of FK-506 is described: This synthesis was performed by previously constructing three building fragments (XX), (XXXII) and (XLVI), which later were coupled sequentially. First the synthesis of these fragments will be presented, and afterwards their sequential coupling will be described. 1) (2RS,4R,6S,7R,8S,10R)-2-(Bis(dimethylamino)phosphono)-7-(tert-butyldimethylsilyloxy)-6,8-dimethoxy-10-(1,3-dithian-2-yl)-4-methylundecane (XX). The reaction of L-arabitol (I) with 2-acetoxyisobutyryl chloride in acetonitrile gives the diacetoxycompound (II), which by treatment with sodium methoxide in THF yields (2S,4S)-1,2:4,5-diepoxy-3-pentanol (III). The protection of (III) with TBS-Cl in THF affords the protected compound (IV), which is condensed with ethoxyacetylene (V) by means of butyllithium and boron trifluoride ethearate in THF giving the diacetylenic alcohol (VI). Cyclization of (VI) by means of HgCl2 and p-toluenesulfonic acid in refluxing ethanol yields the dilactone (VII), which is methylated by means of methyl iodide and lithium diisopropylamide in THF affording the methylated dilactone (VIII). The deprotection of (VIII) with HF in acetonitrile gives the hydroxydilactone (IX), which is benzylated with benzyl trichloroacetimidate and trifluoromethanesulfonic acid in dichloromethane-cyclohexane yielding the benzyl protected dilactone (X). The methanolysis of (X), followed by methylation with NaH and methyl iodide in DMF affords the nonanedioic ester (XI), which is debenzylated by hydrogenolysis with H2 over Pd/C in ethyl acetate giving the hydroxy diester (XII). The lactonization of (XII) with pyridinium p-toluenesulfonate in dichloromethane yields the lactone-methyl ester (XIII), which is selectively reduced with L-Selectride in THF affording the lactol-methyl ester (XIV). The reaction of (XIV) with propane-1,3-dithiol and boron trifluoride ethearate in dichloromethane gives the 1,3-dithiane derivative (XV), which by reduction of its lactone group with LiAlH4 in THF yields (2R,4S,5R,6S,8R)-8-(1,3-dithian-2-yl)-4,6-dimethoxy-2-methylnonane-1,5-diol (XVI). The reaction of (XVI) with I2, pyridine and triphenylphosphine in benzene affords the 1-iodo derivative (XVII), which is protected with TBS trifluoromethanesulfonate and triethylamine in dichloromethane giving the protected iodide (XVIII). Finally, this compound is condensed with ethylphosphonic acid bis(dimethylamide) (XIX) by means of butyllithium in THF to afford the first building fragment (XX).SEE..................Isolation and PurificationThe cultured broth thus obtained was filtered with an aid of diatomaseous earth (5 kg). The mycelial cake was extracted with acetone (50 liters), yielding 50 liters of the extract. The acetone extract from mycelium and the filtrate (135 liters) were combined and passed through a column of a non-ionic adsorption resin "Diaion HP-20" (Trade Mark, maker Mitsubishi Chemical Industries Ltd.) (10 liters). After washing with water (30 liters) and 50% aqueous acetone (30 liters), elution was carried out with 75% aqueous acetone. The eluate (30 liters) was evaporated under reduced pressure to give residual water (2 liters). This residue was extracted with ethyl acetate (2 liters) three times. The ethyl acetate extract was concentrated under reduced pressure to give an oily residue. The oily residue was mixed with twice weight of acidic silica gel (special silica gel grade 12, maker Fuji Devision Co.), and this mixture was slurried in ethyl acetate. After evaporating the solvent, the resultant dry powder was subjected to column chromatography of the same acidic silica gel (800 ml) which was packed with n-hexane. The column was developed with n-hexane (3 liters), a mixture of n-hexane and ethyl acetate (4:1 v/v, 3 liters) and ethyl acetate (3 liters). The fractions containing the object compound were collected and concentrated under reduced pressure to give an oily residue. The oily residue was dissolved in a mixture of n-hexane and ethyl acetate (1:1 v/v, 30 ml) and subjected to column chromatography of silica gel (maker Merck Co., Ltd. 230-400 mesh) (500 ml) packed with the same solvents system. Elution was carried out with a mixture of n-hexane and ethyl acetate (1:1 v/v, 2 liters and 1:2 v/v, 1.5 liters) and ethyl acetate (1.5 liters).Fractions containing the first object compound were collected and concentrated under reduced pressure to give crude FR-900506 substance (3 g) in the form of yellowish powder.............
The total synthesis of FK-506 is described: This synthesis was performed by previously constructing three building fragments (XX), (XXXII) and (XLVI), which later were coupled sequentially. First the synthesis of these fragments will be presented, and afterwards their sequential coupling will be described. 1) (2RS,4R,6S,7R,8S,10R)-2-(Bis(dimethylamino)phosphono)-7-(tert-butyldimethylsilyloxy)-6,8-dimethoxy-10-(1,3-dithian-2-yl)-4-methylundecane (XX). The reaction of L-arabitol (I) with 2-acetoxyisobutyryl chloride in acetonitrile gives the diacetoxycompound (II), which by treatment with sodium methoxide in THF yields (2S,4S)-1,2:4,5-diepoxy-3-pentanol (III). The protection of (III) with TBS-Cl in THF affords the protected compound (IV), which is condensed with ethoxyacetylene (V) by means of butyllithium and boron trifluoride ethearate in THF giving the diacetylenic alcohol (VI). Cyclization of (VI) by means of HgCl2 and p-toluenesulfonic acid in refluxing ethanol yields the dilactone (VII), which is methylated by means of methyl iodide and lithium diisopropylamide in THF affording the methylated dilactone (VIII). The deprotection of (VIII) with HF in acetonitrile gives the hydroxydilactone (IX), which is benzylated with benzyl trichloroacetimidate and trifluoromethanesulfonic acid in dichloromethane-cyclohexane yielding the benzyl protected dilactone (X). The methanolysis of (X), followed by methylation with NaH and methyl iodide in DMF affords the nonanedioic ester (XI), which is debenzylated by hydrogenolysis with H2 over Pd/C in ethyl acetate giving the hydroxy diester (XII). The lactonization of (XII) with pyridinium p-toluenesulfonate in dichloromethane yields the lactone-methyl ester (XIII), which is selectively reduced with L-Selectride in THF affording the lactol-methyl ester (XIV). The reaction of (XIV) with propane-1,3-dithiol and boron trifluoride ethearate in dichloromethane gives the 1,3-dithiane derivative (XV), which by reduction of its lactone group with LiAlH4 in THF yields (2R,4S,5R,6S,8R)-8-(1,3-dithian-2-yl)-4,6-dimethoxy-2-methylnonane-1,5-diol (XVI). The reaction of (XVI) with I2, pyridine and triphenylphosphine in benzene affords the 1-iodo derivative (XVII), which is protected with TBS trifluoromethanesulfonate and triethylamine in dichloromethane giving the protected iodide (XVIII). Finally, this compound is condensed with ethylphosphonic acid bis(dimethylamide) (XIX) by means of butyllithium in THF to afford the first building fragment (XX).SEE..................Isolation and PurificationThe cultured broth thus obtained was filtered with an aid of diatomaseous earth (5 kg). The mycelial cake was extracted with acetone (50 liters), yielding 50 liters of the extract. The acetone extract from mycelium and the filtrate (135 liters) were combined and passed through a column of a non-ionic adsorption resin "Diaion HP-20" (Trade Mark, maker Mitsubishi Chemical Industries Ltd.) (10 liters). After washing with water (30 liters) and 50% aqueous acetone (30 liters), elution was carried out with 75% aqueous acetone. The eluate (30 liters) was evaporated under reduced pressure to give residual water (2 liters). This residue was extracted with ethyl acetate (2 liters) three times. The ethyl acetate extract was concentrated under reduced pressure to give an oily residue. The oily residue was mixed with twice weight of acidic silica gel (special silica gel grade 12, maker Fuji Devision Co.), and this mixture was slurried in ethyl acetate. After evaporating the solvent, the resultant dry powder was subjected to column chromatography of the same acidic silica gel (800 ml) which was packed with n-hexane. The column was developed with n-hexane (3 liters), a mixture of n-hexane and ethyl acetate (4:1 v/v, 3 liters) and ethyl acetate (3 liters). The fractions containing the object compound were collected and concentrated under reduced pressure to give an oily residue. The oily residue was dissolved in a mixture of n-hexane and ethyl acetate (1:1 v/v, 30 ml) and subjected to column chromatography of silica gel (maker Merck Co., Ltd. 230-400 mesh) (500 ml) packed with the same solvents system. Elution was carried out with a mixture of n-hexane and ethyl acetate (1:1 v/v, 2 liters and 1:2 v/v, 1.5 liters) and ethyl acetate (1.5 liters).Fractions containing the first object compound were collected and concentrated under reduced pressure to give crude FR-900506 substance (3 g) in the form of yellowish powder.............

References
- Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, Kohsaka M, Aoki H, Imanaka H (1987). "FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics.". J Antibiot (Tokyo) 40 (9): 1249–55. PMID 2445721.
- Pritchard D (2005). "Sourcing a chemical succession for cyclosporin from parasites and human pathogens.". Drug Discov Today 10 (10): 688–91. doi:10.1016/S1359-6446(05)03395-7.PMID 15896681. Supports source organism, but not team information
- Ponner, B, Cvach, B (Fujisawa Pharmaceutical Co.): Protopic Update 2005
- Healthy Ontario: Tacrolimus topical ointment
- Alloway RR, Germain M, Osama Gaber, A, Bodziak KA, Mulgaonkar SP, Gohh RY, Kaplan B, Katz E, Beckert M, Gordon RD, A Phase II Open-Label, Multi-Center Prospective, Conversion Study in Stable Kidney Transplant Patients to Compare the Pharmacokinetics of LCP-Tacro Tablets Once-A-Day to Prograf Capsules Twice-A-Day. American Transplant Congress, 2008
- http://files.shareholder.com/downloads/ABEA-4J4LWA/1008134289x0x477697/e60eb3d4-849c-41e2-95f3-d8a1eaea3b56/LCP_News_2011_6_21_English_Releases.pdf
- Clinicaltrials.gov identifier: NCT01187953
- William F. Ganong. Review of medical physiology (22nd ed.). Lange medical books. p. 530. ISBN 0-07-144040-2.
- Liu J, Farmer J, Lane W, Friedman J, Weissman I, Schreiber S (1991). "Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes.". Cell 66 (4): 807–15.doi:10.1016/0092-8674(91)90124-H. PMID 1715244.
- McCauley, Jerry (2004-05-19). "Long-Term Graft Survival In Kidney Transplant Recipients". Slide Set Series on Analyses of Immunosuppressive Therapies. Medscape. Retrieved 2006-06-06.
- M.M. Abou-Jaoude, R. Naim, J. Shaheen, N. Naufal, S. Abboud, M. AlHabash, M. Darwish, A. Mulhem, A. Ojjeh, and W.Y. Almawi (2005). "Tacrolimus (FK506) versus cyclosporin microemulsion (Neoral) as maintenance immunosuppresion therapy in kidney transplant recipients.". Transplantation Proceedings 37 (7): 3025–3028. doi:10.1016/j.transproceed.2005.08.040. PMID 16213293.
- Elizabeth Haddad, Vivian McAlister, Elizabeth Renouf, Richard Malthaner, Mette S. Kjaer, and Lise Lotte Gluud (2006). "Cyclosporin versus Tacrolimus for Liver Transplanted Patients". In McAlister, Vivian. Cochrane Database of Systematic Reviews 4 (CD005161): CD005161. doi:10.1002/14651858.CD005161.pub2. PMID 17054241.
- J.G. O'Grady, A. Burroughs, P. Hardy, D. Elbourne, A. Truesdale, and The UK and Ireland Liver Transplant Study Group (2002). "Tacrolimus versus emulsified cyclosporin in liver transplantation: the TMC randomised controlled trial". Lancet 360 (9340): 1119–1125. doi:10.1016/S0140-6736(02)11196-2. PMID 12387959.
- Baumgart DC, Pintoffl JP, Sturm A, Wiedenmann B, Dignass AU (2006). "Tacrolimus is safe and effective in patients with severe steroid-refractory or steroid-dependent inflammatory bowel disease--a long-term follow-up". Am J Gastroenterol 101 (5): 1048–1056. doi:10.1111/j.1572-0241.2006.00524.x. PMID 16573777.
- Baumgart DC, MacDonald JK, Feagan BG (2008). "Tacrolimus (FK506) for induction of remission in refractory ulcerative colitis". In Baumgart, Daniel C. Cochrane Database Syst Rev 16 (3): CD007216. doi:10.1002/14651858.CD007216. PMID 18646177.
- Silverberg, NB; Lin, P; Travis, L; Farley-Li, J; Mancini, AJ; Wagner, AM; Chamlin, SL; Paller, AS (2004). "Tacrolimus ointment promotes repigmentation of vitiligo in children: a review of 57 cases.".Journal of the American Academy of Dermatology 51 (5): 760–6. doi:10.1016/j.jaad.2004.05.036. PMID 15523355.
- Naesens M, Kuypers DR, Sarwal M (2009). "Calcineurin inhibitor nephrotoxicity". Clin. J. Am. Soc. Nephrol. 4 (2): 481–509. doi:10.2215/CJN.04800908. PMID 19218475.
- Miwa Y, Isozaki T, Wakabayashi K, et al. (2008). "Tacrolimus-induced lung injury in a rheumatoid arthritis patient with interstitial pneumonitis". Mod Rheumatol 18 (2): 208–11. doi:10.1007/s10165-008-0034-3. PMID 18306979.
- O'Donnell MM, Williams JP, Weinrieb R, Denysenko L (2007). "Catatonic mutism after liver transplant rapidly reversed with lorazepam". Gen Hosp Psychiatry 29 (3): 280–1.doi:10.1016/j.genhosppsych.2007.01.004. PMID 17484951.
- Hanifin JM, Paller AS, Eichenfield L, Clark RA, Korman N, Weinstein G, Caro I, Jaracz E, Rico MJ; US Tacrolimus Ointment Study Group (2005). "Efficacy and safety of tacrolimus ointment treatment for up to 4 years in patients with atopic dermatitis". J Am Acad Derm 53 (2 suppl 2): S186–94. doi:10.1016/j.jaad.2005.04.062. PMID 16021174.
- N H Cox and Catherine H Smith (December 2002). "Advice to dermatologists re topical tacrolimus" (PDF). Therapy Guidelines Committee. British Association of Dermatologists.
- Fukatsu S, Fukudo M, Masuda S, Yano I, Katsura T, Ogura Y, Oike F, Takada Y, Inui K (2006). "Delayed effect of grapefruit juice on pharmacokinetics and pharmacodynamics of tacrolimus in a living-donor liver transplant recipient". Drug Metab Pharmacokinet 21 (2): 122–5. doi:10.2133/dmpk.21.122. PMID 16702731.
- Fegan, A; White, B; Carlson, JC; Wagner, CR (Jun 9, 2010). "Chemically controlled protein assembly: techniques and applications.". Chemical reviews 110 (6): 3315–36. doi:10.1021/cr8002888.PMID 20353181.
- Tacrolimus, which is also called FK-506, has first discovered by Tanaka, Kuroda and their colleague in Japan see, J. Am. Chem. Soc., 1987, 109, 5031
- and U.S. Pat. No. 4,894,366 issued on Jan. 16, 1990!.
- Total synthesis of FK506 and an FKBP probe reagent, (C8,C9-13C2)-FK506
J Am Chem Soc 1990, 112(14): 5583 - A diastereospecific, non-racemic synthesis of the C.10-C.18 segment of FK-506
Tetrahedron Lett 1988, 29(3): 277
- Tacrolimus levels in Liver Transplants-Indian Study by Dr.Pradeep Naik,Dr.Dharmesh Kapoor, Dr.DCS Reddy
- Prograf prescribing information at Fujisawa
- Pimecrolimus (Elidel Cream) FDA adivisory page (for eczema treatment)
- Tacrolimus (FK506) product page from Fermentek
- U.S. National Library of Medicine: Drug Information Portal - Tacrolimus
Hey Guys,
ReplyDeleteGreeting of day!
Not doubts, it is really good, and knowledgeable an articles post . We are online cancer and anticancer drugs supplier, distributor, wholesaler, exporter and importer from India to worldwide.
Afinitor 10MG and 5MG, Call me at +91 9982034844
You may search for our products through the search bar on our website. If you would like to receive a copy of our product catalog, please contact us at info@alfa-chemistry.com. 1-dodecyl-2,3-methylimidazolium trifluoromethanesulfonate
ReplyDeleteNice blog..! I really loved reading through this article... Thanks for sharing such an amazing post with us and keep blogging. Zoledronic Acid Injection Manufacturers,Suppliers & Exporters in India | Bendamustine Injection Manufacturers,Suppliers & Exporters in India | Pemetrexed Injection Manufacturers,Suppliers & Exporters in India
ReplyDeleteCongratulation for the great post. Those who come to read your Information will find lots of helpful and informative tips. METHYL CARBAZATE
ReplyDeletebuy lsd vials 250ug/drop online.Everyone who uses drugs always wants a common platform where they can buy drugs without any issue. So we are here to help you with that. And when buying products like LSD liquid vials online you should have basic knowledge about that product. Few are given below.
ReplyDeletebuy lsd vials 250ug/drop online.Everyone who uses drugs always wants a common platform where they can buy drugs without any issue. So we are here to help you with that. And when buying products like LSD liquid vials online you should have basic knowledge about that product. Few are given below.
ReplyDeletebuy lsd vials 250ug/drop online.Everyone who uses drugs always wants a common platform where they can buy drugs without any issue. So we are here to help you with that. And when buying products like LSD liquid vials online you should have basic knowledge about that product. Few are given below.
ReplyDelete