FLOZIN SERIES

FULL LENGTH ARTICLES
1 TOFOGLIFLOZIN
2 SERGLIFLOZIN
3 DAPAGLIFLOZIN
4 IPRAGLIFLOZIN
5 EMPAGLIFLOZIN
6 LUSEOGLIFLOZIN
7 REMOGLIFLOZIN
8 ERTUGLIFLOZIN
9 SOTAGLIFLOZON
10 CANAGLIFLOZIN
SEE PART 2 AT
http://apisynthesisint.blogspot.in/p/flozin-series-22.html
11 SBM-TFC-039
12 LIK 066
13 BEXAGLIFLOZIN
14
15

1 TOFOGLIFLOZIN















2 Sergliflozin

Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate


Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
- See more at: http://worlddrugtracker.blogspot.in/search?q=SERGLIFLOZIN&x=0&y=0#sthash.eFhhfbh4.dpuf
Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
- See more at: http://worlddrugtracker.blogspot.in/search?q=SERGLIFLOZIN&x=0&y=0#sthash.eFhhfbh4.dpuf
................................................................................................................................
2 DAPAGLIFLOZIN
..............
4 IPRAGLIFLOZIN










Empagliflozin









FULL LENGTH ARTICLES
1 TOFOGLIFLOZIN
2 SERGLIFLOZIN
3 DAPAGLIFLOZIN
4 IPRAGLIFLOZIN
5 EMPAGLIFLOZIN
6 LUSEOGLIFLOZIN
7 REMOGLIFLOZIN
8 ERTUGLIFLOZIN
9 SOTAGLIFLOZON
10 CANAGLIFLOZIN
SEE PART 2 AT
http://apisynthesisint.blogspot.in/p/flozin-series-22.html
11 SBM-TFC-039
12 LIK 066
13 BEXAGLIFLOZIN
14
15
will be updated .........
1 TOFOGLIFLOZIN
TOFOGLIFLOZIN
CSG-452
R-7201
RG-7201
R-7201
RG-7201
CAS..1201913-82-7
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol hydrate (1:1)
THERAPEUTIC CLAIM Treatment of diabetes mellitus
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3′R,4′S,5′S,6′R)-
2. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol monohydrate
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3′R,4′S,5′S,6′R)-
2. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol monohydrate
MOLECULAR WEIGHT 404.5
SPONSOR Chugai Pharmaceuticals
CODE DESIGNATION CSG452
CODE DESIGNATION CSG452
Tofogliflozin (USAN, codenamed CSG452) is an experimental drug for the treatment ofdiabetes mellitus and is being developed by Chugai Pharma in collaboration with Kowa andSanofi.[1] It is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. As of September 2012, the drug is in Phase III clinical trials.[2][3]
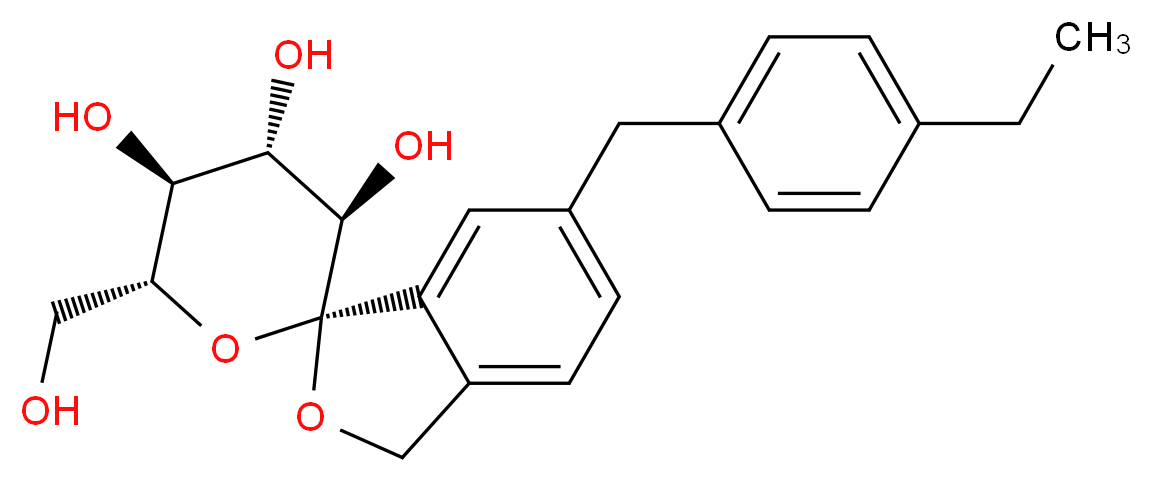
Tofogliflozin is the name of the monohydrate, which is the form used as a drug. The active moiety or anhydrous form (ChemSpider ID: 28530778, CHEMBL2110731) has the chemical formula C22H26O6 and a molecular mass of 386.44 g/mol.[4]
- Chugai Pharmaceutical: Development Pipeline
- Nagata, T.; Fukazawa, M.; Honda, K.; Yata, T.; Kawai, M.; Yamane, M.; Murao, N.; Yamaguchi, K.; Kato, M.; Mitsui, T.; Suzuki, Y.; Ikeda, S.; Kawabe, Y. (2012). “Selective SGLT2 inhibition by tofogliflozin reduces renal glucose reabsorption under hyperglycemic but not under hypo- or euglycemic conditions in rats”. AJP: Endocrinology and Metabolism 304 (4): E414–E423. doi:10.1152/ajpendo.00545.2012.PMID 23249697. edit
- Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. (2012). “Discovery of Tofogliflozin, a NovelC-Arylglucoside with anO-Spiroketal Ring System, as a Highly Selective Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes”. Journal of Medicinal Chemistry 55 (17): 7828–7840.doi:10.1021/jm300884k. PMID 22889351. edit
- Statement on a nonproprietary name adopted by the USAN council: Tofogliflozin.
PATENTS
papers
Chinese Chemical Letters, 2013 , vol. 24, 2 pg. 131 – 133
Journal of Medicinal Chemistry, 2012 , vol. 55, 17 pg. 7828 – 7840
1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
MS (ESI +): 387 [M +1] +.
second set
J. Med. Chem., 2012, 55 (17), pp 7828–7840
DOI: 10.1021/jm300884k
1H NMR (400 MHz, CD3OD) δ: 1.20 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42–3.47 (1H, m), 3.63–3.67 (1H, m), 3.75–3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.3 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07–7.14 (4H, m), 7.17–7.23 (3H, m).
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2.
MS (ESI): 387 [M + H]+. HRMS (ESI), m/z calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801.
……………………………
prepn
[Example 1] (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro- -6′-(hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran] -3 ‘, 4′, one of the preparation step [compound of formula (IX)] 5′-triol Preparation of methanol (2 – hydroxymethyl-phenyl – bromo-4)
To the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.1kg) in – bromoterephthalic was added at below 30 ℃ solution (7.5kg, 30.6mol) of the acid, and the mixture was stirred for 1 hour at 25 ℃. Then cooled to 19 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). In addition to methanol (15.0kg) in the mixture was kept for a while.
Again, to the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.0kg) in – was added at below 30 ℃ solution (7.5kg, 30.6mol) of bromo terephthalic acid, and the reaction was carried out for 1 hour at 25 ℃. Then cooled to 18 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). After addition of methanol (15.0kg) in the mixture is combined with the reaction mixture obtained in the previous reaction, and then the solvent was distilled off under reduced pressure. After addition of methanol (36kg) residue was obtained, and the solvent was evaporated under reduced pressure. Furthermore, (54 ℃ dissolved upon confirmation) which was dissolved by warming was added to methanol (36kg) to the residue. After cooling to room temperature the solution was stirred for 30 minutes added water (60kg). After addition of water (165kg) In addition to this mixture was cooled to 0 ℃, and the mixture was stirred for one hour. Centrifuge the obtained crystals were washed twice with water (45kg), and dried for 2 hours under reduced pressure to give (11.8kg, 54.4mol, 89% yield) of the title compound.
1 H-NMR (DMSO-d 6) δ: 4.49 (4H, t, J = 5.8Hz), 5.27 (1H, t, J = 5.8Hz), 5.38 (1H, t, J = 5.8Hz), 7.31 (1H, d, J = 7.5Hz), 7.47 (1H, d, J = 7.5Hz), 7.50 (1H, s).
Preparation of benzene (ethoxy methyl – methyl – - methoxy-1 1) – bromo-1 ,4 – 2:2 process bis
(- Bromo-4 – 2-hydroxyethyl methyl phenyl) in tetrahydrofuran (57kg) in the solution (8.0kg, 36.9mol) of methanol, I added (185.12g, 0.74mol) of pyridinium p-toluenesulfonate. After cooling to -15 ℃ below the mixture, 2 – was added at -15 ℃ or less (7.70kg, 106.8mol) methoxy propene, and the mixture was stirred 1 h at -15 ~ 0 ℃. Was added aqueous potassium carbonate (25 wt%, 40kg) and the reaction mixture was warmed to room temperature and separate the organic layer was added toluene (35kg). After washing with water (40kg) The organic layer was evaporated under reduced pressure. Was dissolved in toluene (28kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 1.42 (6H, s), 1.45 (6H, s), 3.24 (3H, s), 3.25 (3H, s), 4.45 ( 2H, s), 4.53 (2H, s), 7.28 (1H, dd, J = 1.5,8.0 Hz), 7.50 (1H, d, J = 8.0Hz), 7. 54 (1H, d, J = 1.5Hz).
MS (ESI +): 362 [M +2] +.
MS (ESI +): 362 [M +2] +.
Preparation of on – (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy)-6 – trimethylsilyloxy methyl – tetrahydropyran-2: Step 3
Glucono -1,5 – - D-(+) in tetrahydrofuran (70kg) in the solution (35.8kg, 353.9mol) of N-methylmorpholine (7.88kg, 44.23mol) and lactone, chlorotrimethylsilane ( was added at 40 ℃ less 29.1kg, and 267.9mol), and the mixture was stirred for 2 hours at 30 ~ 40 ℃ resulting mixture. Was cooled to 0 ℃ the reaction mixture was added toluene (34kg) water (39kg), and the organic layer was separated. Twice sodium dihydrogen phosphate aqueous solution (5 wt%, 39.56kg) in, washed once with water (39kg) the organic layer the solvent was evaporated under reduced pressure. Was dissolved in toluene (34.6kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74- 3.83 (3H, m), 3.90 (1H, t, J = 8.0Hz), 3.99 (1H, d, J = 8.0Hz), 4.17 (1H, dt, J = 2 .5,8.0 Hz).
Step 4: (1S, 3′R, 4′S, 5′S, 6′R) -3 ‘, 4′, 5 ‘, 6′-tetrahydro -6,6′ – bis (hydroxymethyl) – spiro [ (3H), 2'-[2H] pyran] -3 ‘, 4′, 5′-Preparation of triol isobenzofuran-1
(Methyl – - – methoxy 1-ethoxy-methyl) – bromo-1 ,4 – 2 prepared in step 2 bis cooled to below -10 ℃ toluene solution of benzene, hexane solution to (15 wt% n-butyl lithium , was added at below 0 ℃ 18.2kg, and 42.61mol), and the mixture was stirred 1.5 h at 5 ℃ resulting mixture. (10.5kg, 40.7mol), was added tetrahydrofuran (33.4kg) then magnesium bromide diethyl ether complex in the mixture, and the mixture was stirred for 1 hour at 25 ℃. Was added at below -10 ℃ toluene solution of the on – tris (trimethylsilyloxy) -6 – - 3,4,5 cooled to -15 ℃ below the mixture prepared in step 3 trimethylsilyloxy methyl – tetrahydropyran-2 was. After stirring 0.5 h at -15 ℃ or less, poured into 20% aqueous ammonium chloride solution to (80kg) of this solution, and the organic layer was separated. After washing with water (80kg) and the organic layer obtained, and the solvent was evaporated under reduced pressure. I was dissolved in methanol (43kg) residue was obtained. Was stirred for 1 hour at 20 ℃ was added (1.4kg, 7.4mol) and p-toluenesulfonic acid monohydrate in the mixture. Thereafter, it was stirred for another hour and cooled to 0 ℃, centrifuged crystals obtained was washed with methanol (25kg), and dried for 8 hours at reduced pressure under 40 ℃, (5.47kg, yield the title compound I got 50%) rate.
1 H-NMR (DMSO-d 6) δ :3.20-3 .25 (1H, m) ,3.41-3 .45 (1H, m) ,3.51-3 .62 (4H, m) , 4.39 (1H, t, J = 6.0Hz) ,4.52-4 .54 (3H, m), 4.86 (1H, d, J = 4.5Hz), 4.93 (1H, d, J = 5.5Hz), 4.99 (1H, d, J = 12.5Hz), 5.03 (1H, d, J = 12.5Hz), 5.23 (1H, t, J = 5 .8 Hz) ,7.24-7 .25 (2H, m), 7.29 (1H, dd, J = 1.5,8.0 Hz).
Step 5: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(methoxycarbonyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3′ , 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2'-[2H] pyran isobenzofuran] spiro
(1S, 3′R, 4′S, 5′S, 6′R) – tetrahydro -6,6 ‘- bis (hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran ] -3 ‘, 4′, 5′-triol 4 (5.3kg, 17.8mol) and – dissolved in acetonitrile (35kg) (13.7kg, 112.1mol) a chloroformate, in the solution of dimethylaminopyridine I was added at 12 ℃ or less (10.01kg, 105.9mol) methyl. Heated to 20 ℃, After stirring for 1 h, was added ethyl acetate (40kg) and water (45kg), and the organic layer was separated and the mixture. Once (45.4kg) aqueous solution consisting of (9.01kg) sodium chloride and potassium hydrogen sulfate (1.35kg), sodium chloride aqueous solution (weight 10%, 44.5kg), sodium chloride aqueous solution (the organic layer was washed successively 20% by weight, in 45.0kg), and the solvent was evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (18kg) and the residue obtained was then evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (13.2kg) again and the residue obtained was obtained as ethylene glycol dimethyl ether solution of the title compound. I was used as it was in the six step.
1 H-NMR (CDCl 3) δ: 3.54 (3H, s), 3.77 (6H, s), 3.811 (3H, s), 3.812 (3H, s), 4.23 ( 1H, dd, J = 2.8,11.9 Hz), 4.32 (1H, dd, J = 4.0,11.9 Hz) ,4.36-4 .40 (1H, m), 5.11 -5.24 (5H, m), 5.41 (1H, d, J = 9.8Hz), 5.51 (1H, t, J = 9.8Hz), 7.25 (1H, d, J = 7.5Hz), 7.42 (1H, d, J = 7.5Hz), 7.44 (1H, s).
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
Step 6: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3 ’4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2'-[2H] pyran isobenzofuran] spiro
[(Methoxycarbonyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro – (1S, 3′R, 4′S, 5′S, 6′R) -6 which had been prepared in Step 5 – 3 ‘, 4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran] Ethylene glycol dimethyl ether in solution, 2 – (2.46kg, 17.8mol), 4 butanol (25kg), anhydrous potassium carbonate – - methyl-2 were sequentially added (3.73kg, 24.9mol) ethyl phenyl boronic acid, in the reaction vessel was replaced with argon atmosphere, was bubbled with argon mixture. To the mixture – after the addition (0.72kg, 0.88mol) and palladium (II) chloride dichloromethane adduct [1,1 '-bis (diphenylphosphino) ferrocene], it was replaced with argon again inside of the vessel, one at 80 ℃ I was stirring time. After cooling, I added sequentially (0.859kg, 5.3mol) of ethylene glycol dimethyl ether (9.85kg), ethyl acetate (19kg), N-acetyl-L-cysteine in the mixture. After stirring for 2.5 h the mixture was filtered and added Celite (5.22kg), and washed with ethyl acetate (78kg) and the filter residue. The combined washings and filtrate, and the solvent is evaporated off under reduced pressure, and in addition (0.58kg, 3.6mol) and ethanol (74kg), N-acetyl-L-cysteine residue was obtained, which is heated to 70 ℃ or I was dissolved residue is then. After addition of water (9.4kg) in the solution, cooled to 60 ℃, and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more The mixture was stirred for 1 hour or more at 5 ℃ less. Centrifuge the resulting solid was washed twice with a mixture of water (35kg) and ethanol (55kg). Was dissolved at 70 ℃ ethanol (77kg) again, wet powder was obtained (10.21kg), cooled to 60 ℃ added water (9.7kg), and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more, and the mixture was stirred for 1 hour or more at 5 ℃ less. (9.45kg, dry powder rate 8.47kg, 13.7mol which was centrifuged obtained crystals were washed with a mixture of water (32kg) and ethanol (51kg), was obtained as a moist powder the title compound, 77% overall yield from the previous step).
1 H-NMR (CDCl 3) δ: 1.20 (3H, t, J = 7.5Hz), 2.60 (2H, q, J = 7.5Hz), 3.50 (3H, s), 3 .76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J = 2.8,11 .9 Hz), 4.33 (1H, dd, J = 4.5,11.9 Hz) ,4.36-4 .40 (1H, m) ,5.11-5 .20 (3H, m), 5 .41 (1H, d, J = 10.0Hz), 5.51 (1H, t, J = 10.0Hz) ,7.07-7 .11 (4H, m), 7.14 (1H, d, J = 7.8Hz), 7.19 (1H, dd, J = 1.5,7.8 Hz), 7.31 (1H, d, J = 1.5Hz).
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
Step 7: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-6 , 4 ‘, 5′-Preparation of triol’ – -3 [(3H), 2'-[2H] pyran isobenzofuran] spiro – (hydroxymethyl) ‘
(1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3′, 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – wet powder spiro [(3H), 2'-[2H] pyran isobenzofuran -1] (8.92kg, In addition at 20 ℃ (4mol / L, 30.02kg, the 104.2mol) aqueous solution of sodium hydroxide, 1 hour the reaction mixture to a solution of (28kg) ethylene glycol dimethyl ether dry end conversion 8.00kg, of 12.9mol) the mixture was stirred. And the organic layer was separated by addition of water (8.0kg) in the mixture. The ethyl acetate aqueous sodium chloride solution (25 wt%, 40kg) and a (36kg) in the organic layer and the aqueous layer was removed after washing. The washed again aqueous sodium chloride solution (25 wt%, 40kg) in the organic layer was evaporated under reduced pressure. Were added and acetone (32.0kg) water (0.8kg) residue was obtained. After the solvent was evaporated under reduced pressure, dissolved in acetone (11.7kg) in water (15.8kg) and the residue obtained was cooled to below 5 ℃. Was added below 10 ℃ water (64kg) to the mixture, and the mixture was stirred for 1 hour at below 10 ℃. Centrifuge the resulting crystals were washed with a mixture of water (8.0kg) and (1.3kg) acetone. For 8 hours through-flow drying 13 ~ 16 ℃ temperature ventilation, under the conditions of 24-33% relative humidity the wet powder, the monohydrate crystal (3.94kg, 9.7mol, 75% yield) of the title compound I was obtained as: (4.502 wt% water content).
Method of measuring the amount of water:
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
MS (ESI +): 387 [M +1] +.
………………………..
Example 1 Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose Step 1: Synthesis of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one
To a solution of D-(+)-glucono-1,5-lactone (7.88 kg) and N-methylmorpholine (35.8 kg) in tetrahydrofuran (70 kg) was added trimethylsilyl chloride (29.1 kg) at 40° C. or below, and then the mixture was stirred at a temperature from 30° C. to 40° C. for 2 hours. After the mixture was cooled to 0° C., toluene (34 kg) and water (39 kg) were added thereto. The organic layer was separated and washed with an aqueous solution of 5% sodium dihydrogen phosphate (39.56 kg×2) and water (39 kg×1). The solvent was evaporated under reduced pressure to give the titled compound as an oil. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74-3.83 (3H, m), 3.90 (1H, t, J=8.0 Hz), 3.99 (1H, d, J=8.0 Hz), 4.17 (1H, dt, J=2.5, 8.0 Hz).
Step 2: Synthesis of 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene
Under a nitrogen atmosphere, to a solution of 2,4-dibromobenzyl alcohol (40 g, 0.15 mol) in tetrahydrofuran (300 ml) was added 2-methoxypropene (144 ml, 1.5 mol) at room temperature, and then the mixture was cooled to 0° C. At the same temperature, pyridinium p-toluenesulfonic acid (75 mg, 0.30 mmol) was added and the mixture was stirred for 1 hour. The reaction mixture was poured into a saturated aqueous solution of sodium hydrogen carbonate cooled to 0° C., and extracted with toluene. The organic layer was washed with a saturated aqueous solution of sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to give the titled compound as an oil in quantitative yield. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 1.44 (6H, s), 3.22 (3H, 4.48 (2H, s), 7.42 (1H, d, J=8.0 Hz), 7.44 (1H, dd, J=1.5, 8.0 Hz), 7.68 (1H, d, J=1.5 Hz).
Step 3: Synthesis of 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran
Under a nitrogen atmosphere, 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene (70 g, 207 mmol), which was obtained in the previous step, was dissolved in toluene (700 mL) and t-butylmethyl ether (70 ml), and n-butyllithium in hexane (1.65 M, 138 ml, 227 mmol) was added dropwise at 0° C. over 30 minutes. After the mixture was stirred for 1.5 hours at 0° C., the mixture was added dropwise to a solution of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one (Example 1, 108 g, 217 mol) in tetrahydrofuran (507 ml) at −78° C., and the reaction mixture was stirred for 2 hours at the same temperature. Triethylamine (5.8 ml, 41 mmol) and trimethylsilyl chloride (29.6 ml, 232 mmol) were added thereto, and the mixture was warmed to 0° C. and stirred for 1 hour to give a solution containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-bromo-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran.
The resulting solution was cooled to −78° C., and n-butyllithium in hexane (1.65 M, 263 ml, 434 mmol) was added dropwise thereto at the same temperature. After the mixture was stirred at −78° C. for 30 minutes, 4-ethylbenzaldehyde (62 ml, 455 mmol) was added dropwise at −78° C., and the mixture was stirred at the same temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the organic layer was separated, and washed with water. The solvent was evaporated under reduced pressure to give a product containing the titled compound as an oil (238 g). The product was used in the next step without further purification.
A portion of the oil was purified by HPLC (column: Inertsil ODS-3, 20 mm I.D.×250 mm; acetonitrile, 30 mL/min) to give four diastereomers of the titled compound (two mixtures each containing two diastereomers).
Mixture of Diastereomers 1 and 2:
1H-NMR (500 MHz, CDCl3) δ: −0.47 (4.8H, s), −0.40 (4.2H, s), −0.003-0.004 (5H, m), 0.07-0.08 (1314, m), 0.15-0.17 (18H, m), 1.200 and 1.202 (3H, each t, J=8.0 Hz), 1.393 and 1.399 (3H, each s), 1.44 (3H, s), 2.61 (2H, q, J=8.0 Hz), 3.221 and 3.223 (3H, each s), 3.43 (1H, t, J=8.5 Hz), 3.54 (1H, dd, J=8.5, 3.0 Hz), 3.61-3.66 (1H, m), 3.80-3.85 (3H, m), 4.56 and 4.58 (1H, each d, J=12.4 Hz), 4.92 and 4.93 (1H, each d, J=12.4 Hz), 5.80 and 5.82 (1H, each d, J=3.0 Hz), 7.14 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.50-7.57 (2H, m).
MS (ESI+): 875 [M+Na]+.
Mixture of Diastereomers 3 and 4:
1H-NMR (500 MHz, toluene-d8, 80° C.) δ: −0.25 (4H, s), −0.22 (5H, s), 0.13 (5H, s), 0.16 (4H, s), 0.211 and 0.214 (9H, each s), 0.25 (9H, s), 0.29 (9H, s), 1.21 (3H, t, J=7.5 Hz), 1.43 (3H, s), 1.45 (3H, s), 2.49 (2H, q, J=7.5 Hz), 3.192 and 3.194 (3H, each s), 3.91-4.04 (4H, m), 4.33-4.39 (2H, m), 4.93 (1H, d, J=14.5 Hz), 5.10-5.17 (1H, m), 5.64 and 5.66 (1H, each s), 7.03 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.59-7.64 (1H, m), 7.87-7.89 (1H, m).
MS (ESI+): 875 [M+Na]+.
Step 4: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose
Under a nitrogen atmosphere, the oil containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran (238 g), which was obtained in the previous step, was dissolved in acetonitrile (693 ml). Water (37 ml) and 1N HCl aq (2.0 ml) were added and the mixture was stirred at room temperature for 5.5 hours. Water (693 ml) and n-heptane (693 ml) were added to the reaction mixture and the aqueous layer was separated. The aqueous layer was washed with n-heptane (693 ml×2), and water was evaporated under reduced pressure to give a product containing water and the titled compound (a diastereomer mixture) as an oil (187 g). The product was used in the next step without further purification.
1H-NMR (500 MHz, CD3OD) δ: 1.200 (3H, t, J=7.7 Hz), 1.201 (3H, t, J=7.7 Hz), 2.61 (2H, q, J=7.7 Hz), 3.44-3.48 (1H, m), 3.63-3.68 (111, m), 3.76-3.84 (4H, m), 5.09 (1H, d, J=12.8 Hz), 5.15 (1H, d, J=12.8 Hz), 5.79 (1H, s), 7.15 (2H, d, J=7.7 Hz), 7.24 and 7.25 (1H, each d, J=8.4 Hz), 7.28 (2H, d, J=7.7 Hz), 7.36 (1H, dd, J=8.4, 1.5 Hz), 7.40-7.42 (114, m).
MS (ESI+): 425 [M+Na]+.
Step 5: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (crude product)
To a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (187 g), which was obtained in the previous step, in 1,2-dimethoxyethane (693 ml) was added 5% Pd/C (26 g, 6.2 mmol, water content ratio: 53%), and the mixture was stirred in the atmosphere of hydrogen gas at room temperature for 4 hours. After filtration, the filtrate was evaporated under reduced pressure to give an oil containing the titled compound (59 g). The purity of the resulting product was 85.7%, which was calculated based on the area ratio measured by HPLC. The product was used in the next step without further purification.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (3H, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Cadenza CD-C18 50 mm P/NCD032
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Gradient operation: Eluent B: 5% to 100% (6 min), 100% (2 min)
Flow rate: 1.0 mL/min
Temperature: 35.0° C.
Detection wavelength: 210 nm
Step 6: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose
Under a nitrogen atmosphere, to a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (59 g) and 4-(dimethylamino)pyridine (175 g, 1436 mmol) in acetonitrile (1040 ml) was added dropwose methyl chloroformate (95 ml, 1231 mmol) at 0° C. The mixture was allowed to warm to room temperature while stirred for 3 hours. After addition of water, the mixture was extracted with isopropyl acetate. The organic layer was washed with an aqueous solution of 3% potassium hydrogensulfate and 20% sodium chloride (three times) and an aqueous solution of 20% sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. To the resulting residue was added ethanol (943 mL) and the mixture was heated to 75° C. to dissolve the residue. The mixture was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (472 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound (94 g). To the product (91 g) was added ethanol (1092 ml), and the product was dissolved by heating to 75° C. The solution was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (360 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound [83 g, total yield from 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene used in Step 3: 68%].
1H-NMR (CDCl3) δ: 1.20 (3H, t, J=7.5 Hz), 2.60 (2H, q, J=7.5 Hz), 3.50 (3H, s), 3.76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J=2.5, 11.8 Hz), 4.33 (1H, dd, J=4.5, 12.0 Hz), 4.36-4.40 (1H, m), 5.11-5.20 (3H, m), 5.41 (1H, d, J=10.0 Hz), 5.51 (1H, t, J=10.0 Hz), 7.07-7.11 (4H, m), 7.14 (1H, d, J=7.5 Hz), 7.19 (1H, dd, J=1.5, 7.8 Hz), 7.31 (1H, d, J=1.5 Hz).
MS (ESI+): 619 [M+1]+, 636 [M+18]+.
Another preparation was carried out in the same manner as Step 6, except that a seed crystal was not used, to give the titled compound as a crystal.
Step 7: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose
To a solution of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose (8.92 kg as wet powder, corresponding to 8.00 kg of dry powder) in 1,2-dimethoxyethane (28 kg) was added a solution of sodium hydroxide (4 mol/L, 30.02 kg) at 20° C., and the mixture was stirred for 1 hour. Water (8.0 kg) was added to the mixture and the layers were separated. To the organic layer were added an aqueous solution of 25% sodium chloride (40 kg) and ethyl acetate (36 kg). The organic layer was separated, washed with an aqueous solution of 25% sodium chloride (40 kg), and the solvent was evaporated under reduced pressure. The purity of the resulting residue was 98.7%, which was calculated based on the area ratio measured by HPLC. To the resulting residue were added acetone (32.0 kg) and water (0.8 kg), and the solvent was evaporated under reduced pressure. To the resulting residue were added acetone (11.7 kg) and water (15.8 kg), and the solution was cooled to 5° C. or below. Water (64 kg) was added to the solution at 10° C. or below, and the mixture was stirred at the same temperature for 1 hour. The resulting crystal was collected by centrifugation, and washed with a mixture of acetone (1.3 kg) and water (8.0 kg). The resulting wet powder was dried by ventilation drying under a condition at air temperature of 13 to 16° C. and relative humidity of 24% to 33% for 8 hours, to give a monohydrate crystal (water content: 4.502%) of the titled compound (3.94 kg). The purity of the resulting compound was 99.1%, which was calculated based on the area ratio measured by HPLC.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (311, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Capcell pack ODS UG-120 (4.6 mm I.D.×150 mm, 3 μm, manufactured by Shiseido Co., Ltd.)
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Mobile phase sending: Concentration gradient was controlled by mixing Eluent A and Eluent B as indicated in the following table.
| TABLE 1 | ||||
| Time from | ||||
| injection (min) | Eluent A (%) | Eluent B (%) | ||
| 0 to 15 | 90→10 | 10→90 | ||
| 15 to 17.5 | 10 | 90 | ||
| 17.5 to 25 | 90 | 10 | ||
Flow rate: 1.0 mL/min
Temperature: 25.0° C.
Detection wavelength: 220 nm
Method for Measurement of Water Content:
Analysis method: coulometric titration method
KF analysis apparatus: Type KF-100 (trace moisture measuring apparatus manufactured by Mitsubishi Chemical Corporation)
Anode solution: Aquamicron AX (manufactured by Mitsubishi Chemical Corporation)
Cathode solution: Aquamicron CXU (manufactured by Mitsubishi Chemical Corporation)
…………………..
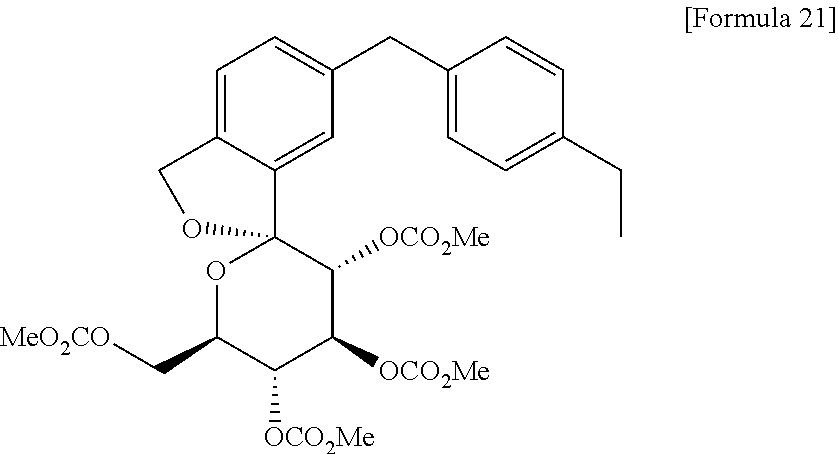
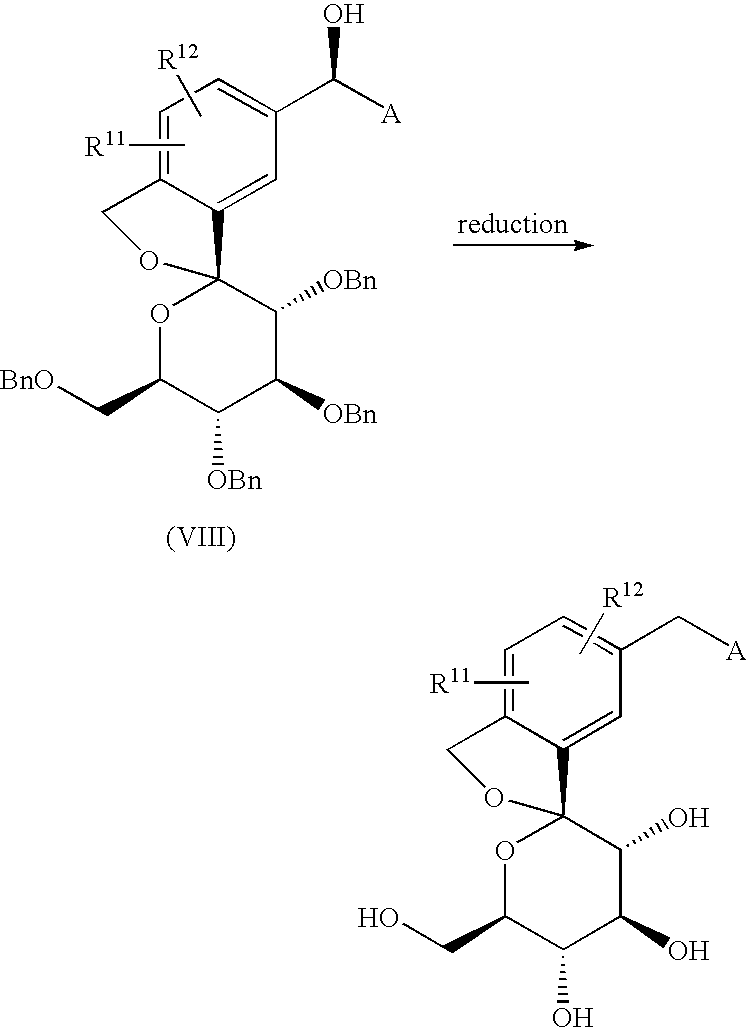
The compound of the present invention can be synthesized as shown in Scheme 1:
wherein R11 and R12 have the same meaning as defined above for substituents on Ar1, A is as defined above, and P represents a protecting group for a hydroxyl group.
2 Sergliflozin
Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2..........TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.

MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.

Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
 Sergliflozin
Sergliflozin
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.

[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of ...
onlinelibrary.wiley.com974 × 740Search by image
Design, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
- World Health Organization (2008). "International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59". WHO Drug Information 22 (1): 66.
- "Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate". American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). "Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level". J Pharmacol Exp Ther 320 (1): 323–30.doi:10.1124/jpet.106.110296. PMID 17050778.
- Prous Science: Molecule of the Month November 2007
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
Sergliflozin
Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2..........TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Sergliflozin
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of ...
onlinelibrary.wiley.com974 × 740Search by image
Design, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
- World Health Organization (2008). "International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59". WHO Drug Information 22 (1): 66.
- "Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate". American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). "Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level". J Pharmacol Exp Ther 320 (1): 323–30.doi:10.1124/jpet.106.110296. PMID 17050778.
- Prous Science: Molecule of the Month November 2007
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
Sergliflozin
Sergliflozin
408504-26-7 cas no
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]oxan-2-yl]methyl carbonate
2-(4-methoxybenzyl)phenyl 6-O-ethoxycarbonyl-beta-D-glucopyranoside
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-[2-[(4-methoxyphenyl)methyl]phenoxy]tetrahydropyran-2-yl]methyl carbonate
ethyl [(2R,3S,4S,5R,6S)-3,4,5-trihydroxy-6-{2-[(4-methoxyphenyl)methyl]phenoxy}oxan-2-yl]methyl carbonate
PHASE 2..........TYPE 3 DIABETES AND OBESITY
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes and obesity.
GW-869682; GW-869682X; KGT-1251
MW 448.4, C23H28O9
KISSEI INNOVATOR
GSK DEVELOPER
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
Sergliflozin etabonate (INN/USAN,[1][2] codenamed GW869682X) is an investigational anti-diabetic drug being developed by GlaxoSmithKline. It did not undergo further development after phase II.
Sergliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3][4]
Sergliflozin
Chemistry
Etabonate refers to the ethyl carbonate group. The remaining structure, which is the active substance, is called sergliflozin.
[PDF] Design, Syntheses, and SAR Studies of Carbocyclic Analogues of ...
onlinelibrary.wiley.com974 × 740Search by image
Design, Syntheses, and SAR Studies of Carbocyclic Analogues of Sergliflozin as Potent SodiumDependent Glucose Cotransporter 2 In
Sergliflozin Etabonate is a benzylphenol glucoside and selective sodium-glucose co-transporter subtype 2 (SGLT2) inhibitor with antihyperglycemic activity. Its prodrug form, sergliflozin etabonate, is orally available and is converted to sergiflozin upon absorption.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
- World Health Organization (2008). "International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59". WHO Drug Information 22 (1): 66.
- "Statement on a nonproprietary name adopted by the USAN council: Sergliflozin etabonate". American Medical Association. Retrieved 2008-08-10.
- Katsuno K, Fujimori Y, Takemura Y, et al. (January 2007). "Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level". J Pharmacol Exp Ther 320 (1): 323–30.doi:10.1124/jpet.106.110296. PMID 17050778.
- Prous Science: Molecule of the Month November 2007
| Patent | Submitted | Granted |
|---|---|---|
| Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat [US2008045466] | 2008-02-21 | |
| NOVEL SUBSTITUTED TETRAHYDRONAPHTHALENES, PROCESS FOR THE PREPARATION THEREOF AND THE USE THEREOF AS MEDICAMENTS [US2010249097] | 2010-09-30 | |
| (CARBOXYLALKYLENEPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF AND USE OF SAME AS A MEDICAMENT [US2010261645] | 2010-10-14 | |
| (CYCLOPROPYLPHENYL)PHENYLOXAMIDES, METHOD FOR THE PRODUCTION THEREOF, AND USE OF SAME AS A MEDICAMENT [US8148375] | 2010-10-14 | 2012-04-03 |
| Crystals of glucopyranosyloxybenzyl benzene derivative [US7371730] | 2005-06-02 | 2008-05-13 |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US2005065098] | 2005-03-24 | |
| Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same [US6872706] | 2004-01-29 | 2005-03-29 |
| Patent | Submitted | Granted |
|---|---|---|
| PROGRESSION INHIBITOR FOR DISEASE ATTRIBUTED TO ABNORMAL ACCUMULATION OF LIVER FAT [US2009286751] | 2009-11-19 | |
| THERAPEUTIC USES OF SGLT2 INHIBITORS [US2011077212] | 2011-03-31 | |
| PHARMACEUTICAL COMPOSITION COMPRISING A SGLT2 INHIBITOR IN COMBINATION WITH A DPP-IV INHIBITOR [US2011098240] | 2011-04-28 | |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Heterocycle-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising them and use thereof [US2011046105] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011046185] | 2011-02-24 | |
| Arylchalcogenoarylalkyl-substituted imidazolidine-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011053947] | 2011-03-03 | |
| Novel aromatic fluoroglycoside derivatives, pharmaceuticals comprising said compounds and the use thereof [US2011059910] | 2011-03-10 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 |
|
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(4-methoxybenzyl)phenyl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 408504-26-7 |
| ATC code | None |
| PubChem | CID: 9824918 |
| IUPHAR/BPS | 4587 |
| ChemSpider | 21234810 |
| ChEMBL | CHEMBL450044 |
| Chemical data | |
| Formula | C23H28O9 |
| Molecular mass | 448.463 g/mol |
//////////
CCOC(=O)OCC1C(C(C(C(O1)OC2=CC=CC=C2CC3=CC=C(C=C3)OC)O)O)O
CCOC(=O)OCC1C(C(C(C(O1)Oc2ccccc2Cc3ccc(cc3)OC)O)O)O
2 DAPAGLIFLOZIN
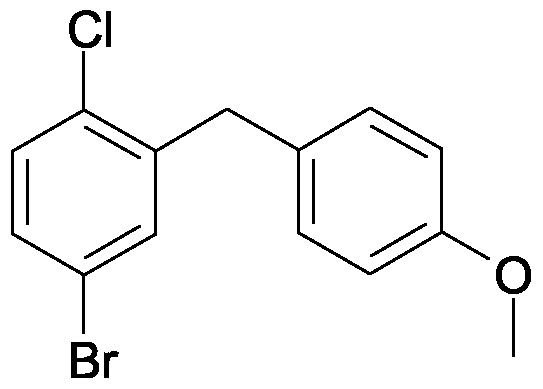
 DAPAGLIFLOZIN, BMS-512148(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,cas 461432-26-8
DAPAGLIFLOZIN, BMS-512148(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,cas 461432-26-8Molecular Formula: C21H25ClO6 Molecular Weight: 408.87 Bristol-Myers Squibb (Originator)
AstraZenecaTYPE 2 DIABETES,SGLT-2 Inhibitorslaunched 2012, as forxiga in EU Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.US-------In 2011, the product was not recommended for approval by the FDA's Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.US-------In 2011, the product was not recommended for approval by the FDA's Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.SWILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013--
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration's (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.The FDA is not bound by the Advisory Committee's recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014. Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia...........................................................................PATENTSWO 2010138535WO 2011060256WO 2012041898WO 2012163990WO 2013068850WO 2012163546WO 2013068850WO 2013079501
Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia...........................................................................PATENTSWO 2010138535WO 2011060256WO 2012041898WO 2012163990WO 2013068850WO 2012163546WO 2013068850WO 2013079501 Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin's method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions.
Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin's method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions. The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- "FDA panel advises against approval of dapagliflozin". 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4'-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ; Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,. WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,. WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,. WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ; Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 - ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,. WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;.. U.S. Pat Appl Publ,. 20,040,138,439
 dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabeticcomplications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.Approximately 100 million people worldwide suffer from type II diabetes (NIDDM - non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .
dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabeticcomplications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.Approximately 100 million people worldwide suffer from type II diabetes (NIDDM - non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 . formula 1WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2
formula 1WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2 formula 2The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in apharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystallinedapagliflozin solvates which additionally contain water molecules (see e.g.Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiralsubstance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps......WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesityExample 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystallinedapagliflozin solvates which additionally contain water molecules (see e.g.Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiralsubstance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided....................................
formula 2The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in apharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystallinedapagliflozin solvates which additionally contain water molecules (see e.g.Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiralsubstance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps......WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesityExample 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystallinedapagliflozin solvates which additionally contain water molecules (see e.g.Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiralsubstance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided....................................In Vitro Characterization and Pharmacokinetics of Dapagliflozin ...
dmd.aspetjournals.org/content/.../DMD29165_supplemental_data_.doc
 Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,64.5, 63.1, 39.2, 15.2.HRMS calculated for C21H25ClNaO6 (M+Na)+For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.: 431.1237; found 431.1234. Anal. CalcdSECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58............................HPLC
Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,64.5, 63.1, 39.2, 15.2.HRMS calculated for C21H25ClNaO6 (M+Na)+For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.: 431.1237; found 431.1234. Anal. CalcdSECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58............................HPLC- HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
- Eluent A: 0.1 % formic acid in water
- Eluent B: 0.1 % formic acid in acetonitrile
- Injection volume: 3 mL
- Flow: 0.7 mL/min
- Gradient:Time [min] [%] B 0.0 25 25.0 65 26.0 70 29.0 70 29.5 25 35.0 25 ..........................
4 IPRAGLIFLOZIN
Ipragliflozin
ASP-1941 , 1(S)-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-1-deoxy-beta-D-glucopyranose L-proline cocrystal
Kotobuki (Originator)
| (1S)-1,5-Anhydro-1-C-[3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl]-D-glucitol |
| Molecular Formula | C21H21FO5S | |
| Molecular Weight | 404.45 | |
| CAS Registry Number | 761423-87-4 |
Ipragliflozin (formerly ASP1941) has been filed in Japan on the back of phase III trials which showed that it could provide significant reductions in glycated haemoglobin levels (HbA1c) levels - a marker of glucose control over time - compared to placebo
According to Astellas' latest R&D pipeline update in February 2013, Astellas is developing ipragliflozin only in Japan. The same document in August 2012 indicated it was also carrying out phase II studies with the drug in the US and Japan.
Astellas Pharma Inc.: Submits Application for Marketing Approval of
Ipragliflozin (ASP1941), SGLT2 Inhibitor for Treatment of
Type 2 Diabetes, in Japan
TOKYO, March 13, 2013 - Astellas Pharma Inc. (“Astellas”; Tokyo:4503; President and CEO:
Yoshihiko Hatanaka) announced today that it has submitted a market authorization application for aSGLT2 inhibitoripragliflozin (generic name; development code: ASP1941) to the Ministry of Health, Labour and Welfare in Japan seeking an approval forthe indication of type 2 diabetes.
Ipragliflozin is a selective SGLT2 (sodium-glucose co-transporter 2)inhibitor discovered through research collaboration with Kotobuki Pharmaceutical Co., Ltd. SGLTs are membrane proteins that
exist on the cell surface and transfer glucose into cells. SGLT2 is a subtype of the sodium-glucose co-transporters and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.
Ipragliflozin reduces blood glucose levels by inhibiting the reuptake of glucose.
In the Phase III pivotal study in monotherapy for type 2 diabetesin Japan, ipragliflozin
demonstrated significant decreases of HbA1c, an index of glycemic control, in change from baseline compared to placebo. Based on the safety resultsin this study, ipragliflozin was safe and well tolerated. Patients with type 2 diabetes generally need combination therapy, so it is important
for a novel oral hypoglycemic agent to be safe to use with existing diabetes therapies. In this regard, Astellas has conducted six Phase III studies to investigate the safety and efficacy of ipragliflozin
used in combination with other hypoglycemic agentsfor a long term period. In these Phase IIIstudies, effectiveness and favorable safety of ipragliflozin was confirmed even in combination with
other hypoglycemic agents.
Astellas expects to provide an additional therapeutic option and further contribute to the treatment of type 2 diabetes by introducing ipragliflozin, an oral hypoglycemic agent with a novel mechanism
of action, into the Japanese market.
About Type 2 Diabetes
Diabetes (medically known as diabetes mellitus) is a disorder in which the body has difficulty regulating its blood glucose (sugar) level. There are two major types of diabetes: type 1 and type 2.
Type 2 diabetes (formerly called non-insulin-dependent diabetes mellitus or adult-onset diabetes) is a disorder that is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. Patients are instructed to increase exercise and diet restrictions, but most
require treatment with an anti-diabetic agent to control blood glucose.
Ipragliflozin (ASP1941), SGLT2 Inhibitor for Treatment of
Type 2 Diabetes, in Japan
TOKYO, March 13, 2013 - Astellas Pharma Inc. (“Astellas”; Tokyo:4503; President and CEO:
Yoshihiko Hatanaka) announced today that it has submitted a market authorization application for aSGLT2 inhibitoripragliflozin (generic name; development code: ASP1941) to the Ministry of Health, Labour and Welfare in Japan seeking an approval forthe indication of type 2 diabetes.
Ipragliflozin is a selective SGLT2 (sodium-glucose co-transporter 2)inhibitor discovered through research collaboration with Kotobuki Pharmaceutical Co., Ltd. SGLTs are membrane proteins that
exist on the cell surface and transfer glucose into cells. SGLT2 is a subtype of the sodium-glucose co-transporters and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.
Ipragliflozin reduces blood glucose levels by inhibiting the reuptake of glucose.
In the Phase III pivotal study in monotherapy for type 2 diabetesin Japan, ipragliflozin
demonstrated significant decreases of HbA1c, an index of glycemic control, in change from baseline compared to placebo. Based on the safety resultsin this study, ipragliflozin was safe and well tolerated. Patients with type 2 diabetes generally need combination therapy, so it is important
for a novel oral hypoglycemic agent to be safe to use with existing diabetes therapies. In this regard, Astellas has conducted six Phase III studies to investigate the safety and efficacy of ipragliflozin
used in combination with other hypoglycemic agentsfor a long term period. In these Phase IIIstudies, effectiveness and favorable safety of ipragliflozin was confirmed even in combination with
other hypoglycemic agents.
Astellas expects to provide an additional therapeutic option and further contribute to the treatment of type 2 diabetes by introducing ipragliflozin, an oral hypoglycemic agent with a novel mechanism
of action, into the Japanese market.
About Type 2 Diabetes
Diabetes (medically known as diabetes mellitus) is a disorder in which the body has difficulty regulating its blood glucose (sugar) level. There are two major types of diabetes: type 1 and type 2.
Type 2 diabetes (formerly called non-insulin-dependent diabetes mellitus or adult-onset diabetes) is a disorder that is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. Patients are instructed to increase exercise and diet restrictions, but most
require treatment with an anti-diabetic agent to control blood glucose.
structure:
The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor "redox economy" (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction
Glucose Gluconoloctone Hemiketal a-anomer β-anomer
R = protecting group
The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.
R = protecting group; R' = H or alkyl
The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.
or Zn
The glycosyl leaving group substitution method: U.S. Patent 7,847,074, also disclosed a method comprising the substitution of a leaving group located at CI of a hydroxy-protected glucosyl species, such as a glycosyl halide, with a metalated aryl compound to prepare SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar method to prepare the SGLT2 inhibitor canagliflozin and preferably diarylzinc complexes are used as nucleophiles along with tetra- >-pivaloyl protected glucosylbromide.
Glucose Glucosyl bromide β-anomer
Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
- The C-glycoside derivative represented by the formula (1) and its salt [hereinafter, they are referred to as "compound (1)" or "compound of formula (1)" in some cases] is known to be useful for treatment and prevention of diabetes such as insulin-dependent diabetes (type 1 diabetes), non-insulin-dependent diabetes (type 2 diabetes) and the like and various diabetes-related diseases including insulin-resistant diseases and obesity (Patent Literature 1).

- The method for producing the C-glycoside derivative represented by the formula (1), described in the Patent Literature 1 is understood to be represented by the below-shown reaction formula (I), by referring to the Examples and Reference Examples, described in the Patent Literature 1. Roughly explaining, it is a method which comprises reacting [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy]tert-butyl)dimethylsilane (synthesized in accordance with Reference Example 37 of the Literature) in a manner shown in Example 65 of the Literature, to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol and then reacting the obtained compound in accordance with Example 100 of the Literature to synthesize intended (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol.

- However, the method for producing the C-glycoside derivative of the formula (1), disclosed in the Patent Literature 1 is not industrially satisfactory in yield and cost, as is seen in later-shown Reference Example 1 of the present Description.
- For example, as described later, the method includes a step of low product yield (for example, a step of about 50% or lower yield) and the overall yield of the C-glycoside derivative (final product) represented by the formula (1) from the compound (8) (starting raw material) is below 7%; therefore, the method has problems in yield and cost from the standpoint of medicine production and has not been satisfactory industrially. In addition, the method includes an operation of purification by column chromatography which uses chloroform as part of purification solvents; use of such a solvent poses a problem in environmental protection and there are various restrictions in industrial application of such an operation; thus, the method has problems in providing an effective medicine.
- Also, an improved method of conducting an addition reaction with trimethylsilyl carbohydrate instead of benzyl carbohydrate and then conducting deprotection for acetylation, is known for a compound which has a structure different from that of the compound of the formula (1) but has a structure common to that of the compound of the formula (1) (Patent Literature 2). It is described in the Patent Literature 2 that the improved method enhances the overall yield to 6.2% from 1.4%. Even in the improved method, however, the yield is low at 6.2% which is far from satisfaction in industrial production.
- Patent Literature 1: WO 2004/080990 Pamphlet
- Patent Literature 2: WO 2006/006496 Pamphlet
- Into a tetrahydrofuran (20 ml) solution of benzo[b]thiophene (5.0 g) was dropwise added a n-hexane solution (25 ml) of n-butyl lithium (1.58 M) at -78°C in an argon atmosphere, followed by stirring at -78°C for 10 minutes. Into this solution was dropwise added a tetrahydrofuran (80 ml) solution of 5-bromo-2-fluorobenzaldehyde (8.0 g), followed by stirring at -78°C for 2.5 hours. The temperature of the reaction mixture was elevated to room temperature. Water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (10.5 g, yield: 83.6%).
1H-NMR (CDCl3): δ
2.74 (1H, d), 6.35 (1H, d), 6.93 (1H, dd), 7.14 (1H, s), 7.27-7.38 (2H, m), 7.39 (1H, m), 7.68 (1H, dd), 7.74 (2H, m)
- First step: synthesis of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol
Second step: synthesis of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane
- To a dimethylformamide (20 ml) solution of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (5.0 g) were added imidazole (1.3 g), a catalytic amount of 4-(dimethylamino)pyridine and tert-butyldimethylchlorosilane (2.7 g), followed by stirring at room temperature for 7 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (5.22 g, yield: 78.0%).
MS: 451 (M+)
1H-NMR (CDCl3): δ
0.05 (3H, s), 0.11 (3H, s), 0.95 (9H, s), 6.34 (1H, s), 6.91 (1H, t), 7.08 (1H, d), 7.23-7.38 (2H, m), 7.64-7.68 (1H, m), 7.75-7.28 (2H, m)
Third step: Synthesis of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose
- Into a tetrahydrofuran (15 ml) solution of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (1.5 g) was dropwise added a n-hexane solution (2.2 ml) of n-butyl lithium (1.58 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 30 minutes. Into the solution was dropwise added a tetrahydrofuran (20 ml) solution of 2,3,4,6-tetra-O-benzyl-D-glucono-1,5-lactone (1.9 g), followed by stirring at -78°C for 15 minutes and then at 0°C for 1.5 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/chloroform/acetone) to obtain 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g, yield: 50.2%). MS: 933 (M+Na)
Fourth step: Synthesis of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose
- To a tetrahydrofuran (15 ml) solution of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g) was added a tetrahydrofuran solution (2.0 ml) of tetrabutylammonium fluoride (1.0 M), followed by stirring at room temperature for 1 hour. The reaction mixture was concentrated per se. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (0.99 g, yield: 74.7%). MS: 819 (M+Na), 779 (M+H-H2O)
Fifth step: Synthesis of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol
- To an acetonitrile (5.0 ml) solution of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (500 mg) were added triethylsilane (175 mg) and boron trifluoride-diethyl ether complex (196 mg) in an argon atmosphere at -20°C, followed by stirring at -20°C for 5 hours. To the reaction mixture was added a saturated aqueous sodium bicarbonate solution, followed by extraction with chloroform. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (150 mg, yield: 30.2%) MS: 787 (M+Na)
1H-NMR (CDCl3): δ
3.42-3.48 (1H, m), 3.55-3.58 (1H, m), 3.72-3.78 (4H, m), 3.83 (1H, d), 4.14-4.30 (3H, m), 4.39 (1H, d), 4.51-4.67 (4H, m), 4.83-4.94 (2H, m), 6.86-6.90 (1H, m), 6.98 (1H, brs), 7.06-7.37 (24H, m), 7.57-7.60 (1H, m), 7.66-7.69 (1H, m)
Sixth step: Synthesis of (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol
- To a dichloromethane (10 ml) solution of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (137 mg) were added pentamethylbenzene (382 mg) and a n-heptane solution (0.75 ml) of boron trichloride (1.0 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 3 hours. Methanol was added to the reaction mixture, the temperature of the resulting mixture was elevated to room temperature, and the mixture was concentrated per se. The residue was purified by silica gel column chromatography (chloroform/methanol) to obtain (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN (63 mg, yield: 87.8%).
1H-NMR (CD3OD): δ
3.29-3.48 (4H, m), 3.68 (1H, dd), 3.87 (1H, dd), 4.11 (1H, d), 4.20-4.29 (2H, m), 7.03 (1H, s), 7.08 (1H, dd), 7.19-7.29 (2H, m), 7.35 (1H, m), 7.42 (1H, dd), 7.64 (1H, d), 7.72 (1H, d)
(1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN
.........................................................
5 EMPAGLIFLOZIN
Empagliflozin
BI-10773
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
M.Wt: 450.91
: C23H27ClO7
: C23H27ClO7
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral treatment of adults with type 2 diabetes (Phase III, expected to conclude by year’s end); Oral treatment of adults with type 2 diabetes plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. Empagliflozin is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra. The Empagliflozin phase III clinical trial program will include about 14,500 patients. The program consists of twelve ongoing international phase III clinical trials, including a large cardiovascular outcomes trial.
Empagliflozin is a novel SGLT2 inhibitor that is described for the treatment or improvement in glycemic control in patients with type 2 diabetes mellitus, for example in WO 05/092877, WO 06/117359, WO 06/120208, WO 2010/092126, WO 2010/092123, WO 2011/039107, WO 2011/039108. The use of a SGLT2 inhibitor in a method for treating obesity is described in WO 08/116,195
WO2005/092877
WO2006/117359 MP 149 DEG CENT
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment oftype 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in theproximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
As of December 2013, empagliflozin is in phase III clinical trials.[2]

When taken in dosages of 10 or 25 mg once a day, the incidence of adverse events was similar to placebo. However, there was a higher frequency of genital infections at both the 10 mg and the 25 mg dosages.
1-chloro-4-(β-D-glucopyranos-1-yl)-2-[4-((S)-tetrahydrofuran-3-yloxy)-benzyl]-benzene of the formula
as described for example in WO 2005/092877. Methods of synthesis are described in the literature, for example WO 06/120208 and WO 2011/039108. According to this invention, it is to be understood that the definition of empagliflozin also comprises its hydrates, solvates and polymorphic forms thereof, and prodrugs thereof. An advantageous crystalline form of empagliflozin is described in WO 2006/117359 and WO 2011/039107 which hereby are incorporated herein in their entirety. This crystalline form possesses good solubility properties which enables a good bioavailability of the SGLT2 inhibitor. Furthermore, the crystalline form is physico-chemically stable and thus provides a good shelf-life stability of the pharmaceutical composition. Preferred pharmaceutical compositions, such as solid formulations for oral administration, for example tablets, are described in WO 2010/092126,
Example 1 : Synthesis of the fluoride VIII.1
Oxalylchloride (176kg; 1386mol; 1 ,14eq) is added to a mixture of 2-chloro-5-iodo benzoic acid (343kg; 1214mol) (compound IX.1 ), fluorobenzene (858kg) and N,N-dimethylformamide (2kg) within 3 hours at a temperature in the range from about 25 to 30°C (gas formation). After completion of the addition, the reaction mixture is stirred for additional 2 hours at a temperature of about 25 to 30°C. The solvent (291 kg) is distilled off at a temperature between 40 and 45°C (p=200mbar). Then the reaction solution (91 1 kg) is added to aluminiumchloride AICI3 (181 kg) and fluorobenzene (192kg) at a temperature between about 25 and 30°C within 2 hours. The reaction solution is stirred at the same temperature for about an additional hour. Then the reaction mixture is added to an amount of 570 kg of water within about 2 hours at a temperature between about 20 and 30°C and stirred for an additional hour. After phase separation the organic phase (1200kg) is separated into two halves (600kg each). From the first half of the organic phase solvent (172kg) is distilled off at a temperature of about 40 to 50°C (p=200mbar). Then 2-propanole (640kg) is added. The solution is heated to about 50°C and then filtered through a charcoal cartouche (clear filtration). The cartouche may be exchanged during filtration and washed with a
fluorobenzene/2-propanole mixture (1 :4; 40kg) after filtration. Solvent (721 kg) is distilled off at a temperature of about 40 to 50°C and p=200mbar. Then 2-propanole (240kg) is added at a temperature in the range between about 40 to 50°C. If the content of fluorobenzene is greater than 1 % as determined via GC, another 140kg of solvent are distilled off and 2- propanole (140kg) is added. Then the solution is cooled from about 50°C to 40°C within one hour and seeding crystals (50g) are added. The solution is further cooled from about 40°C to 20°C within 2 hours. Water (450kg) is added at about 20°C within 1 hour and the suspension is stirred at about 20°C for an additional hour before the suspension is filtered. The filter cake is washed with 2-propanole/water (1 :1 ; 800kg). The product is dried until a water level of <0.06%w/w is obtained. The second half of the organic phase is processed identically. A total of 410kg (94%yield) of product which has a white to off-white crystalline appearance, is obtained. The identity of the product is determined via infrared spectrometry.
Example 2: Synthesis of the ketone VII.1
To a solution of the fluoride VIII.1 (208kg), tetrahydrofuran (407kg) and (S)-3- hydroxytetrahydrofuran (56kg) is added potassium-ie f-butanolate solution (20%) in tetrahydrofuran (388kg) within 3 hrs at 16 to 25°C temperature. After completion of the addition, the mixture is stirred for 60min at 20°C temperature. Then the conversion is determined via HPLC analysis. Water (355kg) is added within 20 min at a temperature of 21 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature: 20°C). The stirrer is switched off and the mixture is left stand for 60 min (temperature: 20°C). The phases are separated and solvent is distilled off from the organic phase at 19 to 45°C temperature under reduced pressure. 2-Propanol (703kg) is added to the residue at 40 to 46°C temperature and solvent is distilled off at 41 to 50°C temperature under reduced pressure. 2-Propanol (162kg) is added to the residue at 47°C temperature and solvent is distilled off at 40 to 47°C temperature under reduced pressure. Then the mixture is cooled to 0°C within 1 hr 55 min. The product is collected on a centrifuge, washed with a mixture of 2- propanol (158kg) and subsequently with ie f.-butylmethylether (88kg) and dried at 19 to 43°C under reduced pressure. 227kg (91 ,8%) of product are obtained as colourless solid. The identity of the product is determined via infrared spectrometry.
Example 3: Synthesis of the iodide V.1
To a solution of ketone VII.1 (217,4kg) and aluminium chloride (AICI3; 81 ,5kg) in toluene (366,8kg) is added 1 ,1 ,3,3-tetramethyldisiloxane (TMDS, 82,5kg) within 1 hr 30 min
(temperature: 18-26°C). After completion of the addition, the mixture is stirred for additional 1 hr at a temperature of 24°C. Then the conversion is determined via HPLC analysis.
Subsequently the reaction mixture is treated with acetone (15,0kg), stirred for 1 hr 5 min at 27°C temperature and the residual TMDS content is analyzed via GC. Then a mixture of water (573kg) and concentrated HCI (34kg) is added to the reaction mixture at a temperature of 20 to 51 °C (aqueous quench). The reaction mixture is stirred for 30 min (temperature:
51 °C). The stirrer is switched off and the mixture is left stand for 20 min (temperature: 52°C). The phases are separated and solvent is distilled off from the organic phase at 53-73°C temperature under reduced pressure. Toluene (52,8kg) and ethanol (435,7kg) are added to the residue at 61 to 70°C temperature. The reaction mixture is cooled to 36°C temperature and seeding crystals (0,25kg) are added. Stirring is continued at this temperature for 35 min. Then the mixture is cooled to 0 to 5°C and stirred for additional 30 min. The product is collected on a centrifuge, washed with ethanol (157kg) and dried at 15 to 37°C under reduced pressure. 181 kg (82,6%) of product are obtained as colourless solid. The identity of the product is determined via the HPLC retention time.
Example 4: Synthesis of the lactone IV.1
A suspension of the D-(+)-gluconic acid-delta-lactone IVa.1 (42,0kg), tetrahydrofuran (277,2kg), 4-methylmorpholine (NMM; 152,4kg) and 4-dimethylaminopyridine (DMAP;
1 ,44kg) is treated with chlorotrimethylsilane (TMSCI; 130,8kg) within 50 min at 13 to 19°C. After completion of the addition stirring is continued for 1 hr 30 min at 20 to 22°C and the conversion is determined via HPLC analysis. Then n-heptane (216,4kg) is added and the mixture is cooled to 5°C. Water (143kg) is added at 3 to 5°C within 15 min. After completion of the addition the mixture is heated to 15°C and stirred for 15 min. The stirrer is switched off and the mixture is left stand for 15 min. Then the phases are separated and the organic layer is washed in succession two times with water (143kg each). Then solvent is distilled off at 38°C under reduced pressure and n-heptane (130kg) is added to the residue. The resulting solution is filtered and the filter is rinsed with n-heptane (63kg) (filter solution and product solution are combined). Then solvent is distilled off at 39 to 40°C under reduced pressure. The water content of the residue is determined via Karl-Fischer analysis (result: 0,0%).
1 12,4kg of the product is obtained as an oil (containing residual n-heptane, which explains the yield of >100%). The identity of the product is determined via infrared spectrometry.
Example 5a: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (267kg) in tetrahydrofuran (429kg) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 - 1 .1 mol/mol) (472kg) at -21 to -15°C temperature within 1 hr 50 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 3,45%. Then the lactone IV.1 (320kg) is added at -25 to -18°C temperature within 1 hr 25 min. The resulting mixture is stirred for further 1 hr 30 min at -13 to -18°C. On completion the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (938L; concentration: 10 %-weight) is added to the reaction mixture of a volume of about 2500L at -13 to 19°C within 1 hr 25 min.
The solvent is partially distilled off from the reaction mixture (residual volume: 1816-1905L) at 20 to 30°C under reduced pressure and 2-methyltetrahydrofuran (532kg) is added. Then the stirrer is switched off and the phases are separated at 29°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 2 to 3. Then solvent is distilled off from the organic phase at 30 to 33°C under reduced pressure and methanol (1202kg) is added followed by the addition of a solution of 1 ,25N HCI in methanol (75kg) at 20°C (pH = 0). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 20 to 32°C under reduced pressure and addition of methanol (409kg).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. 2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of 111.1 is greater or equal to 97,0% to 3,0%.
In this particular case both criteria are met. Triethylamin (14kg) is added (pH = 7,4) and solvent is distilled off under reduced pressure, acetonitrile (835kg) is added and further distilled under reduced pressure. This procedure is repeated (addition of acetonitrile: 694kg) and methylene chloride (640kg) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,27%).
The reaction mixture is then added within 1 hr 40 min at 10 to 19°C to a preformed mixture of AICI3 (176kg), methylene chloride (474kg), acetonitrile (340kg), and triethylsilane (205kg). The resulting mixture is stirred at 18 to 20°C for 70 min. After completion of the reaction, water (1263L) is added at 20 to 30°C within 1 hr 30 min and the mixture is partially distilled at 30 to 53°C under atmospheric pressure and the phases are separated. Toluene (698kg) is added to the organic phase and solvent is distilled off under reduced pressure at 22 to 33°C. The product is then crystallized by addition of seeding crystals (0,5kg) at 31 °C and water (267kg) added after cooling to 20°C. The reaction mixture is cooled to 5°C within 55 min and stirred at 3 to 5°C for 12 hrs. Finally the product is collected on a centrifuge as colourless, crystalline solid, washed with toluene (348kg) and dried at 22 to 58°C. 21 1 kg (73%) of product are obtained. The identity of the product is determined via the HPLC retention time.
Example 5b: Synthesis of the glucoside 11.1
To a solution of the iodide V.1 (30g) in tetrahydrofuran (55ml_) is added Turbogrignard solution (isopropylmagnesium chloride/lithium chloride solution, 14 weight-% iPrMgCI in THF, molar ratio LiCI : iPrMgCI = 0,9 - 1 .1 mol/mol) (53g) at -14 to -13°C temperature within 35 min. On completion of the addition the conversion is determined via HPLC analysis. The reaction is regarded as completed when the area of the peak corresponding to the iodide V.1 is smaller than 5,0% of the total area of both peaks, iodide V.1 and the corresponding desiodo compound of iodide V.1 . If the reaction is not completed, additional Turbogrignard solution is added until the criterion is met. In this particular case the result is 0,35%. Then the lactone IV.1 (36g) is added at -15 to -6°C temperature within 15 min. The resulting mixture is stirred for further 1 hr at -6 to -7°C. On completion, the conversion is determined via HPLC analysis (for information). On completion, a solution of citric acid in water (105mL;
concentration: 10 %-weight) is added to the reaction mixture at -15 to 10°C within 30 min. The solvent is partially distilled off from the reaction mixture (residual volume: 200mL) at 20 to 35°C under reduced pressure and 2-methyltetrahydrofuran (71 mL) is added. Then the mixture is stirred for 25min at 30°C. Then the stirrer is switched off and the phases are separated at 30°C. After phase separation the pH value of the organic phase is measured with a pH electrode (Mettler Toledo MT HA 405 DPA SC) or alternatively with pH indicator paper (such as pH-Fix 0-14, Macherey and Nagel). The measured pH value is 3. Then solvent is distilled off from the organic phase at 35°C under reduced pressure and methanol (126ml_) is added followed by the addition of a solution of 1 ,25N HCI in methanol (10,1 ml_) at 25°C (pH = 1 -2). Full conversion to the acetale 111.1 is achieved by subsequent distillation at 35°C under reduced pressure and addition of methanol (47ml_).
Completion of the reaction is obtained when two criteria are fulfilled:
1 ) The ratio of the sum of the HPLC-area of the alpha-form + beta-form of intermediate 111.1 relative to the area of intermediate llla.1 is greater or equal to 96,0% : 4,0%. In this particular case the ratio is 99,6% : 0,43%.
2) The ratio of the HPLC-area of the alpha-form of intermediate 111.1 to the beta-form of III.1 is greater or equal to 97,0% to 3,0%. In this particular case the ratio is 98,7% : 1 ,3%.
Triethylamin (2,1 mL) is added (pH = 9) and solvent is distilled off at 35°C under reduced pressure, acetonitrile (120ml_) is added and further distilled under reduced pressure at 30 to 35°C. This procedure is repeated (addition of acetonitrile: 102ml_) and methylene chloride (55ml_) is added to the resulting mixture to yield a mixture of the acetale 111.1 in acetonitrile and methylene chloride. The water content of the mixture is determined via Karl Fischer titration (result: 0,04%).
The reaction mixture is then added within 1 hr 5 min at 20°C to a preformed mixture of AICI3 (19,8g), methylene chloride (49ml_), acetonitrile (51 mL), and triethylsilane (23g). The resulting mixture is stirred at 20 to 30°C for 60 min. After completion of the reaction, water (156mL) is added at 20°C within 25 min and the mixture is partially distilled at 55°C under atmospheric pressure and the phases are separated at 33°C. The mixture is heated to 43°C and toluene (90mL) is added and solvent is distilled off under reduced pressure at 41 to 43°C. Then acetonitrile (1 OmL) is added at 41 °C and the percentage of acetonitrile is determined via GC measurement. In this particular case, the acetonitrile percentage is 27%- weight. The product is then crystallized by addition of seeding crystals (0,1 g) at 44°C and the mixture is further stirred at 44°C for 15min. The mixture is then cooled to 20°C within 60min and water (142mL) is added at 20°C within 30min. The reaction mixture is cooled to 0 to 5°C within 60 min and stirred at 3°C for 16 hrs. Finally the product is collected on a filter as colourless, crystalline solid, washed with toluene (80mL) and dried at 20 to 70°C. 20, 4g (62,6%) of product are obtained. The identity of the product is determined via the HPLC retention time.
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). "Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors". Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- "Empagliflozin". clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). "Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus". J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). "From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus". Curr Med Res Opin25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). "Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus". Endocr Pract 14 (6): 782–90. PMID 18996802.
[1]. Grempler R, Thomas L, Eckhardt M et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012 Jan;14(1):83-90.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
Abstract
AIMS: Empagliflozin is a selective sodium glucose cotransporter-2 (SGLT-2) inhibitor in clinical development for the treatment of type 2 diabetes mellitus. This study assessed pharmacological properties of empagliflozin in vitro and pharmacokinetic properties in vivo and compared its potency and selectivity with other SGLT-2 inhibitors. METHODS: [(14)C]-alpha-methyl glucopyranoside (AMG) uptake experiments were performed with stable cell lines over-expressing human (h) SGLT-1, 2 and 4. Two new cell lines over-expressing hSGLT-5 and hSGLT-6 were established and [(14)C]-mannose and [(14)C]-myo-inositol uptake assays developed. Binding kinetics were analysed using a radioligand binding assay with [(3)H]-labelled empagliflozin and HEK293-hSGLT-2 cell membranes. Acute in vivo assessment of pharmacokinetics was performed with normoglycaemic beagle dogs and Zucker diabetic fatty (ZDF) rats. RESULTS: Empagliflozin has an IC(50) of 3.1 nM for hSGLT-2. Its binding to SGLT-2 is competitive with glucose (half-life approximately 1 h). Compared with other SGLT-2 inhibitors, empagliflozin has a high degree of selectivity over SGLT-1, 4, 5 and 6. Species differences in SGLT-1 selectivity were identified. Empagliflozin pharmacokinetics in ZDF rats were characterised by moderate total plasma clearance (CL) and bioavailability (BA), while in beagle dogs CL was low and BA was high. CONCLUSIONS: Empagliflozin is a potent and competitive SGLT-2 inhibitor with an excellent selectivity profile and the highest selectivity window of the tested SGLT-2 inhibitors over hSGLT-1. Empagliflozin represents an innovative therapeutic approach to treat diabetes.
[2]. Thomas L, Grempler R, Eckhardt M et al. Long-term treatment with empagliflozin, a novel, potent and selective SGLT-2 inhibitor, improves glycaemic control and features of metabolic syndrome in diabetic rats. Diabetes Obes Metab. 2012 Jan;14(1):94-6.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
Abstract
Empagliflozin is a potent, selective sodium glucose co-transporter-2 inhibitor that is in development for the treatment of type 2 diabetes. This series of studies was conducted to assess the in vivo pharmacological effects of single or multiple doses of empagliflozin in Zucker diabetic fatty rats. Single doses of empagliflozin resulted in dose-dependent increases in urinary glucose excretion and reductions in blood glucose levels. After multiple doses (5 weeks), fasting blood glucose levels were reduced by 26 and 39% with 1 and 3 mg/kg empagliflozin, respectively, relative to vehicle. After 5 weeks, HbA1c levels were reduced (from a baseline of 7.9%) by 0.3 and 1.1% with 1 and 3 mg/kg empagliflozin, respectively, versus an increase of 1.1% with vehicle. Hyperinsulinaemic-euglycaemic clamp indicated improved insulin sensitivity with empagliflozin after multiple doses versus vehicle. These findings support the development of empagliflozin for the treatment of type 2 diabetes.
[3]. Luippold G, Klein T, Mark M, Grempler R. Empagliflozin, a novel potent and selective SGLT-2 inhibitor, improves glycaemic control alone and in combination with insulin in streptozotocin-induced diabetic rats, a model of type 1 diabetes mellitus. Diabetes Obes Metab. 2012 Jul;14(7):601-7.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
Abstract
AIM: Sodium glucose cotransporter-2 (SGLT-2) is key to reabsorption of glucose in the kidney. SGLT-2 inhibitors are in clinical development for treatment of type 2 diabetes mellitus (T2DM). The mechanism may be of value also in the treatment of type 1 diabetes mellitus (T1DM). This study investigated effects of the SGLT-2 inhibitor, empagliflozin, alone and in combination with insulin, on glucose homeostasis in an animal model of T1DM. METHODS: Sprague-Dawley rats were administered a single intraperitoneal injection of streptozotocin (STZ; 60 mg/kg). Acutely, STZ rats received two doses of insulin glargine with or without empagliflozin, and blood glucose was measured. In a subchronic study, STZ rats received empagliflozin alone, one or two insulin-releasing implants or a combination of one implant and empagliflozin over 28 days; blood glucose and HbA(1c) were measured. RESULTS: In the acute setting, empagliflozin in combination with 1.5 IU insulin induced a similar glucose-lowering effect as 6 IU insulin. Both interventions were more efficacious than monotherapy with 1.5 IU insulin. In the subchronic study, 12-h blood glucose profile on day 28 in the combination group was lower than with one implant, and similar to two implants. Plasma HbA(1c) was improved in the combination group and in animals with two implants. CONCLUSIONS: Empagliflozin reduced blood glucose levels in a T1DM animal model. Empagliflozin combined with low-dose insulin showed comparable glucose-lowering efficacy to treatment with high-dose insulin. Our data suggest that empagliflozin is an efficacious adjunctive-to-insulin therapy with the clinical potential for the treatment of T1DM.
[4]. Macha S, Rose P, Mattheus M et al. Lack of drug-drug interaction between empagliflozin, a sodium glucose cotransporter-2 inhibitor, and warfarin in healthy volunteers. Diabetes Obes Metab. 2012 Oct 24. doi: 10.1111/dom.12028. [Epub ahead of print]
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin's anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
Abstract
AIM: To investigate potential drug-drug interactions between empagliflozin and warfarin. MATERIALS AND METHODS: Healthy subjects (n=18) received empagliflozin 25 mg qd for 5 days (treatment A), followed by empagliflozin 25 mg qd for 7 days (days 6-12) with a single 25 mg dose of warfarin on day 6 (B), and a single 25 mg dose of warfarin alone (C), in an open-label, crossover study. Subjects received treatments in sequence AB_C or C_AB with a washout period of ≥14 days between AB and C or C and AB. RESULTS: Warfarin had no effect on empagliflozin area under concentration-time curve or maximum plasma concentration at steady-state (AUC(τ) (,ss) or C(max,ss) ): geometric mean ratios (GMRs) (90% confidence intervals [CI]) were 100.89% (96.86, 105.10) and 100.64% (89.79, 112.80), respectively. Empagliflozin had no effect on AUC from 0 hours to infinity (AUC(0) (-∞) ) or C(max) for R-warfarin or S-warfarin (GMRs [90% CI] for AUC(0) (-∞) : 98.49% [95.29, 101.80] and 95.88% [93.40, 98.43], respectively; C(max) : 97.89% [91.12, 105.15] and 98.88% [91.84, 106.47], respectively). Empagliflozin had no clinically relevant effects on warfarin's anticoagulant activity (international normalised ratio [INR]) (GMR [95% CI] for peak INR: 0.87 [0.73, 1.04]; area under the effect-time curve from 0 to 168 hours: 0.88 [0.79, 0.98]. No drug-related adverse events were reported for empagliflozin after monotherapy or combined administration. The combination of empagliflozin and warfarin was well tolerated. CONCLUSIONS: No drug-drug interactions were observed between empagliflozin and warfarin, indicating that empagliflozin and warfarin can be co-administered without dosage adjustments of either drug.
[5]. Sarashina A, Koiwai K, Seman LJ et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of Empagliflozin, a Sodium Glucose Cotransporter-2 (SGLT-2) Inhibitor, in Healthy Japanese Subjects. Drug Metab Pharmacokinet. 2012 Nov 13. [Epub ahead of print]
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
Abstract
This randomized, placebo-controlled within dose groups, double-blind, single rising dose study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of 1 mg to 100 mg doses of empagliflozin in 48 healthy Japanese male subjects. Empagliflozin was rapidly absorbed, reaching peak levels in 1.25 to 2.50 hours; thereafter, plasma concentrations declined in a biphasic fashion, with mean terminal elimination half-life ranging from 7.76 to 11.7 hours. Increase in empagliflozin exposure was proportional to dose. Oral clearance was dose independent and ranged from 140 to 172 mL/min. In the 24 hours following 100 mg empagliflozin administration, the mean (%CV) amount of glucose excreted in urine was 74.3 (17.1) g. The amount and the maximum rate of glucose excreted via urine increased with dose of empagliflozin. Nine adverse events, all of mild intensity, were reported by 8 subjects (7 with empagliflozin and 1 with placebo). No hypoglycemia was reported. In conclusion, 1 mg to 100 mg doses of empagliflozin had a good safety and tolerability profile in healthy Japanese male subjects. Exposure to empagliflozin was dose-proportional. The amount and rate of urinary glucose excretion were higher with empagliflozin than with placebo, and increased with empagliflozin dose.
..................................
formula A
Example I
(5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone
38.3 ml oxalyl chloride and 0.8 ml of dimethylformamide are added to a mixture of
100 g of 5-bromo-2-chloro-benzoic acid in 500 ml dichloromethane. The reaction mixture is stirred for 14 h, then filtered and separated from all volatile constituents in the rotary evaporator. The residue is dissolved in 150 ml dichloromethane, the solution is cooled to -5 0C, and 46.5 g of anisole are added. Then 51.5 g of aluminum trichloride are added batchwise so that the temperature does not exceed 5 0C. The solution is stirred for another 1 h at 1 to 5 0C and then poured onto crushed ice. The organic phase is separated, and the aqueous phase is extracted another three times with dichloromethane. The combined organic phases are washed with aqueous 1 M hydrochloric acid, twice with aqueous 1 M sodium hydroxide solution and with brine. Then the organic phase is dried, the solvent is removed and the residue is recrystallised in ethanol. Yield: 86.3 g (64% of theory)
Mass spectrum (ESI+): m/z = 325/327/329 (Br+CI) [M+H]+
Example Il
4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene
A solution of 86.2 g (5-bromo-2-chloro-phenyl)-(4-methoxy-phenyl)-methanone and 101.5 ml triethylsilane in 75 ml dichloromethane and 150 ml acetonitrile is cooled to 1O0C. Then with stirring 50.8 ml of boron trifluoride etherate are added so that the temperature does not exceed 2O0C. The solution is stirred for 14 h at ambient temperature, before another 9 ml triethylsilane and 4.4 ml boron trifluoride etherate are added. The solution is stirred for a further 3 h at 45 to 5O0C and then cooled to ambient temperature. A solution of 28 g potassium hydroxide in 70 ml of water is added, and the resulting mixture is stirred for 2 h. Then the organic phase is separated off and the aqueous phase is extracted another three times with diisopropylether. The combined organic phases are washed twice with aqueous 2 M potassium hydroxide solution and once with brine and then dried over sodium sulfate. After the solvent has been removed the residue is washed in ethanol, separated again and dried at 6O0C. Yield: 50.0 g (61 % of theory)
Mass spectrum (ESI+): m/z = 310/312/314 (Br+CI) [M+H]+
Example III
4-(5-bromo-2-chloro-benzyl)-phenol
A solution of 14.8 g 4-bromo-1-chloro-2-(4-methoxy-benzyl)-benzene in 150 ml dichloromethane is cooled in an ice bath. Then 50 ml of a 1 M solution of boron tribromide in dichloromethane are added, and the solution is stirred for 2 h at ambient temperature. The solution is then cooled in an ice bath again, and saturated aqueous potassium carbonate solution is added dropwise. At ambient temperature the mixture is adjusted with aqueous 1 M hydrochloric acid to a pH of 1 , the organic phase is separated, and the aqueous phase is extracted another three times with ethyl acetate. The combined organic phases are dried over sodium sulphate, and the solvent is removed completely. Yield: 13.9 g (98% of theory) Mass spectrum (ESI ): m/z = 295/297/299 (Br+CI) [M-HV
Example IV
r4-(5-bromo-2-chloro-benzyl)-phenoxyl-tert-butyl-dimethyl-silane
A solution of 13.9 g 4-(5-bromo-2-chloro-benzyl)-phenol in 140 ml dichloromethane is cooled in an ice bath. Then 7.54 g tert-butyldimethylsilylchlorid in 20 ml dichloromethane are added followed by 9.8 ml triethylamine and 0.5 g 4- dimethylaminopyridine. The solution is stirred for 16 h at ambient temperature and then diluted with 100 ml dichloromethane. The organic phase is washed twice with aqueous 1 M hydrochloric acid and once with aqueous sodium hydrogen carbonate solution and then dried over sodium sulfate. After the solvent has been removed the residue is filtered through silica gel (cyclohexane/ethyl acetate 100:1 ). Yield: 16.8 g (87% of theory) Mass spectrum (El): m/z = 410/412/414 (Br+CI) [M]+
Example V
2.3.4.6-tetrakis-O-(trimethylsilyl)-D-glucopyranone
A solution of 20 g D-glucono-1 ,5-lactone and 98.5 ml Λ/-methylmorpholine in 200 ml of tetrahydrofuran is cooled to -5 0C. Then 85 ml trimethylsilylchloride are added dropwise so that the temperature does not exceed 5 0C. The solution is then stirred for 1 h at ambient temperature, 5 h at 35 0C and again for 14 h at ambient temperature. After the addition of 300 ml of toluene the solution is cooled in an ice bath, and 500 ml of water are added so that the temperature does not exceed 100C. The organic phase is then separated and washed in each case once with aqueous sodium dihydrogen phosphate solution, water and brine. The solvent is removed, the residue is taken up in 250 ml of toluene, and the solvent is again removed completely. Yield: 52.5 g (approx. 90% pure)
Mass spectrum (ESI+): m/z = 467 [M+H]+
Example Vl
1-chloro-4-(β-D-qlucopyranos-1-yl)-2-(4-hvdroxybenzyl)-benzene
A solution of 4.0 g [4-(5-bromo-2-chloro-benzyl)-phenoxy]-te/Tf-butyl-dimethyl-silane in 42 ml dry diethyl ether is cooled to -800C under argon. 11.6 ml of a 1.7 M solution of te/if-butyllithium in pentane are slowly added dropwise to the cooled solution, and then the solution is stirred for 30 min at -80 0C. This solution is then added dropwise through a transfer needle, which is cooled with dry ice, to a solution of 4.78 g
2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone in 38 ml diethyl ether chilled to - 80 0C. The resulting solution is stirred for 3 h at -78 0C. Then a solution of 1.1 ml methanesulphonic acid in 35 ml of methanol is added and the solution is stirred for 16 h at ambient temperature. The solution is then neutralised with solid sodium hydrogen carbonate, ethyl acetate is added and the methanol is removed together with the ether. Aqueous sodium hydrogen carbonate solution is added to the remaining solution, and the resulting mixture is extracted four times with ethyl acetate. The organic phases are dried over sodium sulphate and evaporated down. The residue is dissolved in 30 ml acetonitrile and 30 ml dichloromethane and the solution is cooled to -10 0C. After the addition of 4.4 ml triethylsilane 2.6 ml boron trifluoride etherate are added dropwise so that the temperature does not exceed -5 0C. After the addition is complete the solution is stirred for another 5 h at -5 to -10 0C and then quenched by the addition of aqueous sodium hydrogen carbonate solution. The organic phase is separated, and the aqueous phase is extracted four times with ethyl acetate. The combined organic phases are dried over sodium sulfate, the solvent is removed, and the residue is purified by chromatography on silica gel (dichoromethane/methanol 1 :0->3:1 ). The product then obtained is an approx. 6:1 mixture of β/α which can be converted into the pure β-anomer by global acetylation of the hydroxy groups with acetic anhydride and pyridine in dichloromethane and recrystallization of the product from ethanol. The product thus obtained is converted into the title compound by deacetylation in methanol with aqueous 4 M potassium hydroxide solution. Yield: 1.6 g (46% of theory)
Mass spectrum (ESI+): m/z = 398/400 (Cl) [M+H]+
Preparation of the compound A:
1-chloro-4-(β-D-qlucopyranos-1-yl)-2-r4-(<fS)-tetrahvdrofuran-3-yloxy)-benzyll- benzene
0.19 g (f?)-3-(4-methylphenylsulfonyloxy)-tetrahydrofuran are added to a mixture of 0.20 g 1-chloro-4-(β-D-glucopyranos-1-yl)-2-(4-hydroxybenzyl)-benzene and 0.29 g cesium carbonate in 2.5 ml dimethylformamide. The mixture is stirred at 75 0C for 4 h, before another 0.29 g caesium carbonate and 0.19 g (f?)-3-(4-methylphenyl- sulfonyloxy)-tetrahydrofuran are added. After an additional 14 h stirring at 75 0C the mixture is cooled to ambient temperature and brine is added. The resulting mixture is extracted with ethyl acetate, the combined organic extracts are dried over sodium sulfate, and the solvent is removed. The residue is purified by chromatography on silica gel (dichloromethane/methanol 1 :0 -> 5:1 ). Yield: 0.12 g (49% of theory) Mass spectrum (ESI+): m/z = 451/453 (Cl) [M+H] +
................................................
6 LUSEOGLIFLOZIN



..................................................
7 REMOGLIFLOZIN

...................................................
9 SOTAGLIFLOZIN
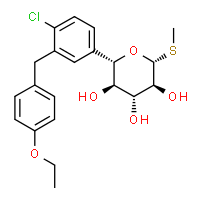





















..............................................
6 LUSEOGLIFLOZIN
LUSEOGLIFLOZIN, CAS 898537-18-3
An antidiabetic agent that inhibits sodium-dependent glucose cotransporter 2 (SGLT2).
An antidiabetic agent that inhibits sodium-dependent glucose cotransporter 2 (SGLT2).
Taisho (Originator), PHASE 3
TS-071
TS-071, an SGLT-2 inhibitor, is in phase III clinical development at Taisho for the oral treatment of type 1 and type 2 diabetes
In 2012, the product was licensed to Novartis and Taisho Toyama Pharmaceutical by Taisho in Japan for comarketing for the treatment of type 2 diabetes.
Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels. The number of people with diabetes worldwide is expected to rise from 285 million in 2010 to 438 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic β-cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
Diabetes which results from complete deficiency in insulin secretion is Type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is Type 2 diabetes. Type 2 diabetes (Non insulin dependent diabetes) accounts for 90-95 % of all diabetes. An early defect in Type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes. A key component of the pathophysiology of Type 2 diabetes mellitus involves an impaired pancreatic β-cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The β-cell compensates for insulin resistance by increasing the insulin secretion, eventually resulting in reduced β-cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in Type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith. Various antidiabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity). These therapies have various side effects: biguanides cause lactic acidosis, sulfonylurea compounds cause significant hypoglycemia, a-glucosidase inhibitors cause abdominal bloating and diarrhea, and thiazolidinediones cause edema and weight gain. Recently introduced line of therapy includes inhibitors of dipeptidyl peptidase-IV (DPP-IV) enzyme, which may be useful in the treatment of diabetes, particularly in Type 2 diabetes. DPP-IV inhibitors lead to decrease in inactivation of incretins glucagon like peptide- 1 (GLP-1) and gastric inhibitory peptide (GIP), thus leading to increased production of insulin by the pancreas in a glucose dependent manner. All of these therapies discussed, have an insulin dependent mechanism.
Another mechanism which offers insulin independent means of reducing glycemic levels, is the inhibition of sodium glucose co-transporters (SGLTs). In healthy individuals, almost 99% of the plasma glucose filtered in the kidneys is reabsorbed, thus leading to only less than 1% of the total filtered glucose being excreted in urine. Two types of SGLTs, SGLT-1 and SGLT-2, enable the kidneys to recover filtered glucose. SGLT-1 is a low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose ” transporter, located mainly in the S 1 segment of the proximal tubule of the kidney. SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases. The inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
Humans lacking a functional SGLT-2 gene appear to live normal lives, other than exhibiting copious glucose excretion with no adverse effects on carbohydrate metabolism. However, humans with SGLT-1 gene mutations are unable to transport glucose or galactose normally across the intestinal wall, resulting in condition known as glucose-galactose malabsorption syndrome.
Hence, competitive inhibition of SGLT-2, leading to renal excretion of glucose represents an attractive approach to normalize the high blood glucose associated with diabetes. Lower blood glucose levels would, in turn, lead to reduced rates of protein glycation, improved insulin sensitivity in liver and peripheral tissues, and improved cell function. As a consequence of progressive reduction in hepatic insulin resistance, the elevated hepatic glucose output which is characteristic of Type 2 diabetes would be expected to gradually diminish to normal values. In addition, excretion of glucose may reduce overall caloric load and lead to weight loss. Risk of hypoglycemia associated with SGLT-2 inhibition mechanism is low, because there is no interference with the normal counter regulatory mechanisms for glucose.
The first known non-selective SGLT-2 inhibitor was the natural product phlorizin
(glucose, 1 -[2-P-D-glucopyranosyloxy)-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)- 1 – propanone). Subsequently, several other synthetic analogues were derived based on the structure of phlorizin. Optimisation of the scaffolds to achieve selective SGLT-2 inhibitors led to the discovery of several considerably different scaffolds.
C-glycoside derivatives have been disclosed, for example, in PCT publications
W.O20040131 18, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033, WO2007136116, WO2007000445, WO2007093610, WO2008069327, WO2008020011, WO2008013321, WO2008013277, WO2008042688, WO2008122014, WO2008116195, WO2008042688, WO2009026537, WO2010147430, WO2010095768, WO2010023594, WO2010022313, WO2011051864, WO201 1048148 and WO2012019496 US patents US65151 17B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630. The compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb’s “Dapagliflozin” with Formula A, Mitsubishi Tanabe and Johnson & Johnson’s “Canagliflozin” with Formula B, Lexicon’s “Lx-421 1″ with Formula C, Boehringer Ingelheim and Eli Lilly’s “Empagliflozin” with Formula D, Roche and Chugai’s “Tofogliflozin” with Formula E, Taisho’s “Luseogliflozin” with Formula F, Pfizer’ s “Ertugliflozin” with Formula G and Astellas and Kotobuki’s “Ipragliflozin” with Formula H.
Formula G Formula H
In spite of all these molecules in advanced stages of human clinical trials, there is still no drug available in the market as SGLT-2 inhibitor. Out of the potential candidates entering the clinical stages, many have been discontinued, emphasizing the unmet need. Thus there is an ongoing requirement to screen more scaffolds useful as SGLT-2 inhibitors that can have advantageous potency, stability, selectivity, better half-life, and/ or better pharmacodynamic properties. In this regard, a novel class of SGLT-2 inhibitors is provided herein
SYNTHESIS
- Example 5

Synthesis of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose
- Five drops of 1,2-dibromoethane were added to a mixture of magnesium (41 mg, 1.67 mmol), 1-bromo-3-(4-ethoxybenzyl)-6-methoxy-4-methylbenzene (0.51 g, 1.51 mmol) and tetrahydrofuran (2 mL). After heated to reflux for one hour, this mixture was allowed to stand still to room temperature to prepare a Grignard reagent. A tetrahydrofuran solution (1.40 mL) of 1.0 M i-propyl magnesium chloride and the prepared Grignard reagent were added dropwise sequentially to a tetrahydrofuran (5 mL) solution of 2,3,4,6-tetra-O-benzyl-5-thio-D-glucono-1,5-lactone (0.76 g, 1.38 mmol) while cooled on ice and the mixture was stirred for 30 minutes. After the reaction mixture was added with a saturated ammonium chloride aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate =4:1) to obtain (0.76 g, 68%) a yellow oily title compound.
1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.37 (t, J=6.92 Hz, 3 H) 2.21 (s, 3 H) 3.51 – 4.20 (m, 12 H) 3.85 – 3.89 (m, 3 H) 4.51 (s, 2 H) 4.65 (d, J=10.72 Hz, 1 H) 4.71 (d, J=5.75 Hz, 1 H) 4.78 – 4.99 (m, 3 H) 6.59 – 7.43 (m, 26 H)
Example 6
- [0315]

Synthesis of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol
- An acetonitrile (18 mL) solution of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose (840 mg, 1.04 mmol) was added sequentially with Et3SiH (0.415 mL, 2.60 mmol) and BF3·Et2O (0.198 mL, 1.56 mmol) at -18°C and stirred for an hour. After the reaction mixture was added with a saturated sodium bicarbonate aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and then dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=4:1) to obtain the title compound (640 mg, 77%).
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.35 (t, J=6.88 Hz, 3 H) 2.21 (s, 3 H) 3.02 – 3.21 (m, 1 H) 3.55 (t,J=9.40 Hz, 1 H) 3.71 (s, 1 H) 3.74 – 3.97 (m, 10 H) 4.01 (s, 1 H) 4.45 – 4.56 (m, 3 H) 4.60 (d, J=10.55 Hz, 2 H) 4.86 (s, 2 H) 4.90 (d, J=10.55 Hz, 1H) 6.58 – 6.76 (m, 5 H) 6.90 (d, J=7.34 Hz, 1 H) 7.09 – 7.19 (m, 5 H) 7.23 – 7.35 (m, 15 H).
ESI m/z = 812 (M+NH4).
Example 7

Synthesis of (1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
- A mixture of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol (630 mg, 0.792 mmol), 20% palladium hydroxide on activated carbon (650 mg) and ethyl acetate (10 mL) – ethanol (10 mL) was stirred under hydrogen atmosphere at room temperature for 66 hours. The insolubles in the reaction mixture were filtered off with celite and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography (chloroform:methanol =10:1) to obtain a colorless powdery title compound (280 mg, 81%) as 0.5 hydrate. 1H NMR (600 MHz, METHANOL- d4) δ ppm 1.35 (t, J=6.9 Hz, 3 H) 2.17 (s, 3 H) 2.92 – 3.01 (m, 1 H) 3.24 (t, J=8.71 Hz, 1 H) 3.54 – 3.60 (m, 1 H) 3.72 (dd, J=11.5, 6.4 Hz, 1 H) 3.81 (s, 3 H) 3.83 (s, 2 H) 3.94 (dd, J=11.5, 3.7 Hz, 1 H) 3.97 (q, J=6.9 Hz, 2 H) 4.33 (s, 1 H) 6.77 (d, J=8.3 Hz, 2 H) 6.76 (s, 1 H) 6.99 (d, J=8.3 Hz, 2 H) 7.10 (s, 1 H). ESI m/z = 452 (M+NH4+), 493 (M+CH3CO2-). mp 155.0-157.0°C. Anal. Calcd for C23H30O6S·0.5H2O: C, 62.28; H, 7.06. Found: C, 62.39; H, 7.10.
..................................................
7 REMOGLIFLOZIN
REMOGLIFLOZIN ETABONATE
5-methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl 6-O-(ethoxycarbonyl)-β-D-glucopyranoside, CAS 442201-24-3
189075 BHV-091009 GSK-189075 GSK-189075A KGT-1681
BHV Pharma Kissei (Originator) , GlaxoSmithKline
Remogliflozin etabonate (INN/USAN)[1] is a proposed drug for the treatment of type 2diabetes being investigated by GlaxoSmithKline.[2] Remogliflozin is now being developed by BHV Pharma.

Remogliflozin inhibits the sodium-glucose transport proteins, which are responsible for glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[3]

- Statement on a nonproprietory name adopted by the USAN council
- Fujimori Y, Katsuno K, Nakashima I, Ishikawa-Takemura Y, Fujikura H, Isaji M (June 2008). “Remogliflozin etabonate, in a Novel Category of Selective Low-Affinity / High-Capacity Sodium Glucose Cotransporter (SGLT2) Inhibitors, Exhibits Antidiabetic Efficacy in Rodent Models”. J. Pharmacol. Exp. Ther. 327 (1): 268–276.doi:10.1124/jpet.108.140210. PMID 18583547.
- Prous Science: Molecule of the Month November 2007
DPP IV inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. For example, DPP IV inhibitors and their uses are disclosed in WO 2002/068420, WO 2004/018467, WO 2004/018468, WO 2004/018469, WO 2004/041820, WO 2004/046148, WO 2005/051950, WO 2005/082906, WO 2005/063750, WO 2005/085246, WO 2006/027204, WO 2006/029769, WO2007/014886; WO 2004/050658, WO 2004/1 1 1051 , WO 2005/058901 , WO 2005/097798; WO 2006/068163, WO 2007/071738, WO 2008/017670; WO 2007/054201 or WO 2007/128761.
Chemical structures of remogliflozin etabonate (A), remogliflozin (B), sergliflozin (C), phlorizin (D), and T-1095 (E). Remogliflozin etabonate is metabolized to remogliflozin, its active form.
...................................................
8 ERTUGLIFLOZIN








 ERTUGLIFLOZIN
ERTUGLIFLOZIN




...................................................
8 ERTUGLIFLOZIN

ERTUGLIFLOZIN, PFIZER
THERAPEUTIC CLAIM Treatment of type 2 diabetes
CHEMICAL NAMES
1. β-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-(hydroxymethyl)-
2. (1S,2S,3S,4R,5S)-5-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
CHEMICAL NAMES
1. β-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-(hydroxymethyl)-
2. (1S,2S,3S,4R,5S)-5-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
PF-04971729, MK 8835
M. Wt: 436.88
Formula: C22H25ClO7
CAS No:. 1210344-57-2
Formula: C22H25ClO7
CAS No:. 1210344-57-2
Diabetes looms as a threat to human health worldwide. As a result, considerable research efforts are devoted to identify new and efficacious anti-diabetic agents lacking the side effects associated with some of the current drugs (hypoglycemia, weight gain).Inhibition of sodium-dependent glucose cotransporter 2 (SGLT2), a transporter located in the kidney, is a mechanism that promotes glucosuria and therefore, reduction of plasma glucose concentration. Since the mechanism operates in a glucose-dependent and insulin-independent manner, and is associated with weight loss, it has emerged as a very promising approach to the pathophysiologic treatment of type 2 diabetes. Ertugliflozin (PF-04971729), an anti-diabetic agent currently in development (Phase 3 clinical trials) and belonging to a new class of SGLT2 inhibitors bearing a dioxa-bicyclo[3.2.1]octane bridged ketal motif.
SYNTHESIS
Scheme 1 outlines the general procedures one could use to provide compounds of the present invention.
Scheme 1 AIIyI 2,3,4-tιϊ-O-benzyl-D-glucopyranoside (La, where Pg1 is a benzyl group) can be prepared by procedures described by Shinya Hanashima, et al., in Bioorganic & Medicinal Chemistry, 9, 367 (2001 ); Patricia A. Gent et al. in Journal of the Chemical Society, Perkin 1, 1835 (1974); Hans Peter Wessel in the Journal of Carbohydrate Chemistry, 7, 263, (1988); or Yoko Yuasa, et al., in Organic Process Research & Development, 8, 405-407
(2004). In step 1 of Scheme 1 , the hydroxymethylene group can be introduced onto the glycoside by means of a Swern oxidation followed by treatment with formaldehyde in the presence of an alkali metal hydroxide (e.g., sodium hydroxide). This is referred to as an aldol-Cannizzaro reaction. The Swern oxidation is described by Kanji Omura and Daniel Swern in Tetrahedron, 34, 1651 (1978). Modifications of this process known to those of skill in the art may also be used. For example, other oxidants, like stabilized 2- iodoxybenzoic acid described by Ozanne, A. et al. in Organic Letters, 5, 2903 (2003), as well as other oxidants known by those skilled in the art can also be used. The aldol Cannizzaro sequence has been described by Robert Schaffer in the Journal of The American Chemical Society, 81 , 5452 (1959) and Amigues, E.J., et al., in Tetrahedron, 63,10042 (2007).
In step 2 of Scheme 1 , protecting groups (Pg2) can be added by treating intermediate (MD) with the appropriate reagents and procedures for the particular protecting group desired. For example, p-methoxybenzyl (PMB) groups may be introduced by treatment of intermediate (MD) with p-methoxybenzyl bromide or p-methoxybenzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide (DMF). Conditions involving para-methoxybenzyltrichloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. Benzyl (Bn) groups may be introduced by treatment of intermediate (MD) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide. Conditions involving benzylthchloroacetimidate in presence of a catalytic amount of acid (e.g., trifluoromethanesulfonic acid, methanesulfonic acid, or camphorsulfonic acid) in a solvent such as dichloromethane, heptane or hexanes can also be used. In step 3 of Scheme 1 , the allyl protection group is removed (e.g., by treatment with palladium chloride in methanol; cosolvent like dichloromethane may also be used; other conditions known by those skilled in the art could also be used, see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991 ) to form the lactol (Ld).
In step 4 of Scheme 1 , oxidation of the unprotected hydroxyl group to an oxo group (e.g., Swern oxidation) then forms the lactone (l-e).
In step 5 of Scheme 1 , the lactone (Le) is reacted with Λ/,O-dimethyl hydroxylamine hydrochloride to form the corresponding Weinreb amide which may exist in equilibrium in a closed/opened form, (l-f/l-g). The “Weinreb amide” (LgJ can be made using procedures well known to those of skill in the art. See, Nahm, S., and S. M. Weinreb, Tetrahedron Letters. 22 (39), 3815-1818 (1981 ). For example, intermediate (l-f/l-α) can be prepared from the commercially available Λ/,O-dimethylhydroxylamine hydrochloride and an activating agent (e.g., trimethylaluminum). In step 6 of Scheme 1 , the arylbenzyl group (Ar) is introduced using the desired organometallic reagent (e.g., organo lithium compound (ArLi) or organomagnesium compound (ArMgX)) in tetrahydrofuran (THF) at a temperature ranging from about -780C to about 2O0C followed by hydrolysis (upon standing in protic conditions) to the corresponding lactol (N) which may be in equilibrium with the corresponding ketone (Ni). The bridged ketal motif found in (A) and (B) can be prepared by removing the protecting groups (Pg2) using the appropriate reagents for the protecting groups employed. For example, the PMB protecting groups may be removed by treatment with trifluoroacetic acid in the presence of anisole and dichloromethane (DCM) at about O0C to about 230C (room temperature). The remaining protecting groups (Pg1) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final products (A) and (B). When R1 is CN, the use of a Lewis acid like boron trichloride at a temperature ranging from about -780C to about room temperature in a solvent like dichloromethane or 1 ,2-dichloroethane may also be used to remove benzyl protective and/or para- methoxybenzyl protective groups. When R1 is CN and R2 is (Ci-C4)alkoxy in intermdediate (l-i) or in products (A) or (B), upon treatment with a Lewis acid such as boron trichloride or boron tribomide, partial to complete de-alkylation to the corresponding phenol may occur to lead to the corresponding compound (A) or (B) where R1 is CN and R2 is OH. If this occurs, the (d- C4)alkoxy group may be re-introduced via selective alkylation using a (CrC4) alkyl iodide under mildly basic conditions, for example, potassium carbonate in acetone at a temperature ranging from about room temperature to about 56 degrees Celsius.
When R1 and/or R2 is (CrC4)alkyl-SO2- it is understood by one skilled in the art that the organometallic addition step 6 (Scheme 1 ) will be carried out on the corresponding (d- C4)alkyl-S- containing organometallic reagent. The thio-alkyl is then oxidized at a later stage to the corresponding sulfone using conventional methods known by those skilled in the art.
The compounds of the present invention may be prepared as co-crystals using any suitable method. A representative scheme for preparing such co-crystals is described in Scheme 2.
Scheme 2
In Scheme 2, wherein Me is methyl and Et is ethyl, in step 1 , 1-(5-bromo-2- chlorobenzyl)-4-ethoxybenzene is dissolved in 3:1 , toluene: tetrahydrofuran followed by cooling the resulting solution to <-70°C. To this solution is added hexyllithium while maintaining the reaction at <-65°C followed by stirring for 1 hour. (3R,4S,5R,6R)-3,4,5- ths(thmethylsilyloxy)-6-((trimethylsilyloxy)methyl)-tetrahydropyran-2-one (ll-a) is dissolved in toluene and the resulting solution is cooled to -150C. This solution is then added to the – 7O0C aryllithium solution followed by stirring for 1 hour. A solution of methanesulfonic acid in methanol is then added followed by warming to room temperature and stirring for 16 to 24 hours. The reaction is deemed complete when the α-anomer level is < 3%. The reaction is then basified by the addition of 5 M aqueous sodium hydroxide solution. The resulting salts are filtered off followed by concentration of the crude product solution. 2- methyltetrahydrofuran is added as a co-solvent and the organic phase is extracted twice with water. The organic phase is then concentrated to 4 volumes in toluene. This concentrate is then added to a 5:1 , heptane: toluene solution causing precipitate to form. The solids are collected and dried under vacuum to afford a solid.
In step 2 of Scheme 2, to (ll-b) in methylene chloride is added imidazole followed by cooling to O0C and then addition of trimethylsilylchlohde to give the persilylated product.
The reaction is warmed to room temperature and quenched by the addition of water, and the organic phase is washed with water. This crude methylene chloride solution of (ll-c) is dried over sodium sulfate and then taken on crude into the next step.
In step 3 of Scheme 2, the crude solution of (ll-c) in methylene chloride is concentrated to low volume and then the solvent is exchanged to methanol. The methanol solution of (ll-c) is cooled to O0C, then 1 mol% of potassium carbonate is added as a solution in methanol followed by stirring for 5 hours. The reaction is then quenched by addition of 1 mol% acetic acid in methanol, followed by warming to room temperature, solvent exchange to ethyl acetate, and then filtration of the minor amount of inorganic solids. The crude ethyl acetate solution of (ll-d) is taken directly into the next step.
In step 4 of Scheme 2, the crude solution of (ll-d) is concentrated to low volume, then diluted with methylene chloride and dimethylsulfoxide. Triethylamine is added followed by cooling to 1O0C and then sulfur trioxide pyridine complex is added in 3 portions as a solid at 10 minute intervals. The reaction is stirred an additional 3 hours at 1O0C before quenching with water and warming to room temperature. The phases are separated followed by washing the methylene chloride layer with aqueous ammonium chloride. The crude methylene chloride solution of (ll-e) is taken directly into the next step.
In step 5 of Scheme 2, the crude solution of (ll-e) is concentrated to low volume and then the solvent is exchanged to ethanol. Thirty equivalents of aqueous formaldehyde is added followed by warming to 550C. An aqueous solution of 2 equivalents of potassium phosphate, tribasic is added followed by stirring for 24 hours at 550C. The reaction temperature is then raised to 7O0C for an additional 12 hours. The reaction is cooled to room temperature, diluted with te/t-butyl methyl ether and brine. The phases are separated followed by solvent exchange of the organic phase to ethyl acetate. The ethyl acetate phase is washed with brine and concentrated to low volume. The crude concentrate is then purified by silica gel flash chromatography eluting with 5% methanol, 95% toluene. Product containing fractions are combined and concentrated to low volume.
Methanol is added followed by stirring until precipitation occurs. The suspension is cooled and the solids are collected and rinsed with heptane followed by drying. Product (ll-f) is isolated as a solid.
In step 6 of Scheme 2, compound (ll-f) is dissolved in 5 volumes of methylene chloride followed by the addition of 1 mol% SiliaBonc/® tosic acid and stirring for 18 hours at room temperature. The acid catalyst is filtered off and the methylene chloride solution of (ll-g) is taken directly into the next step co-crystallization procedure.
In step 7 of Scheme 2, the methylene chloride solution of (ll-g) is concentrated and then the solvent is exchanged to 2-propanol. Water is added followed by warming to 550C. An aqueous solution of L-pyroglutamic acid is added followed by cooling the resulting solution to room temperature. The solution is then seeded and granulated for 18 hours. After cooling, the solids are collected and rinsed with heptane followed by drying. Product (ll-h) is isolated as a solid.
An alternative synthesis route for compounds (A) of the present invention is depicted in Scheme 3 and described below.
Scheme 3
The synthesis of (lll-a), where R3 is an alkyl or fluoro substituted alkyl (except for the carbon adjacent to the oxygen atom) can be prepared in a similar way as described in step 1 of Scheme 2. In step 1 of Scheme 3, the primary hydroxyl group is selectively protected by an appropriate protective group. For example, a trityl group (Pg3 = Tr) can be introduced by treatment of intermediate (lll-a) with chlorotriphenylmethane in presence of a base like pyridine in a solvent like toluene, tetrahydrofuran or dichloromethane at a temperature ranging from about 0 degrees Celsius to about room temperature. Additional examples of such protective groups and experimental conditions are known by those skilled in the art and can be found in T. W. Greene, Protective Groups in Organic Synthesis. John Wiley & Sons, New York, 1991.
In step 2 of Scheme 3, the secondary hydroxyl groups can be protected by the appropriate protecting groups. For example, benzyl groups (Pg4 is Bn) can be introduced by treatment of intermediate (lll-b) with benzyl bromide or benzyl chloride in the presence of sodium hydride, potassium hydride, potassium te/t-butoxide in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or Λ/,Λ/-dimethylformamide at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius. Acetyl or benzoyl groups (Pg4 = Ac or Bz) may be introduced by treatment of intermediate (lll-b) with acetyl chloride, acetyl bromide or acetic anhydride or benzoyl chloride or benzoic anhydride in the presence of a base like triethylamine, Λ/,Λ/-diisopropylethylamine or 4-
(dimethylamino)pyridine in a solvent like tetrahydrofuran, 1 ,2-dimethoxyethane or dichloromethane at a temperature ranging from about 0 degrees Celsius to about 80 degrees Celsius.
In step 3 of Scheme 3, the primary hydroxyl group is deprotected to lead to intermediate (lll-d). When Pg3 is Tr, intermediate (lll-c) is treated in the presence of an acid like para-toluenesulfonic acid in a alcoholic solvent like methanol at a temperature ranging from about -20 degrees Celsius to about room temperature to provide intermediate (lll-d). Cosolvents like chloroform may be used.
In step 4 of Scheme 3, a hydroxymethylene group is introduced through a process similar to the one already described in Scheme 1 (step 1 ) and Scheme 2 (steps 4 and 5).
Other sources of formaldehyde, like paraformaldehyde in a solvent like ethanol at a temperature ranging from about room temperature to about 70 degrees Celsius in the presence of an alkali metal alkoxide can also be used in this step. When Pg4is Bn, this step provides intermediate (lll-e) and when Pg4 is Ac or Bz, this step provides intermediate (lll-f).
In step 5 of Scheme 3, intermediate (lll-e) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (lll-g).
In step 6 of Scheme 3, the remaining protecting groups (Pg4) may then be removed using the appropriate chemistry for the particular protecting groups. For example, benzyl protecting groups may be removed by treating with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature to produce the final product (A).
In step 7 of Scheme 3, intermediate (lll-f) is treated with an acid like trifluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A). Another alternative scheme for synthesizing product (A) is depicted in Scheme 4 and described below.
Scheme 4 In step 1 of Scheme 4, intermediate (lll-a) is treated with the appropriate arylsulfonyl chloride R4SO2CI or arylsulfonic anhydride R4S(O)2OS(O)2R4 (wherein R4 is an optionally substituted aryl group, such as found in the arylsulfonyl chlorides 4-methyl-benzenesulfonyl chloride, 4-nitro-benzenesulfonyl chloride, 4-fluoro-benzenesulfonyl chloride, 2,6-dichloro- benzenesulfonyl chloride, 4-fluoro-2-methyl-benzenesulfonyl chloride, and 2,4,6-trichloro- benzenesulfonyl chloride, and in the arylsulfonic anhydride, p-toluenesulfonic anhydride) in presence of a base like pyridine, triethylamine, Λ/,Λ/-diisopropylethylamine in a solvent like tetrahydrofuran, 2-methyltetrahydrofuran at a temperature ranging from about -20 degrees Celsius to about room temperature. Some Lewis acids like zinc(ll) bromide may be used as additives. In step 2 of Scheme 4, intermediate (IV-a) is submitted to a Kornblum-type oxidation
(see, Kornblum, N., et al., Journal of The American Chemical Society, 81 , 4113 (1959)) to produce the corresponding aldehyde which may exist in equilibrium with the corresponding hydrate and/or hemiacetal form. For example intermediate (IV-a) is treated in the presence of a base like pyridine, 2,6-lutidine, 2,4,6-collidine, Λ/,Λ/-diisopropylethylamine, A- (dimethylamino)pyridine in a solvent like dimethyl sulfoxide at a temperature ranging from about room temperature to about 150 degrees Celsius. The aldehyde intermediate produced is then submitted to the aldol/Cannizzaro conditions described for step 1 (Scheme 1 ) and step 5 (Scheme 2) to produce intermediate (IV-b). In step 3 of Scheme 4, intermediate (IV-b) is treated with an acid like thfluoroacetic acid or an acidic resin in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce the final product (A).
When R2 is (C2-C4)alkynyl the process may be performed using Scheme 5, wherein R6 is H or (CrC2)alkyl.
Scheme 5
In step 1 of Scheme 5, which provides intermediate (V-i), the organometallic addition step is carried out in a similar way to the one described in Schemel , step 6, using the organometallic reagent derived from (V-a), where Pg5 is a suitable protective group for the hydroxyl group. For instance Pgs can be a te/t-butyldimethylsilyl group (TBS) (see
US2007/0054867 for preparation of for instance {4-[(5-bromo-2-chloro-phenyl)-methyl]- phenoxy}-te/t-butyl-dimethyl-silane).
In step 2 of Scheme 5, when Pg2 = PMB, intermediate (V-i) is treated with an acid like trifluoroacetic acid, methanesulfonic acid or an acidic resin in presence of anisole in a solvent like dichloromethane at a temperature ranging from about -10 degrees Celsius to about room temperature to produce intermediate (V-j).
In step 3 of Scheme 5, protecting groups (Pg5) and (Pg1) can be removed to provide (V-k). Typically (Pg5) is TBS and Pg1 is Bn. In this circumstance, the protecting groups are removed by sequential treatment of (V-j) with 1 ) tetrabutylammonium fluoride in a solvent like tetrahydrofuran or 2-methyltetrahydrofuran at a temperature ranging from 0 degrees
Celsius to about 40 degrees Celsius and 2) treatment with formic acid in the presence of palladium (Pd black) in a protic solvent (e.g., ethanol/THF) at about room temperature. In this sequence, the order of the 2 reactions is interchangeable.
In step 4 of Scheme 5, intermediate (V-k) is treated with N,N-bis- (trifluoromethanesulfonyl)-aniline in presence of a base like triethylamine or 4- dimethyaminopyridine in a solvent like dichloromethane or 1 ,2-dichloroethane at a temperature ranging from 0 degrees Celsius to about 40 degrees Celsius to produce intermediate (V-I).
In step 5 of Scheme 5, intermediate (V-I) is subjected to a Sonogashira-type reaction (see, Sonogashira, K. Coupling Reactions Between sp2 and sp Carbon Centers. In
Comprehensive Organic Synthesis (eds. Trost, B. M., Fleming, I.), 3, 521-549, (Pergamon, Oxford, 1991 )).
IS ERTUGLIFLOZIN
Example 4
(1 S.2S.3S.4R.5S)-5-[4-chloro-3-(4-ethoxy-benzyl)-Dhen yll- 1 -h vdroxymeth yl-6.8-dioxa- bicvclo[3.2.1loctane-2,3Λ-triol (4A) and (1S,2S,3SΛS,5S)-5-[4-chloro-3-(4-ethoxy- benzvD-phen yll- 1 -h vdroxymeth yl-6, 8-dioxa-bicvclo[3.2.1 loctane-2, 3, 4-triol (4B):
To a solution of {(2S,3S)-2,3,4-tris-benzyloxy-5-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6,8- dioxa-bicyclo[3.2.1]oct-1-yl}-methanol (l-4k: 335 mg) in ethanol/tetrahydrofuran (10 ml_, 4/1 volume) was added successively formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) and the resulting mixture was stirred at room temperature. After 1 hour, additional formic acid (420 microL, 22 equivalents) and palladium black (208 mg, 4 equivalents) were added and the mixture was allowed to stir for an additional hour at room temperature. The palladium was filtered and the crude mixture obtained after evaporation of solvent was purified by HPLC preparative.
HPLC preparative: reverse phase C18 Gemini column 5 micrometer 30 x 100 mm, 40 mL/minute, gradient of acetonitrile/0.1 % formic acid : water/0.1 % formic acid; 25 to 50% of acetonitrile/0.1 % formic acid over 18 minutes; UV detection: 220 nm. The HPLC indicated a ratio of diastereomers of 1.1 :1 (4A:4B).
4A: (60 mg, 29% yield); Rt = 12.4 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode); 481.3 (M+HCO2 ~; negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.43 (d, 1 H, J = 1.9 Hz), 7.36 (dd, 1 H, J = 8.3 and 2Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.04 (m, 2H), 6.79-6.75 (m, 2H), 4.12 (d, 1 H, J = 7.5 Hz), 4.00 (s, 2H), 3.96 (q, 2H, J = 7.0 Hz), 3.81 (d, 1 H, J = 12.5 Hz), 3.75 (dd, 1 H, J = 8.3 and 1.3 Hz), 3.65 (d, 1 H, J = 12.5 Hz), 3.63 (t, 1 H, J = 8.2 Hz), 3.57 (dd, 1 H, J = 7.5 and 1.3 Hz), 3.52 (d, 1 H, J = 8.0 Hz), 1.33 (t, 3H, J = 6.9 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1360.
4B: (30 mg, 15% yield); Rt = 13.2 minutes; the fractions containing the product were concentrated under reduced pressure. The crude material was precipitated from ethyl acetate and heptane. The resulting white solid was washed with heptane 2 times and dried under reduced pressure.
MS (LCMS) 437.3 (M+H+; positive mode) 481.3 (M+HCO2 “, negative mode). 1H NMR (400 MHz, methanol-d4) delta 7.48 (d, 1 H, J = 1.9 Hz) 7.40 (dd, 1 H, J = 8.1 and 1.9 Hz), 7.32 (d, 1 H, J = 8.3 Hz), 7.08-7.03 (m, 2H), 6.80-6.74 (m, 2H), 4.04-3.99 (m, 3H), 3.95 (q, 2H, J = 7 Hz), 3.89-3.81 (m, 4H), 3.73 (d, 1 H, J = 12.5 Hz), 3.49 (d, 1 H, J = 7.3 Hz), 1.32 (t, 3H, J = 7 Hz). HRMS calculated for C22H26O7CI (M+H+) 437.1361 , found 437.1358.
Merck & Co., Inc. and Pfizer Enter Worldwide Collaboration Agreement to Develop and Commercialize Ertugliflozin, an Investigational Medicine for Type 2 Diabetes
Merck & Co., Inc. and Pfizer Enter Worldwide Collaboration Agreement to Develop and Commercialize Ertugliflozin, an Investigational Medicine for Type 2 Diabetes
ERTUGLIFLOZIN
Monday, April 29, 2013 9:23 am EDT
Merck & Co., Inc. (NYSE: MRK), known as MSD outside the United States and Canada (“Merck”), and Pfizer Inc. (NYSE:PFE) today announced that they have entered into a worldwide (except Japan) collaboration agreement for the development and commercialization of Pfizer’s ertugliflozin (PF-04971729), an investigational oral sodium glucose cotransporter (SGLT2) inhibitor being evaluated for the treatment of type 2 diabetes. Ertugliflozin is Phase III ready, with trials expected to begin later in 2013.
“We are pleased to join forces with Merck in the battle against type 2 diabetes and the burden that it poses on global health,” said John Young, president and general manager, Pfizer Primary Care. “Through this collaboration, we believe we can build on Merck’s leadership position in diabetes care with the introduction of ertugliflozin, an innovative SGLT2 inhibitor discovered by Pfizer scientists.”
Under the terms of the agreement, Merck, through a subsidiary, and Pfizer will collaborate on the clinical development and commercialization of ertugliflozin and ertugliflozin-containing fixed-dose combinations with metformin and JANUVIA® (sitagliptin) tablets. Merck will continue to retain the rights to its existing portfolio of sitagliptin-containing products. Pfizer has received an upfront payment and milestones of $60 million and will be eligible for additional payments associated with the achievement of pre-specified future clinical, regulatory and commercial milestones. Merck and Pfizer will share potential revenues and certain costs on a 60/40 percent basis.
“Merck continues to build upon our leadership position in the oral treatment of type 2 diabetes through our own research and business development,” said Nancy Thornberry, senior vice president and Diabetes and Endocrinology franchise head, Merck Research Laboratories. “We believe ertugliflozin has the potential to complement our strong portfolio of investigational and marketed products, and we look forward to collaborating with Pfizer on its development.”
……………….
Development of an Early-Phase Bulk Enabling Route to Sodium-Dependent Glucose Cotransporter 2 Inhibitor Ertugliflozin
David Bernhardson, Thomas A. Brandt, Catherine A. Hulford, Richard S. Lehner, Brian R. Preston, Kristin Price, John F. Sagal, Michael J. St. Pierre, Peter H. Thompson, and Benjamin Thuma
pp 57–65
Publication Date (Web): January 3, 2014 (Article)
DOI: 10.1021/op400289z
The development and optimization of a scalable synthesis of sodium-dependent glucose cotransporter 2 inhibitor, ertugliflozin, for the treatment of type-2 diabetes is described. Highlights of the chemistry are a concise, four-step synthesis of a structurally complex API from known intermediate 4 via persilylation–selective monodesilylation, primary alcohol oxidation, aldol-crossed-Cannizzaro reaction, and solid-phase acid-catalyzed bicyclic ketal formation. The final API was isolated as the l-pyroglutamic acid cocrystal.
1= ertugliflozin
| PF-04971729, a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF- 04971729 also were very weak (IC50 900μM). The disposition of PF-04971729, an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. |
| References on PF-04971729: [1]. 1. Amit S. Kalgutkar, Meera Tugnait, Tong Zhu, et al.Preclinical Species and Human Disposition of PF-04971729, a Selective Inhibitor of the Sodium-Dependent Glucose cotransporter 2 and Clinical Candidate for the Treatment of Type 2 . Diabetes Mellitus Drug Metabolism and Diposition, 2011, 39 (9):. 1609-1619 Abstract (1S, 2S, 3S, 4R, 5S) -5 – [4-Chloro-3-(4-ethoxybenzyl) phenyl] -1 -hydroxymethyl-6 ,8-dioxabicyclo [3.2.1] octane-2 ,3,4-triol (PF-04971729), a potent and selective inhibitor of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials for the treatment of diabetes mellitus. This article describes the preclinical species and in vitro human disposition characteristics of PF-04971729 that were used in experiments performed to support the first-in-human study. Plasma clearance was low in rats (4.04 ml · min? 1 · kg? 1) and dogs (1.64 ml · min? 1 · kg? 1), resulting in half-lives of 4.10 and 7.63 h, respectively. Moderate to good bioavailability in rats (69%) and dogs (94%) was . observed after oral dosing The in vitro biotransformation profile of PF-04971729 in liver microsomes and cryopreserved hepatocytes from rat, dog, and human was qualitatively similar;. prominent metabolic pathways included monohydroxylation, O-deethylation, and glucuronidation No human-specific metabolites of PF-04971729 were detected in in vitro studies. Reaction phenotyping studies using recombinant enzymes indicated a role of CYP3A4/3A5, CYP2D6, and UGT1A9/2B7 in the metabolism of PF-04971729. No competitive or time-dependent inhibition of the major human cytochrome P450 enzymes was discerned with PF-04971729. Inhibitory effects against the organic cation transporter 2-mediated uptake of [14C] metformin by PF-04971729 also were very weak (IC50 =? 900 μM). Single-species allometric scaling of rat pharmacokinetics of PF-04971729 was used to predict human clearance, distribution volume, and oral bioavailability. Human pharmacokinetic predictions were consistent with the potential for a low daily dose. First-in-human studies after oral administration indicated that the human pharmacokinetics / dose predictions for PF -04971729 were in the range that is likely to yield a favorable pharmacodynamic response.. [2] … Timothy Colin Hardman, Simon William Dubrey Development and potential role of type-2 sodium-glucose transporter Inhibitors for Management of type 2 Diabetes Diabetes Ther 2011 September; 2 (3):. 133-145 Abstract There is a recognized need for new treatment options for type 2 diabetes mellitus (T2DM). Recovery of glucose from the glomerular filtrate represents an important mechanism in maintaining glucose homeostasis and represents a novel target for the management of T2DM. Recovery of glucose from the glomerular filtrate is executed principally by the type 2 sodium-glucose cotransporter (SGLT2). Inhibition of SGLT2 promotes glucose excretion and normalizes glycemia in animal models. First reports of specifically designed SGLT2 inhibitors began to appear in the second half of the 1990s. Several candidate SGLT2 inhibitors are currently under development, with four in the later stages of clinical testing. The safety profile of SGLT2 inhibitors is expected to be good, as their target is a highly specific membrane transporter expressed almost exclusively within the renal tubules. One safety concern is that of glycosuria , which could predispose patients to increased urinary tract infections. So far the reported safety profile of SGLT2 inhibitors in clinical studies appears to confirm that the class is well tolerated. Where SGLT2 inhibitors will fit in the current cascade of treatments for T2DM has yet to be established. The expected favorable safety profile and insulin-independent mechanism of action appear to support their use in combination with other antidiabetic drugs. Promotion of glucose excretion introduces the opportunity to clear calories (80-90 g [300-400 calories] of glucose per day) in patients that are generally overweight, and is expected to work synergistically with weight reduction programs. Experience will most likely lead to better understanding of which patients are likely to respond best to SGLT2 inhibitors, and under what circumstances.[3]. Zhuang Miao, Gianluca Nucci, Neeta Amin. Pharmacokinetics, Metabolism and Excretion of the Anti-Diabetic Agent Ertugliflozin (PF-04971729) in Healthy Male the Subjects. Drug Metabolism and Diposition. Abstract The Disposition of ertugliflozin (PF-04971729) , an orally active selective inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a single 25-mg oral dose of [14C]-PF-04971729 to healthy human subjects. Mass balance was achieved with approximately 91% of the administered dose recovered in urine and feces. The total administered radioactivity excreted in feces and urine was 40.9% and 50.2%, respectively. The absorption of PF-04971729 in humans was rapid with a Tmax at ~ 1.0 h. Of the total radioactivity excreted in feces and urine, unchanged PF-04971729 collectively accounted for ~ 35.3% of the dose, suggestive of moderate metabolic elimination in humans. The principal biotransformation pathway involved glucuronidation of the glycoside hydroxyl groups to yield three regioisomeric metabolites M4a, M4b and M4c (~ 39.3% of the dose in urine) of which M4c was the major regioisomer (~ 31.7% of the dose). The structure of M4a and M4c were confirmed to be PF-04971729-4-O-β-and-3-O-β-glucuronide , respectively, via comparison of the HPLC retention time and mass spectra with authentic standards. A minor metabolic fate involved oxidation by cytochrome P450 to yield monohydroxylated metabolites M1 and M3 and des-ethyl PF-04971729 (M2), which accounted for ~ 5.2% of the dose in excreta. In plasma, unchanged PF-04971729 and the corresponding 4-O-β-(M4a) and 3-O-β-(M4c) glucuronides were the principal components, which accounted for 49.9, 12.2 and 24.1% of the circulating radioactivity. Overall, these data suggest that PF-04971729 is well absorbed in humans, and eliminated largely via glucuronidation.. [4] .. Tristan S. Maurer, Avijit Ghosh, Nahor Haddish-Berhane pharmacodynamic Model of Sodium-Glucose Transporter 2 (SGLT2) Inhibition: Implications for Quantitative Translational Pharmacology AAPS J. 2011; 13 (4): 576-584 Abstract Sodium-glucose co-transporter-2 (SGLT2) inhibitors are an emerging class of agents for use in the treatment of type 2 diabetes mellitus (T2DM). Inhibition of SGLT2 leads to improved glycemic control through increased urinary glucose excretion (UGE). In this study, a biologically based pharmacokinetic / pharmacodynamic (PK / PD) model of SGLT2 inhibitor-mediated UGE was developed. The derived model was used to characterize the acute PK / PD relationship of the SGLT2 inhibitor, dapagliflozin, in rats. The quantitative translational pharmacology of dapagliflozin was examined through both prospective simulation and direct modeling of mean literature data obtained for dapagliflozin in healthy subjects. Prospective simulations provided time courses of UGE that were of consistent shape to clinical observations, but were modestly biased toward under prediction. Direct modeling provided an improved characterization of the data and precise parameter estimates which were reasonably consistent with those predicted from preclinical data. Overall, these results indicate that the acute clinical pharmacology of SGLT2 inhibitors in healthy subjects can be reasonably well predicted from preclinical data through rational accounting of species differences in pharmacokinetics, physiology, and SGLT2 pharmacology. Because these data can be generated at the earliest stages of drug discovery, the proposed model is useful in the design and development of novel SGLT2 inhibitors. In addition, this model is expected to serve as a useful foundation for future efforts to understand and predict the effects of SGLT2 inhibition under chronic administration and in other patient populations.[5]. Yoojin Kim, Ambika R Babu Clinical potential of sodium-glucose cotransporter 2 Inhibitors in the Management of type 2 Diabetes Diabetes Obes Metab Syndr 2012; 5:…. 313-327 Abstract Background The Kidney plays an Important role in glucose metabolism, and has been considered a target for therapeutic intervention. The sodium-glucose cotransporter type 2 (SGLT2) mediates most of the glucose reabsorption from the proximal renal tubule. Inhibition of SGLT2 leads to glucosuria and provides a unique mechanism to lower elevated blood glucose levels in diabetes. The purpose of this review is to explore the physiology of SGLT2 and discuss several SGLT2 inhibitors which have clinical data in patients with type 2 diabetes. Methods We performed a PubMed search using the terms “SGLT2″ and “SGLT2 inhibitor” through April 10, 2012. Published articles, press releases, and abstracts presented at national and international meetings were considered. Results SGLT2 inhibitors correct a novel pathophysiological defect, have an insulin-independent action, are efficacious with glycosylated hemoglobin reduction ranging from 0.5% to 1.5%, promote weight loss, have a low incidence of hypoglycemia, complement the action of other antidiabetic agents, and can be used at any stage of diabetes. They are generally well tolerated. However, due to side effects, such as repeated urinary tract and genital infections, increased hematocrit, and decreased blood pressure, appropriate patient selection for drug initiation and close monitoring after initiation will be important. Results of ongoing clinical studies of the effect of SGLT2 inhibitors on diabetic complications and cardiovascular safety are crucial to determine the risk -benefit ratio. A recent decision by the Committee for Medicinal Products for Human Use of the European Medicines Agency has recommended approval of dapagliflozin for the treatment of type 2 diabetes as an adjunct to diet and exercise, in combination with other glucose-lowering medicinal products , including insulin, and as a monotherapy for metformin-intolerant patients. Clinical research also remains to be carried out on the long-term effects of glucosuria and other potential effects of SGLT2 inhibitors, especially in view of the observed increase in the incidence of bladder and breast cancer SGLT2 inhibitors represent a promising approach for the treatment of diabetes, and could potentially be an addition to existing therapies Keywords:.. sodium-glucose cotransporter type 2, SGLT2, inhibitors, kidney, glucosuria, oral diabetes agent, weight loss.[6]. Clinical Trials with PF-04971729 |
Example 6 Manufacturing Process for Tablets US20130137646
 |
...................................................
9 SOTAGLIFLOZIN
LX 4211, Sotagliflozin, LP-802034 , lex 1287
UNII-6B4ZBS263Y
Methyl (5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-thio-beta-L-xylopyranoside
β-L-Xylopyranoside, methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-, (5S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol,
(5S)-Methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-beta-L-xylopyranoside
1018899-04-1
C21H25ClO5S, 424.94, LP-802034
LX-4211 is a dual SGLT2/1 inhibitor; Antidiabetic agents.
LX-4211 is a SGLT-2 inhibitor being evaluated in phase II clinical studies at Lexicon Pharmaceuticals for the oral treatment of type 2 diabetes.
Summary
- Co-administration of LX4211 led to a nearly one-third reduction in mealtime insulin for Type 1 diabetics.
- Although there was no reduction in basal insulin use, the LX4211 group saw better glucose control, lower HbA1c, and weight loss.
- Partnering LX4211 is still management’s top priority but independent development in Type 1 diabetes is at least an option.
Lexicon Pharmaceuticals (LXRX) continues to generate data on its SGLT-1/2 inhibitor LX4211 that suggest this is an effective and promising medication for treating not only Type 2 diabetes (the common target for non-insulin medications for diabetes), but also Type 1 as well. Lexicon’s most recent update, a small short-term Phase II study in Type 1 diabetics is certainly a positive update, but it’s not what investors really want to see. Lexicon still needs to find a development partner for LX4211 and the ongoing delays don’t help sentiment or the long-term prospects for the drug.
A Potentially Meaningful Addition To Type 1 Care
On Monday morning, Lexicon released top-line data from a small (33-patient) Phase II study of LX4211 in Type 1 diabetics on insulin. The results support the notion that SGLT inhibition can play a valuable role in improving glucose control for Type 1 diabetics.
This small study enrolled generally well-controlled patients (HbA1c levels ranging from 7 to 9, with an average of 7.9) and the addition of LX4211 led to 32% reduction in bolus (mealtime) insulin versus a 6% reduction in the placebo group. Even with the lower bolus insulin, patients in the LX4211 group showed a 0.55% reduction in HbA1c versus a 0.06% reduction in the placebo group. Patients taking LX4211 demonstrated better glucose control (more time spent in the target range of 70-180 mg/dL) and saw a 1.7kg weight loss versus a 0.5kg weight gain in the placebo group
……………………..
Scheme 1 :
Scheme 2:
Scheme 3:
3(a) 3(b)
Scheme 4:
4(a) 4(b)
Scheme 3:

…………………
EXAMPLES
- Aspects of this invention can be understood from the following examples.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone
- To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt% water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86% yield, 96.7 area% pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 – 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73.
- To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H).
- To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahvdrofuro[2.3-d][1,3]dioxol-5-yl)(morpholino)methanone
- A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL),∼1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25°C. Additional charges of TEMPO (14.3g, 0.5 mol%) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to < 10°C and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15°C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L x 3). The combined organic layer was concentrated under vacuo (∼100-120 torr) at < 35°C (28-32°C vapor, 45-50°C bath) to low volume (- 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached < 1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5°C and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ5.96 (d, J = 3.6 Hz, 1H), 4.5 8 (d, J = 3.6 Hz, 1H), 4.53 (d, J =3.2Hz,1H), 4.30 (d, J= 3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) 8 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25. 1.
- The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (-80 L) to remove excess morpholine. When final volume reached -12 L, heptane (14 L) was added slowly at 60-70°C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136°C (DSC). 1H NMR (CDCl3), δ 6.02 (d, J = 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
- A 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF = 0.003 wt% water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step.
- A jacketed 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF = 0.003 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 6°C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9°C. The resulting orange solution was diluted with dichloromethane (75mL) and was cooled to -7°C. Then a solution of 2-chloro-5-iodobenzoyl chloride (≤ 0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3°C. The reaction mixture was warmed slightly and held at +5°C for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450mL pre-cooled (∼5°C) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10min with internal temperature remaining below 28°C. The quenched biphasic mixture was stirred at 20°C for 45min and the lower organic phase was washed with 1N aq. HCl (200mL), twice with saturated aq sodium bicarbonate (200mL per wash), and with saturated aq sodium chloride (200mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93g, 99.0 area% by HPLC at 220nm, 1.0 area% regioisomer at 200nm, 98.5 % “as-is” yield).
- A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300mL, KF = 0.004 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 5°C.Then triethylsilane (28mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24mL, 194.46mmo1,2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150mL) followed by saturated aq sodium bicarbonate (150mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq sodium bicarbonate (100 mL), and with saturated aq sodium chloride (50mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45°C was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45°C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
- To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500mag, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5°C, and the mixture was stirred for 1.5 h at 0-5°C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5°C and the mixture was kept stirring for 1h, warmed to 20°C and stirred at 20°C for 2 hours. The reaction was quenched with saturated aq NH4CI, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7. 88 (dd, J= 8.4, 2.0 Hz, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.12 (d, J= 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.07 (d, J = 3.2 Hz, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.16 (d, J = 7.2 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone
- To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4X to the morpholinoamide) at room temperature and cooled to -5°C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at -5°C over 3 hours. This Grignard solution was used in the ketone formation below.
- [0055]To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity = 97 wt%, 2.01 kg, 7.34 mol) and THF (11 L, 5.5X) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30°C. The homogeneous solution was then cooled to – 25°C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at -25°C over 3 hours. Then the above Grignard solution was added to this solution at -20 over 41 minutes. The resulting solution was further stirred at -20°C before quench. The reaction mixture was added to 10 wt% aqueous NH4Cl (10 L, 5X) at 0°C with vigorous stirring, and stirred for 30 minutes at 0°C. To this mixture was added slowly 6 N HCl (4 L, 2X) at 0°C to obtain a clear solution and stirred for 30 minutes at 10°C. After phase split, the organic layer was washed with 25 wt% aq NaCl (5 L, 2.5X). Then the organic layer was concentrated to a 3X solution under the conditions (200 mbar, bath temp 50°C). EtOAc (24 L, 12X) was added, and evaporated to a 3X solution under the conditions (150 mbar, bath temp 50°C). After removed solids by a polish filtration, EtOAc (4 L, 2X) was added and concentrated to dryness (150 mbar, bath temp 50°C). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70°C to obtain a 2.5X homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5X) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5X) was added slowly to a little cloudy solution at 70°C. After stirred for 0.5 h at 70°C, the suspension was slowly cooled to 60°C and stirred for 1 h at 60°C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4X), dried under vacuum at 45°C to give the desired ketone as fluffy solids (2.57 kg, 100 wt% by HPLC, purity-adjusted yield: 81%).
(2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)-((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17X) was added CeCl3.7H2O (118.5g, 1.2 equiv) and the mixture was stirred at 20°C until all solids were dissolved. The mixture was then cooled to -78°C and NaBH4 (12.03g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed -70°C. The mixture was stirred at – 78°C for 1 hour, slowly warmed to 0°C and quenched with saturated aq NH4Cl (550 mL, 5X). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1L, 10X x2) and washed with brine (550 mL, 5X). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100°C and stirred for 15 hours. The mixture was then cooled to room temperature (20°C) and concentrated under vacuum to give a yellow oil (crude, ∼118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0°C. Then, Ac2O (195 mL, -8.0 equiv) was added and the mixture was warmed to 20°C and stirred at 20°C for 2h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, x2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (-133g).
- To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4X) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80°C for 3.5 hours. The mixture was cooled to 20°C and Mel (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20°C for 3h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1.3 L, 10X) and washed with H2O (650 mL, 5X x2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5X) and the mixture was reslurried at 60°C for 2h and then cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 70 mL, x3). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) δ 7.37 (d, J= 8.0 Hz, 1H), 7.20 (dd, J= 8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J = 9.6 Hz, 1H), 5.20 (t, J = 9.6 Hz, 1H), 5.05 (t, J= 9.6 Hz, 1H), 4.51 (d, J=9.6Hz, 1H), 4.38 (d, J= 9.6Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2.11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
- To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19X). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25°C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt% aq NH4Cl (2.5 L, 1X) and the mixture was concentrated under vacuum to 5X, diluted with water (10 L, 4X) and MTBE (12.5L, 5X). The mixture was cooled to 10°C and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2X). The combined aqueous layer was extracted with MTBE (12.5 L, 5X). The combined organic layers were washed with brine (2.5 L, 1X) and concentrated under vacuum to 3X. MeCN (15 L, 6X) was added. The mixture was concentrated again to 10 L (4X) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
- The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol% aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80°C for 2 hours and then cooled to 20°C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2X) and diluted with MTBE (15 L, 6X). The organic layer was separated, washed with brine (5 L, 2X) and concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the mixture was concentrated to 7.5 L (3X).
- The above MeCN solution of (3S,4R,SR,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10°C, added with dimethylaminopyridine (17.53 g, 2.5 mol%), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2X, 6.0 equiv) so that the temperature of the mixture was kept below 20°C. The reaction was then warmed to 20°C and stirred for 1 hour and diluted with MTBE (15 L, 6X). The mixture was slowly quenched with water (7.5 L, 3X). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2X), 1 N NaHSO4 (5 L, 2X), and brine (5 L, 2X) in sequence.
- The organic layer was then concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the solution was concentrated to 7.5 L (3X) (KF = 0.08%). Dioxane (12.5 L, 5X) was added and the solution was concentrated to 7.50 L (3X) (KF = 0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL).
- To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80°C for 3 hours (>97% conversion). The mixture was cooled to 20°C and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20°C for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20°C for 1 hours. The mixture was then diluted with MTBE (25 L, 10X) and washed with water (12.5 L, 5X x2). The organic layer was separated and concentrated under vacuum to -5 L (2X). MeOH (12.5 L, 5X) was added and the mixture was concentrated to 5X to afford a slurry. The mixture was then heated at 60°C for 1 hour and cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 2.5 L, 1X x2, 1.0 L, 0.4X). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
- To a slurry of (2S,3S,4R,SS,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mo1) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 20°C and the mixture was stirred at 20°C for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.0g, 95%). 1H NMR (CDCl3) δ 7.38 (d, J = 8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, 1H), 3.50 (m, 2H), 3.42 (br s, 1H), 2.95 (br s, 1H), 2.57 (br s, 1H), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
…………
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H- pyran-3,4,5-triol:
LEX-1287 The compound is an inhibitor of the sodium glucose co-transporter 2, and may be useful in the treatment of diabetes and a variety of other diseases and conditions. See U.S. patent application no. 11/862,690, filed September 28, 2007.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran-3,4,5-triol To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-
(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mol) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. The mixture was then
18
LEX-1287 concentrated to 300 mL, added to H2O (IL) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.Og, 95%). IH NMR (CDC13) δ 7.38 (d, J = 8.4 Hz, IH), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, IH), 4.15 (d, J = 9.6 Hz, IH), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, IH), 3.50 (m, 2H), 3.42 (br s, IH), 2.95 (br s, IH), 2.57 (br s, IH), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
6.9. Preparation of Crystalline Anhydrous (2S,3R,4R,5S,6R)-2-(4-chloro-
3-f4-ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran- 3,4,5-triol Form 1
Under slightly positive nitrogen pressure, to a 50 L reactor was charged MeOH (12 L) and the triacetate (1.70 Kg, 3.09 mol). Methanol (5L) was added as a rinse. The slurry was then added NaOMe in MeOH (25 wt%, 340 mL, 0.2X) in 15 minutes at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. To the mixture was added slowly water (25.5 L, 15X) in 45 minutes with 5 g seeding (DSC123°C). Solids crashed out and the mixture was stirred at 200C for 1 hour, cooled to 00C and stirred for 30 minutes. The solid was filtered and washed with water (1.7 L, IX, x2) and the cake was dried under vacuum at 45°C overnight to afford the title compound (m.p. ~ 123 0C by DSC peak; 1.28 Kg, 97.7% yield).
…………..
EXAMPLES
Aspects of this invention can be understood from the following examples, which do not limit its scope.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
To a 12 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(−)-xylose (504.40 g, 3.360 mol), acetone (5 L, reagent grade) and anhydrous MgSO4 powder (811.23 g, 6.740 mol/2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol/0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24° C. over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH =8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25 L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt % water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23 g, 86% yield, 96.7 area % pure by GC). 1H NMR (400 MHz, DMSO-d6) δ 1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93-4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 3C NMR (101 MHz, DMSO-d6) δ 26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0 g, 131 mmol) in acetone (375 mL, 15×) and H2O (125 mL, 5×) was added NaHCO3 (33.0 g, 3.0 equiv), NaBr (2.8 g, 20 mol %) and TEMPO (0.40 g, 2 mol %) at 20° C. The mixture was cooled to 0-5° C. and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20° C. for 24h. Methanol (20 mL) was added and the mixture was stirred at 20° C. for 1 h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2×). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12× ×3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5×) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0 g, 79%).1H NMR (methanol-d4), δ 6.00 (d, J=3.2 Hz, 1H), 4.72 d, J=3.2 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.38 (d, J=3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0 g, 24.5 mmol) in THF (100 ML, 20×) was added TBTU (11.8 g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20° C. for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20° C. for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2× ×2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4: 1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10° C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (˜100-120 torr) at <35° C. (28-32° C. vapor, 45-50° C. bath) to low volume (˜6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5° C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) δ 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (˜80 L) to remove excess morpholine. When final volume reached 12 L, heptane (14 L) was added slowly at 60-70° C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136° C. (DSC). 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
A 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF=0.003 wt % water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step. A jacketed 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF=0.003 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 6° C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9° C. The resulting orange solution was diluted with dichloromethane (75 mL) and was cooled to −7° C. Then a solution of 2-chloro-5-iodobenzoyl chloride (<0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3° C. The reaction mixture was warmed slightly and held at +5° C. for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450 mL pre-cooled (˜5° C.) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10 min with internal temperature remaining below 28° C. The quenched biphasic mixture was stirred at 20° C. for 45 min and the lower organic phase was washed with 1N aq. HCl (200 mL), twice with saturated aq. sodium bicarbonate (200 mL per wash), and with saturated aq. sodium chloride (200 mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93 g, 99.0 area % by HPLC at 220 nm, 1.0 area % regioisomer at 200 nm, 98.5 % “as-is” yield). A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300 mL, KF=0.004 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 5° C. Then triethylsilane (28 mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24 mL, 194.46 mmol, 2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30 min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150 mL) followed by saturated aq sodium bicarbonate (150 mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq. sodium bicarbonate (100 mL), and with saturated aq. sodium chloride (50 mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45° C. was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45° C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220 nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500 mg, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5° C., and the mixture was stirred for 1.5 h at 0-5° C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5° C. and the mixture was kept stirring for 1 h, warmed to 20° C. and stirred at 20° C. for 2 hours. The reaction was quenched with saturated aq NH4Cl, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7.88 (dd, J=8.4, 2.0 Hz, 1H), 7.82 (d, J=2.0 Hz, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.12 (d, J=8.4 Hz, 2H), 6.86 (d, J=8.4 Hz, 2H), 6.07 (d, J=3.2 Hz, 1H), 5.21 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 4.56 (d, J=3.2 Hz, 1H), 4.16 (d, J=7.2 Hz, 2H), 4.03 (q, J=7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J=7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d]1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4× to the morpholinoamide) at room temperature and cooled to −5° C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at −5° C. over 3 hours. This Grignard solution was used in the ketone formation below. To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity=97 wt %, 2.01 kg, 7.34 mol) and THF (11 L, 5.5×) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30° C. The homogeneous solution was then cooled to −25° C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at −25° C. over 3 hours. Then the above Grignard solution was added to this solution at −20 over 41 minutes. The resulting solution was further stirred at −20° C. before quench. The reaction mixture was added to 10 wt % aqueous NH4Cl (10 L, 5×) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C. To this mixture was added slowly 6 N HCl (4 L, 2×) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C. After phase split, the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5×). Then the organic layer was concentrated to a 3× solution under the conditions (200 mbar, bath temp 50° C.). EtOAc (24 L, 12×) was added, and evaporated to a 3× solution under the conditions (150 mbar, bath temp 50° C.). After removed solids by a polish filtration, EtOAc (4 L, 2×) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5× homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5×) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5×) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C. and stirred for 1 h at 60° C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4×), dried under vacuum at 45° C. to give the desired ketone as fluffy solids (2.57 kg, 100 wt % by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17×) was added CeCl3.7H2O (118.5 g, 1.2 equiv) and the mixture was stirred at 20° C. until all solids were dissolved. The mixture was then cooled to −78° C. and NaBH4 (12.03 g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed −70° C. The mixture was stirred at −78° C. for 1 hour, slowly warmed to 0° C. and quenched with saturated aq NH4Cl (550 mL, 5×). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1 L, 10× ×2) and washed with brine (550 mL, 5×). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115 g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100° C. and stirred for 15 hours. The mixture was then cooled to room temperature (20° C.) and concentrated under vacuum to give a yellow oil (crude, 118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0° C. Then, Ac2O (195 mL, ˜8.0 equiv) was added and the mixture was warmed to 20° C. and stirred at 20° C. for 2 h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, ×2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (˜133 g). To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4×) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80° C. for 3.5 hours. The mixture was cooled to 20° C. and MeI (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20° C. for 3 h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1. 3 L, 10×) and washed with H2O (650 mL, 5× ×2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5×) and the mixture was reslurried at 60° C. for 2 h and then cooled to 0C and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 70 mL, ×3). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) 6 7.37 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J=9.6 Hz, 1H), 5.20 (t, J=9.6 Hz, 1H), 5.05 (t, J =9.6 Hz, 1H), 4.51 (d, J =9.6 Hz, 1H), 4.38 (d, J=9.6 Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2. 11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J=7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19×). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25° C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt % aq NH4Cl (2.5 L, 1×) and the mixture was concentrated under vacuum to 5×, diluted with water (10 L, 4×) and MTBE (12.5 L, 5×). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2×). The combined aqueous layer was extracted with MTBE (12.5 L, 5×). The combined organic layers were washed with brine (2.5 L, 1×) and concentrated under vacuum to 3×. MeCN (15 L, 6×) was added. The mixture was concentrated again to 10 L (4×) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN. The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80° C. for 2 hours and then cooled to 20° C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2×) and diluted with MTBE (15 L, 6×). The organic layer was separated, washed with brine (5 L, 2×) and concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the mixture was concentrated to 7.5 L (3×). The above MeCN solution of (3S,4R,5R,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10° C., added with dimethylaminopyridine (17.53 g, 2.5 mol %), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2×, 6.0 equiv) so that the temperature of the mixture was kept below 20° C. The reaction was then warmed to 20° C. and stirred for 1 hour and diluted with MTBE (15 L, 6×). The mixture was slowly quenched with water (7.5 L, 3×). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2×), 1 N NaHSO4 (5 L, 2×), and brine (5 L, 2×) in sequence. The organic layer was then concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the solution was concentrated to 7.5 L (3×) (KF=0.08%). Dioxane (12.5 L, 5×) was added and the solution was concentrated to 7.50 L (3×) (KF=0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL). To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80° C. for 3 hours (>97% conversion). The mixture was cooled to 20° C. and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20° C. for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20° C. for 1 hours. The mixture was then diluted with MTBE (25 L, 10×) and washed with water (12.5 L, 5× ×2). The organic layer was separated and concentrated under vacuum to ˜5 L (2×). MeOH (12.5 L, 5×) was added and the mixture was concentrated to 5× to afford a slurry. The mixture was then heated at 60° C. for 1 hour and cooled to 0° C. and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 2.5 L, 1× ×2, 1.0 L, 0.4×). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0. 164 mol) in MeOH (900 mL, 10×) was added NaOMe in MeOH (25 wt %, 18 mL, 0.2×) at 20° C. and the mixture was stirred at 20° C. for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1 L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, ×3) and the cake was dried under vacuum at 45° C. overnight to afford the desired methyl thiolate (67.0 g, 95%). 1H NMR (CDCl3) 6 7.38 (d, J=8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J=8.8 Hz, 2H), 6.83 (d, J=8.8 Hz, 2H), 4.35 (d, J=9.6 Hz, 1H), 4.15 (d, J=9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J=8.8 Hz, 1H), 3.50 (m, 2H), 2.73 (br s, 3H), 2.17 (s, 3H), 1.40 (t, J=7.2 Hz, 3H).
…………..
Kanwal A, Banerjee SK.
Pharm Pat Anal. 2013 Jan;2(1):77-91. doi: 10.4155/ppa.12.78.
clinical trials………..http://clinicaltrials.gov/search/intervention=LX-4211+OR+LX4211
On the importance of synthetic organic chemistry in drug discovery: reflections on the discovery of antidiabetic agent ertugliflozin. , Med. Chem. Commun., 2013, 4, 101
..............................................
10
CANAGLIFLOZIN

CANAGLIFLOZIN
Canagliflozin
Canagliflozin is a highly potent and selective subtype 2 sodium-glucose transport protein (SGLT2) inhibitor to CHO- hSGLT2, CHO- rSGLT2 and CHO- mSGLT2 with IC50 of 4.4 nM, 3.7 nM and 2 nM, respectively.
M.F.C24H25FO5S
M.Wt: 444.52
CAS No: 842133-18-0
(1S)-1,5-Anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
NMR.....http://file.selleckchem.com/downloads/nmr/S276003-Canagliflozin-HNMR-Selleck.pdf
Canagliflozin (INN, trade name Invokana) is a drug of the gliflozin class, used for the treatment of type 2 diabetes.[1][2] It was developed by Mitsubishi Tanabe Pharma and is marketed under license by Janssen, a division of Johnson & Johnson.[3]
U.S. Patent No, 7,943,788 B2 (the '788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US '582 B2 and US '202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US '487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US '487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US '487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US '487 Al is not suitable for pharmaceutical preparations.
The US '487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process. Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Medical use



///////////
11


 Chemical
name of Empagliflozin (BI10773):
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
Chemical
name of Empagliflozin (BI10773):
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
















CANAGLIFLOZIN
CANAGLIFLOZIN
Canagliflozin
Canagliflozin is a highly potent and selective subtype 2 sodium-glucose transport protein (SGLT2) inhibitor to CHO- hSGLT2, CHO- rSGLT2 and CHO- mSGLT2 with IC50 of 4.4 nM, 3.7 nM and 2 nM, respectively.
M.F.C24H25FO5S
M.Wt: 444.52
CAS No: 842133-18-0
(1S)-1,5-Anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
NMR.....http://file.selleckchem.com/downloads/nmr/S276003-Canagliflozin-HNMR-Selleck.pdf
| Canagliflozin Hemihydrate (1S)-1,5-Anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol hydrate (2:1) | 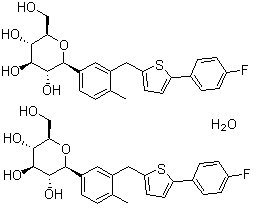 | 928672-86-0 |
Canagliflozin (INN, trade name Invokana) is a drug of the gliflozin class, used for the treatment of type 2 diabetes.[1][2] It was developed by Mitsubishi Tanabe Pharma and is marketed under license by Janssen, a division of Johnson & Johnson.[3]
U.S. Patent No, 7,943,788 B2 (the '788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US '582 B2 and US '202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US '487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US '487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US '487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US '487 Al is not suitable for pharmaceutical preparations.
The US '487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process. Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Medical use
- Canagliflozin
is an antidiabetic drug used to improve glycemic control in people with
type 2 diabetes. In extensive clinical trials, canagliflozin produced a
consistent dose-dependent reduction in HbA1c
of 0.77% to 1.16% when administered as monotherapy, combination with
metformin, combination with metformin & Sulfonyulrea, combination
with metformin & pioglitazone and In combination with insulin from a
baselines of 7.8% to 8.1%, in combination with metformin, or in combination with metformin and a sulfonylurea.
When added to metformin Canagliflozin 100mg was shown to be
non-inferior to both Sitagliptin 100mg and glimiperide in reductions on
HbA1c at one year, whilst canagliflozin 300mg successfully demontrated
statistical superiority over both Sitagliptin and glimiperide in HbA1c
reductions. Secondary efficacy endpoint of superior body weight
reduction and blood pressure reduction (versus Sitagliptin and
glimiperide)) were observed as well. Canagliflozin produces beneficial
effects on HDL cholesterol whilst increasing LDL cholesterol to produce no change in total cholesterol.[4][5]
Contraindications
Canagliflozin has proven to be clinically effective in people with moderate renal failure and treatment can be continued in patients with renal impairment.
Adverse effects
Canagliflozin, as is common with all sglt2 inhibitors, increased (generally mild) urinary tract infections, genital fungal infections, thirst,[6] LDL cholesterol, and was associated with increased urination and episodes of low blood pressure.
There are concerns it may increase the risk of diabetic ketoacidosis.[7]
Cardiovascular problems have been discussed with this class of drugs.[citation needed] The pre-specified endpoint for cardiovascular safety in the canagliflozin clinical development program was Major Cardiovascular Events Plus (MACE-Plus), defined as the occurrence of any of the following events: cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, or unstable angina leading to hospitalization. This endpoint occurred in more people in the placebo group (20.5%) than in the canagliflozin treated group (18.9%).
Nonetheless, an FDA advisory committee expressed concern regarding the cardiovascular safety of canagliflozin. A greater number of cardiovascular events was observed during the first 30 days of treatment in canagliflozin treated people (0.45%) relative to placebo treated people (0.07%), suggesting an early period of enhanced cardiovascular risk. In addition, there was an increased risk of stroke in canagliflozin treated people. However none of these effects were seen as statistically significant. Additional cardiovascular safety data from the ongoing CANVAS trial is expected in 2015.[8]
Interactions
The drug may increase the risk of dehydration in combination with diuretic drugs.
Because it increases renal excretion of glucose, treatment with canagliflozin prevents renal reabsorption of 1,5-anhydroglucitol, leading to artifactual decreases in serum 1,5-anhydroglucitol; it can therefore interfere with the use of serum 1,5-anhydroglucitol (assay trade name, GlycoMark) as a measure of postprandial glucose excursions.[9]
Mechanism of action
Canagliflozin is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the renal glucose reabsorption (SGLT1 being responsible for the remaining 10%). Blocking this transporter causes up to 119 grams of blood glucose per day to be eliminated through the urine,[10] corresponding to 476 kilocalories. Additional water is eliminated by osmotic diuresis, resulting in a lowering of blood pressure.
This mechanism is associated with a low risk of hypoglycaemia (too low blood glucose) compared to other antidiabetic drugs such as sulfonylurea derivatives and insulin.[11]
History
On July 4, 2011, the European Medicines Agency approved a paediatric investigation plan and granted both a deferral and a waiver for canagliflozin (EMEA-001030-PIP01-10) in accordance with EC Regulation No.1901/2006 of the European Parliament and of the Council.[12]
In March 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States.[13]
SYNTHESIS
.............
Canagliflozin
is an API that is an inhibitor of SGLT2 and is being developed for the
treatment of type 2 diabetes mellitus.[0011] The IUPAC systematic name
of canagliflozin is (25,,3/?,4i?,55',6 ?)-2-{3-[5-[4-fluoro-
phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol,
and is also known as
(15)-l,5-anhydro-l-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-
methylphenyl]-D-glucitol and l-(
-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-
thienylmethyl]benzene. Canagliflozin is a white to off-white powder with
a molecular formula of C24H25F05S and a molecular weight of 444.52. The structure of canagliflozin is shown as compound B.
 Compound B - Canagliflozin
Compound B - Canagliflozin
[0012] In US 2008/0146515 Al, a crystalline hemihydrate form of canagliflozin (shown as Compound C) is disclosed, having the powder X-ray diffraction (XRPD) pattern comprising the following 2Θ values measured using CuKa radiation: 4.36±0.2, 13.54±0.2, 16.00±0.2, 19.32±0.2, and 20.80±0.2. The XRPD pattern is shown in Figure 24. A process for the preparation of canagliflozin hemihydrate is also disclosed in US 2008/0146515 Al.
 Compound C - hemihydrate form of canagliflozin
Compound C - hemihydrate form of canagliflozin
[0013] In US 2009/0233874 Al, a crystalline form of canagliflozin is disclosed.
[0012] In US 2008/0146515 Al, a crystalline hemihydrate form of canagliflozin (shown as Compound C) is disclosed, having the powder X-ray diffraction (XRPD) pattern comprising the following 2Θ values measured using CuKa radiation: 4.36±0.2, 13.54±0.2, 16.00±0.2, 19.32±0.2, and 20.80±0.2. The XRPD pattern is shown in Figure 24. A process for the preparation of canagliflozin hemihydrate is also disclosed in US 2008/0146515 Al.
[0013] In US 2009/0233874 Al, a crystalline form of canagliflozin is disclosed.
........
WO
2005/012326 pamphlet discloses a class of compounds that are inhibitors
of sodium-dependent glucose transporter (SGLT) and thus of therapeutic
use for treatment of diabetes, obesity, diabetic complications, and the
like. There is described in WO 2005/012326 pamphlet
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
of formula (I):

Example
1 Crystalline
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
hemihydrate1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
was prepared in a similar manner as described in WO 2005/012326.
 (1)
To a solution of
5-bromo-1-[5-(4-fluorophenyl)-2-thienylmethyl]-2-methylbenzene (1, 28.9
g) in tetrahydrofuran (480 ml) and toluene (480 ml) was added
n-butyllithium (1.6M hexane solution, 50.0 ml) dropwise at −67 to −70°
C. under argon atmosphere, and the mixture was stirred for 20 minutes at
the same temperature. Thereto was added a solution of 2 (34.0 g) in
toluene (240 ml) dropwise at the same temperature, and the mixture was
further stirred for 1 hour at the same temperature. Subsequently,
thereto was added a solution of methanesulfonic acid (21.0 g) in
methanol (480 ml) dropwise, and the resulting mixture was allowed to
warm to room temperature and stirred for 17 hours. The mixture was
cooled under ice—water cooling, and thereto was added a saturated
aqueous sodium hydrogen carbonate solution. The mixture was extracted
with ethyl acetate, and the combined organic layer was washed with brine
and dried over magnesium sulfate. The insoluble was filtered off and
the solvent was evaporated under reduced pressure. The residue was
triturated with toluene (100 ml)—hexane (400 ml) to give
1-(1-methoxyglucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]-benzene
(3) (31.6 g). APCI-Mass m/Z 492 (M+NH4).
(1)
To a solution of
5-bromo-1-[5-(4-fluorophenyl)-2-thienylmethyl]-2-methylbenzene (1, 28.9
g) in tetrahydrofuran (480 ml) and toluene (480 ml) was added
n-butyllithium (1.6M hexane solution, 50.0 ml) dropwise at −67 to −70°
C. under argon atmosphere, and the mixture was stirred for 20 minutes at
the same temperature. Thereto was added a solution of 2 (34.0 g) in
toluene (240 ml) dropwise at the same temperature, and the mixture was
further stirred for 1 hour at the same temperature. Subsequently,
thereto was added a solution of methanesulfonic acid (21.0 g) in
methanol (480 ml) dropwise, and the resulting mixture was allowed to
warm to room temperature and stirred for 17 hours. The mixture was
cooled under ice—water cooling, and thereto was added a saturated
aqueous sodium hydrogen carbonate solution. The mixture was extracted
with ethyl acetate, and the combined organic layer was washed with brine
and dried over magnesium sulfate. The insoluble was filtered off and
the solvent was evaporated under reduced pressure. The residue was
triturated with toluene (100 ml)—hexane (400 ml) to give
1-(1-methoxyglucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]-benzene
(3) (31.6 g). APCI-Mass m/Z 492 (M+NH4).
(2) A solution of 3 (63.1 g) and triethylsilane (46.4 g) in dichloromethane (660 ml) was cooled by dry ice-acetone bath under argon atmosphere, and thereto was added dropwise boron trifluoride•ethyl ether complex (50.0 ml), and the mixture was stirred at the same temperature. The mixture was allowed to warm to 0° C. and stirred for 2 hours. At the same temperature, a saturated aqueous sodium hydrogen carbonate solution (800 ml) was added, and the mixture was stirred for 30 minutes. The organic solvent was evaporated under reduced pressure, and the residue was poured into water and extracted with ethyl acetate twice. The organic layer was washed with water twice, dried over magnesium sulfate and treated with activated carbon. The insoluble was filtered off and the solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (300 ml), and thereto were added diethyl ether (600 ml) and H2O (6 ml). The mixture was stirred at room temperature overnight, and the precipitate was collected, washed with ethyl acetate-diethyl ether (1:4) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (33.5 g) as colorless crystals.
mp 98-100° C. APCI-Mass m/Z 462 (M+NH4). 1H-NMR (DMSO-d6) δ 2.26 (3H, s), 3.13-3.28 (4H, m), 3.44 (1H, m), 3.69 (1H, m), 3.96 (1H, d, J=9.3 Hz), 4.10, 4.15 (each 1H, d, J=16.0 Hz), 4.43 (1H, t, J=5.8 Hz), 4.72 (1H, d, J=5.6 Hz), 4.92 (2H, d, J=4.8 Hz), 6.80 (1H, d, J=3.5 Hz), 7.11-7.15 (2H, m), 7.18-7.25 (3H, m), 7.28 (1H, d, J=3.5 Hz), 7.59 (2H, dd, J=8.8, 5.4 Hz).
Anal. Calcd. for C24H25FO5S.0.5H2O: C, 63.56; H, 5.78; F, 4.19; S, 7.07. Found: C, 63.52; H, 5.72; F, 4.08; S, 7.00.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
Example 2An amorphous powder of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene (1.62 g) was dissolved in acetone (15 ml), and thereto were added H2O (30 ml) and a crystalline seed. The mixture was stirred at room temperature for 18 hours, and the precipitate was collected, washed with acetone—H2O (1:4, 30 ml) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (1.52 g) as colorless crystals. mp 97-100° C.
(2) A solution of 3 (63.1 g) and triethylsilane (46.4 g) in dichloromethane (660 ml) was cooled by dry ice-acetone bath under argon atmosphere, and thereto was added dropwise boron trifluoride•ethyl ether complex (50.0 ml), and the mixture was stirred at the same temperature. The mixture was allowed to warm to 0° C. and stirred for 2 hours. At the same temperature, a saturated aqueous sodium hydrogen carbonate solution (800 ml) was added, and the mixture was stirred for 30 minutes. The organic solvent was evaporated under reduced pressure, and the residue was poured into water and extracted with ethyl acetate twice. The organic layer was washed with water twice, dried over magnesium sulfate and treated with activated carbon. The insoluble was filtered off and the solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (300 ml), and thereto were added diethyl ether (600 ml) and H2O (6 ml). The mixture was stirred at room temperature overnight, and the precipitate was collected, washed with ethyl acetate-diethyl ether (1:4) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (33.5 g) as colorless crystals.
mp 98-100° C. APCI-Mass m/Z 462 (M+NH4). 1H-NMR (DMSO-d6) δ 2.26 (3H, s), 3.13-3.28 (4H, m), 3.44 (1H, m), 3.69 (1H, m), 3.96 (1H, d, J=9.3 Hz), 4.10, 4.15 (each 1H, d, J=16.0 Hz), 4.43 (1H, t, J=5.8 Hz), 4.72 (1H, d, J=5.6 Hz), 4.92 (2H, d, J=4.8 Hz), 6.80 (1H, d, J=3.5 Hz), 7.11-7.15 (2H, m), 7.18-7.25 (3H, m), 7.28 (1H, d, J=3.5 Hz), 7.59 (2H, dd, J=8.8, 5.4 Hz).
Anal. Calcd. for C24H25FO5S.0.5H2O: C, 63.56; H, 5.78; F, 4.19; S, 7.07. Found: C, 63.52; H, 5.72; F, 4.08; S, 7.00.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
Example 2An amorphous powder of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene (1.62 g) was dissolved in acetone (15 ml), and thereto were added H2O (30 ml) and a crystalline seed. The mixture was stirred at room temperature for 18 hours, and the precipitate was collected, washed with acetone—H2O (1:4, 30 ml) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (1.52 g) as colorless crystals. mp 97-100° C.
........
there
are a significant number of other β-C-arylglucoside derived drug
candidates, most of which differ only in the aglycone moiety (i.e.,
these compounds comprise a central 1-deoxy-glucose ring moiety that is
arylated at CI). It is this fact that makes them attractive targets for a
novel synthetic platform technology, since a single methodology should
be able to furnish a plurality of products. Among β-C-arylglucosides
that possess known SGLT2 inhibition also currently in clinical
development are canagliflozin, empagliflozin, and ipragliflozin.
 Dapagliflozin Canagliflozin
Dapagliflozin Canagliflozin
 Ipragliflozin .....................Empagliflozin
Ipragliflozin .....................Empagliflozin
[0007] A series of synthetic methods have been reported in the peer-reviewed and patent literature that can be used for the preparation of β-C-arylglucosides. These methods are described below and are referred herein as the gluconolactone method, the metalated glucal method, the glucal epoxide method and the glycosyl leaving group substitution method.
[0008] The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor "redox economy" (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction
Glucose Gluconoloctone Hemiketal a-anomer β-anomer
R = protecting group
[0009] The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
[0010] U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.
R = protecting group; R' = H or alkyl
[0011] The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.
or
Zn[0012] The glycosyl leaving group substitution method: U.S. Patent
7,847,074, also disclosed a method comprising the substitution of a
leaving group located at CI of a hydroxy-protected glucosyl species,
such as a glycosyl halide, with a metalated aryl compound to prepare
SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar
method to prepare the SGLT2 inhibitor canagliflozin and preferably
diarylzinc complexes are used as nucleophiles along with tetra-
>-pivaloyl protected glucosylbromide.
Glucose Glucosyl bromide β-anomer
[0013] Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
[0014] It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
[0015] In view of the above, there remains a need for a shorter, more efficient and
stereoselective, redox economic process for the preparation of β-C-arylglucosides. A new process should be applicable to the industrial manufacture of SGLT2 inhibitors and their prodrugs,
EXAMPLE 22 - Synthesis of 2,4-di-0-feri-butyldiphenylsUyl-l-C-(3-((5-(4- fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (2,4-di-6>-TBDPS- canagliflozin; (IVi"))
 [0227]
2-(5-Bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (1.5 g, 4.15
mmol) and magnesium powder (0.33 g, 13.7 mmol) were placed in a suitable
reactor, followed by THF (9 mL) and 1,2-dibromoethane (95 μί). The
mixture was heated to reflux. After the reaction was initiated, a
solution of 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (2.5
g, 6.92 mmol) in THF (15mL) was added dropwise. The mixture was stirred
for another 2 hours under reflux, and was then cooled to ambient
temperature and titrated to determine the concentration. The thus
prepared 3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl
magnesium bromide (0.29 M in THF, 17 mL, 5.0 mmol) and A1C13
(0.5 M in THF, 4.0 mL, 2.0 mmol) were mixed at ambient temperature to
give a black solution, which was stirred at ambient temperature for 1
hour. To a solution of l
,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64
g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added rc-BuLi
(0.4 mL, 1.0 mmol, 2.5 M solution in Bu20). After stirring
for about 5 min the solution was then added into the above prepared
aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to
rinse the flask. The mixture was concentrated under reduced pressure (50
torr) at 60 °C (external bath temperature) to remove low-boiling point
ethereal solvents, and PhOMe (6 mL) was then added. The remaining
mixture was heated at 150 °C (external bath temperature) for 5 hours at
which time HPLC assay analysis indicated a 68% yield of
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-
(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-
-D-glucopyranoside. After cooling to ambient temperature, the reaction
was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous
earth at ambient temperature, then the mixture was filtered and the
filter cake was washed with THF. The combined filtrates were
concentrated and the crude product was purified by silica gel column
chromatography (eluting with 1 :20 MTBE/rc-heptane) to give the product
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-
methylphenyl)- -D-glucopyranoside (0.51 g, 56%) as a white powder.
[0227]
2-(5-Bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (1.5 g, 4.15
mmol) and magnesium powder (0.33 g, 13.7 mmol) were placed in a suitable
reactor, followed by THF (9 mL) and 1,2-dibromoethane (95 μί). The
mixture was heated to reflux. After the reaction was initiated, a
solution of 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (2.5
g, 6.92 mmol) in THF (15mL) was added dropwise. The mixture was stirred
for another 2 hours under reflux, and was then cooled to ambient
temperature and titrated to determine the concentration. The thus
prepared 3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl
magnesium bromide (0.29 M in THF, 17 mL, 5.0 mmol) and A1C13
(0.5 M in THF, 4.0 mL, 2.0 mmol) were mixed at ambient temperature to
give a black solution, which was stirred at ambient temperature for 1
hour. To a solution of l
,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64
g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added rc-BuLi
(0.4 mL, 1.0 mmol, 2.5 M solution in Bu20). After stirring
for about 5 min the solution was then added into the above prepared
aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to
rinse the flask. The mixture was concentrated under reduced pressure (50
torr) at 60 °C (external bath temperature) to remove low-boiling point
ethereal solvents, and PhOMe (6 mL) was then added. The remaining
mixture was heated at 150 °C (external bath temperature) for 5 hours at
which time HPLC assay analysis indicated a 68% yield of
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-
(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-
-D-glucopyranoside. After cooling to ambient temperature, the reaction
was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous
earth at ambient temperature, then the mixture was filtered and the
filter cake was washed with THF. The combined filtrates were
concentrated and the crude product was purified by silica gel column
chromatography (eluting with 1 :20 MTBE/rc-heptane) to give the product
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-
methylphenyl)- -D-glucopyranoside (0.51 g, 56%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.65 (d, J= 7.2 Hz, 2H), 7.55 (d, J= 7.2 Hz, 2H), 7.48 (dd, J= 7.6, 5.6 Hz, 2H), 7.44-7.20 (m, 16H), 7.11-6.95 (m, 6H), 6.57 (d, J= 3.2 Hz, IH), 4.25 (d, J= 9.6 Hz, IH), 4.06 (s, 2H), 3.90-3.86 (m, IH), 3.81-3.76 (m, IH), 3.61-3.57 (m, IH), 3.54-3.49 (m, 2H), 3.40 (dd, J= 8.8, 8.8 Hz, IH), 2.31 (s, 3H), 1.81 (dd, J= 6.6, 6.6 Hz, IH, OH), 1.19 (d, J= 4.4 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 162.1 (d, J= 246 Hz, C), 143.1 (C), 141.4 (C), 137.9 (C), 136.8 (C), 136.5 (C), 136.4 (CH x2), 136.1 (CH x2), 135.25 (C), 135.20 (CH x2), 135.0 (CH x2), 134.8 (C), 132.8 (C), 132.3 (C), 130.9 (d, J= 3.5 Hz, C), 130.5 (CH), 130.0 (CH), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.6 (CH x4), 127.5 (CH x2), 127.2 (CH x2), 127.1 (d, J= 8.2 Hz, CH x2), 127.06 (CH), 126.0 (CH), 122.7 (CH), 115.7 (d, J= 21.8 Hz, CH x2), 82.7 (CH), 80.5 (CH), 79.4 (CH), 76.3 (CH), 72.9 (CH), 62.8 (CH2), 34.1(CH2), 27.2 (CH3 x3), 26.7 (CH3 x3), 19.6, (C), 19.3 (CH3),19.2 (C); LCMS (ESI) m/z 938 (100, [M+NH4]+), 943 (10, [M+Na]+).
EXAMPLE 23 - Synthesis of canagliflozin (l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)- 4-methylphenyl)- -D-glucopyranoside; (Ii))
 [0228]
A mixture of the
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-
2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (408 mg, 0.44 mmol)
and TBAF (3.5 mL, 3.5 mmol, 1.0 M in THF) was stirred at ambient
temperature for 4 hours. CaC03 (0.73 g), Dowex 50WX8-400 ion
exchange resin (2.2 g) and MeOH (5mL) were added to the product mixture
and the suspension was stirred at ambient temperature for 1 hour and
then the mixture was filtered through a pad of diatomaceous earth. The
filter cake was rinsed with MeOH and the combined filtrates was
evaporated under vacuum and the resulting residue was purified by column
chromatography (eluting with 1 :20 MeOH/DCM) affording canagliflozin (143 mg, 73%).
[0228]
A mixture of the
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-
2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (408 mg, 0.44 mmol)
and TBAF (3.5 mL, 3.5 mmol, 1.0 M in THF) was stirred at ambient
temperature for 4 hours. CaC03 (0.73 g), Dowex 50WX8-400 ion
exchange resin (2.2 g) and MeOH (5mL) were added to the product mixture
and the suspension was stirred at ambient temperature for 1 hour and
then the mixture was filtered through a pad of diatomaceous earth. The
filter cake was rinsed with MeOH and the combined filtrates was
evaporated under vacuum and the resulting residue was purified by column
chromatography (eluting with 1 :20 MeOH/DCM) affording canagliflozin (143 mg, 73%).
 III II"
III II"
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- "D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
[0007] A series of synthetic methods have been reported in the peer-reviewed and patent literature that can be used for the preparation of β-C-arylglucosides. These methods are described below and are referred herein as the gluconolactone method, the metalated glucal method, the glucal epoxide method and the glycosyl leaving group substitution method.
[0008] The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor "redox economy" (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction
R = protecting group
[0009] The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
[0010] U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.
[0011] The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.
[0013] Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
[0014] It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
[0015] In view of the above, there remains a need for a shorter, more efficient and
stereoselective, redox economic process for the preparation of β-C-arylglucosides. A new process should be applicable to the industrial manufacture of SGLT2 inhibitors and their prodrugs,
EXAMPLE 22 - Synthesis of 2,4-di-0-feri-butyldiphenylsUyl-l-C-(3-((5-(4- fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (2,4-di-6>-TBDPS- canagliflozin; (IVi"))
1H NMR (400 MHz, CDC13) δ 7.65 (d, J= 7.2 Hz, 2H), 7.55 (d, J= 7.2 Hz, 2H), 7.48 (dd, J= 7.6, 5.6 Hz, 2H), 7.44-7.20 (m, 16H), 7.11-6.95 (m, 6H), 6.57 (d, J= 3.2 Hz, IH), 4.25 (d, J= 9.6 Hz, IH), 4.06 (s, 2H), 3.90-3.86 (m, IH), 3.81-3.76 (m, IH), 3.61-3.57 (m, IH), 3.54-3.49 (m, 2H), 3.40 (dd, J= 8.8, 8.8 Hz, IH), 2.31 (s, 3H), 1.81 (dd, J= 6.6, 6.6 Hz, IH, OH), 1.19 (d, J= 4.4 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 162.1 (d, J= 246 Hz, C), 143.1 (C), 141.4 (C), 137.9 (C), 136.8 (C), 136.5 (C), 136.4 (CH x2), 136.1 (CH x2), 135.25 (C), 135.20 (CH x2), 135.0 (CH x2), 134.8 (C), 132.8 (C), 132.3 (C), 130.9 (d, J= 3.5 Hz, C), 130.5 (CH), 130.0 (CH), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.6 (CH x4), 127.5 (CH x2), 127.2 (CH x2), 127.1 (d, J= 8.2 Hz, CH x2), 127.06 (CH), 126.0 (CH), 122.7 (CH), 115.7 (d, J= 21.8 Hz, CH x2), 82.7 (CH), 80.5 (CH), 79.4 (CH), 76.3 (CH), 72.9 (CH), 62.8 (CH2), 34.1(CH2), 27.2 (CH3 x3), 26.7 (CH3 x3), 19.6, (C), 19.3 (CH3),19.2 (C); LCMS (ESI) m/z 938 (100, [M+NH4]+), 943 (10, [M+Na]+).
EXAMPLE 23 - Synthesis of canagliflozin (l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)- 4-methylphenyl)- -D-glucopyranoside; (Ii))
1H NMR (400 MHz, DMSO-J6) δ 7.63-7.57 (m, 2H), 7.28 (d, J= 3.6 Hz, 1H), 7.23-7.18 (m, 3H), 7.17-7.12 (m, 2H), 6.80 (d, J= 3.6 Hz, 1H), 4.93 (br, 2H, OH), 4.73 (br, 1H, OH), 4.44 (br,IH, OH), 4.16 (d, J= 16 Hz, 1H), 4.10 (d, J= 16 Hz, 1H), 3.97 (d, J= 9.2 Hz, 1H), 3.71 (d, J=I I.6 Hz, 1H), 3.47-3.43 (m, 1H), 3.30-3.15 (m, 4H), 2.27 (s, 3H);
13C NMR (100 MHz, DMSO- d6) δ 161.8 (d, J= 243 Hz, C), 144.1 (C), 140.7 (C), 138.7 (C), 137.8 (C), 135.4 (C), 131.0 (d, J= 3.1 Hz, C), 130.1 (CH), 129.5 (CH), 127.4 (d, J= 8.1 Hz, CH x2), 126.8 (CH), 126.7 (CH), 123.9 (CH), 116.4 (d, J= 21.6 Hz, CH x2), 81.8 (CH), 81.7 (CH), 79.0 (CH), 75.2 (CH), 70.9 (CH), 61.9 (CH2), 33.9 (CH2), 19.3 (CH3);
LCMS (ESI) m/z 462 (100, [M+NH4]+), 467 (3, [M+Na]+).
Example 1 - Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II") [0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- "D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
........
FIG. 1:
X-ray powder diffraction pattern of the crystalline of hemihydrate of the compound of formula (I).
X-ray powder diffraction pattern of the crystalline of hemihydrate of the compound of formula (I).
FIG. 2:
Infra-red spectrum of the crystalline of hemihydrate of the compound of formula (I).http://www.google.com/patents/US7943582
Infra-red spectrum of the crystalline of hemihydrate of the compound of formula (I).http://www.google.com/patents/US7943582
.............
FIGS. 3 and 4 provide the XRPD pattern and IR spectrum, respectively, of amorphous canagliflozin.
 | |
| Systematic (IUPAC) name | |
|---|---|
(2S,3R,4R,5S,6R)-2-{3-[5-[4-Fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
| |
| Clinical data | |
| Trade names | Invokana |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 65% |
| Protein binding | 99% |
| Metabolism | Hepatic glucuronidation |
| Biological half-life | 11.8 (10–13) hours |
| Excretion | Fecal and 33% renal |
| Identifiers | |
| CAS Registry Number | 842133-18-0 |
| ATC code | A10BX11 |
| PubChem | CID: 24812758 |
| IUPHAR/BPS | 4582 |
| DrugBank | DB08907 |
| ChemSpider | 26333259 |
| UNII | 6S49DGR869 |
| ChEBI | CHEBI:73274 |
| ChEMBL | CHEMBL2103841 |
| Synonyms | JNJ-28431754; TA-7284; (1S)-1,5-anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol |
| Chemical data | |
| Formula | C24H25FO5S |
| Molecular mass | 444.52 g/mol |
References
- New J&J diabetes drug effective in mid-stage study, Jun 26, 2010
- Edward C. Chao (2011). "Canagliflozin". Drugs of the Future 36 (5): 351–357. doi:10.1358/dof.2011.36.5.1590789.
- http://www.investor.jnj.com/releasedetail.cfm?releaseid=710584
- "Summary Review" (PDF). FDA.gov. March 29, 2013. Retrieved 2014-07-09.
- "HIGHLIGHTS OF PRESCRIBING INFORMATION" (PDF). FDA.gov. 2013. Retrieved 2014-07-09.
- Haberfeld, H (ed.). Austria-Codex (in German) (2013/14, supplement 01/14 ed.). Vienna: Österreichischer Apothekerverlag.
- FDA (2015-05-15). "SGLT2 inhibitors: Drug Safety Communication - FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood". Retrieved 19 May 2015.
- http://www.medscape.com/viewarticle/777503
- Balis, Dainius A; Tong, Cindy; Meininger, Gary (July 2014). "Effect of canagliflozin, a sodium–glucose cotransporter 2 inhibitor, on measurement of serum 1,5-anhydroglucitol". J Diabetes 6 (4): 378–380. doi:10.1111/1753-0407.12116.
- Prous Science: Molecule of the Month November 2007
- A. Klement (20 January 2014). "Tubuläre Senkung des Blutzuckers bei Diabetes mellitus: Invokana". Österreichische Apothekerzeitung (in German) (2/2014): 20f.
- "EMEA-001030-PIP01-10". EMA European Medicines Agency. Retrieved May 6, 2013.
- "U.S. FDA approves Johnson & Johnson diabetes drug, canagliflozin". Reuters. Mar 29, 2013.
U.S. health regulators have approved a new diabetes drug from Johnson & Johnson, making it the first in its class to be approved in the United States.
| WO2005012326A1 | Jul 30, 2004 | Feb 10, 2005 | Tanabe Seiyaku Co | Novel compounds having inhibitory activity against sodium-dependant transporter |
| WO2013064909A2 * | Oct 30, 2012 | May 10, 2013 | Scinopharm Taiwan, Ltd. | Crystalline and non-crystalline forms of sglt2 inhibitors |
| CN103655539A * | Dec 13, 2013 | Mar 26, 2014 | 重庆医药工业研究院有限责任公司 | Oral solid preparation of canagliflozin and preparation method thereof |
| US7943582 | Dec 3, 2007 | May 17, 2011 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyransoyl)-4-methyl-3-[5-(4-fluorophenyl)-2- thienylmethyl]benzene hemihydrate |
| US7943788 | Jan 31, 2005 | May 17, 2011 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US8513202 | May 9, 2011 | Aug 20, 2013 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate |
| US20130237487 | Oct 30, 2012 | Sep 12, 2013 | Scinopharm Taiwan, Ltd. | Crystalline and non-crystalline forms of sglt2 inhibitors |
| WO2008002824A1 * | Jun 21, 2007 | Jan 3, 2008 | Squibb Bristol Myers Co | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| US6774112 * | Apr 8, 2002 | Aug 10, 2004 | Bristol-Myers Squibb Company | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US20090143316 * | Apr 4, 2007 | Jun 4, 2009 | Astellas Pharma Inc. | Cocrystal of c-glycoside derivative and l-proline |
| US20110087017 * | Oct 14, 2010 | Apr 14, 2011 | Vittorio Farina | Process for the preparation of compounds useful as inhibitors of sglt2 |
| US20110098240 * | Aug 15, 2008 | Apr 28, 2011 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor in combination with a dpp-iv inhibitor |
| Reference | ||
|---|---|---|
| 1 | * | OGURA H. ET AL.: '5-FLUOROURACIL NUCLEOSIDES. SYNTHESIS OF A STEREO-CONTROLLED NUCLEOSIDE SYNTHESIS FROM ANHYDRO SUGARS' NUCLEIC ACID CHEM. vol. 4, 1991, pages 109 - 112, XP000607288 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014195966A2 * | May 30, 2014 | Dec 11, 2014 | Cadila Healthcare Limited | Amorphous form of canagliflozin and process for preparing thereof |
| US9006188 | May 23, 2014 | Apr 14, 2015 | Mapi Pharma Ltd. | Co-crystals of dapagliflozin |
11
A Survey of Promising Late-Stage Diabetes Drugs
THIS IS DEC2012 COMPILATION. READER MAY ENCOUNTER AN APPROVED OR DROPPED ENTRY
A
variety of new drugs are in development for the treatment of type 1 or
type 2 diabetes. In addition to new dipeptidyl peptidase-4 (DPP-IV)
inhibitors, glucagon-like peptide (GLP) 1 analogs, basal insulin
analogs, and new insulin formulations, there are also unique dual
peroxisome proliferator-activated receptor (PPAR) α/γ agonist,
G-protein-coupled receptor (GPR) 40 agonist, sodium dependent glucose
transporter 2 (SGLT2) inhibitor, and several other unique agents now in
development.
A
list of SEVERAL drug candidates has been compiled for which diabetes is
at least one proposed or approved indication, and for which one
indication has reached Phase III or Registration phases. Each entry
includes the name of the drug candidate, the sponsor, and, where
applicable, chemical structures, collaboration partners; method of
action; indication (by market, where applicable); and phase of trial.
Some products are still in clinical trial phases for new indications or
formulations after winning marketing approval for initial indications;
these approvals, where applicable, are listed on the bottom of each
entry.
ASP1941 (ipragliflozin)
Sponsor/Developer: Astellas; co-developed with Kotobuki
Mechanism of action: Sodium dependent glucose transporter 2 (SGLT2) inhibitor
Indication (Phase): Japan—Type 2 diabetes with inadequate glycemic control while on a sulfonylurea or pioglitazone alone (Phase III)
U.S.
and EU—Development discontinued “after comprehensive consideration of
intensified competition for this product, and the prioritization in our
pipeline”
Canagliflozin
Sponsor/Developer: Johnson & Johnson (Janssen Research & Development); licensed from Mitsubishi Tanabe Pharma
Mechanism of action: SGLT2 inhibitor
Indication (Phase): U.S.—Once-daily oral treatment for adults with type 2 diabetes (registration; NDA filed May 2012)
EU—Once-daily oral treatment for adults with type 2 diabetes (registration; MAA filed June 2012)
Canagliflozin is the most advanced diabetes therapy in the pipeline at J&J.
Chemical Structure of Canagliflozin
Chemical Name for Canagliflozin: (2S,3R,4R,5S,6R)-2-{3-[5-[4-Fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
CAS number: 842133-18-0
Empagliflozin (BI10773)
Sponsor/Developer: Eli Lilly and Boehringer Ingelheim
Mechanism of action: SGLT 2 inhibitor
Indication (Phase): Oral
treatment of adults with type 2 diabetes (Phase III, expected to
conclude by year’s end); Oral treatment of adults with type 2 diabetes
plus high blood pressure (Phase IIb; trial results released Oct. 2)
NDA, MAA filings planned for 2013
Chemical Structure of Empagliflozin (BI10773)
 Chemical
name of Empagliflozin (BI10773):
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
Chemical
name of Empagliflozin (BI10773):
(2S,3R,4R,5S,6R)-2-[4-chloro-3-[[4-[(3S)-oxolan-3-yl]oxyphenyl]methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
CAS number:864070-44-0
Forxiga™ (dapagliflozin)
Sponsor/Developer: Bristol-Myers Squibb and AstraZeneca
Mechanism of action: SGLT2 inhibitor
Indication (Phase): EU—Approved Nov. 14 as once-daily oral medication for adults with type 2 diabetes; first SGLT2 drug to gain such approval
U.S.—Once-daily
oral medication for adults with type 2 diabetes (Registration; FDA in
October 2012 postpones decision three months pending submission of
additional clinical trial data; complete response letter issued January
2012; FDA Endocrinologic and Metabolic Drugs Advisory Committee
recommends against approval, July 2011; NDA filed March 2011)
Japan—Diabetes (Registration; NDA expected to be filed in first half of 2013)
Forxiga
is the first medicine in the new SGLT2 class to gain regulatory
approval for the treatment of type 2 diabetes. Dapagliflozin is awaiting
approval in the USA, where the Food and Drug Administration earlier
this year issued a complete response letter requesting additional data.
In addition to dapagliflozin, AstraZeneca and Bristol-Myers Squibb’s diabetes alliance included the approved DPP-4 inhibitor Onglyza (saxagliptin) and a form of the drug combined with metformin called Kombiglyze.
Chemical Structure of Dapagliflozin (Forxiga)

Chemical Name of Dapagliflozin (Forxiga): (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
CAS number: 461432-26-8
Tofogliflozin hydrate (CSG452)
Sponsor/Developer: Roche and Chugai Pharmaceutical
Mechanism of action: SGLT2 inhibitor
Indication (Phase): Oral treatment for type 2 diabetes (Phase III)
Type
II diabetes is the most common form of diabetes accounting for 90% of
diabetes cases. Over 100 million people worldwide have type-2 diabetes
(nearly 17 million in the U.S.) and the prevalence is increasing
dramatically in both the developed and developing worlds. Type-II
diabetes is a lifelong illness, which generally starts in middle age or
later part of life, but can start at any age. Patients with type-2
diabetes do not respond properly to insulin, the hormone that normally
allows the body to convert blood glucose into energy or store it in
cells to be used later. The problem in type-2 diabetes is a condition
called insulin resistance where the body produces insulin, in normal or
even high amounts, but certain mechanisms prevent insulin from moving
glucose into cells. Because the body does not use insulin properly,
glucose rises to unsafe levels in the blood, the condition known as
hyperglycemia.
Hyperglycemia,
that is, elevated plasma glucose, is a hallmark of diabetes. Plasma
glucose is normally filtered in the kidney in the glomerulus but is
actively reabsorbed in the proximal tubule (kidney). Sodium-dependent
glucose co-transporter SGLT2 appears to be the major transporter
responsible for the reuptake of glucose at this site. The SGLT inhibitor
phlorizin, and closely related analogs, inhibit this reuptake process
in diabetic rodents and dogs, resulting in normalization of plasma
glucose levels by promoting glucose excretion without hypoglycemic side
effects. Long term (6 month) treatment of Zucker diabetic rats with an
SGLT2 inhibitor has been reported to improve insulin response to
glycemia, improve insulin sensitivity, and delay the onset of
nephropathy and neuropathy in these animals, with no detectable
pathology in the kidney and no electrolyte imbalance in plasma.
Selective inhibition of SGLT2 in diabetic patients would be expected to
normalize plasma glucose by enhancing the excretion of glucose in the
urine, thereby improving insulin sensitivity and delaying the
development of diabetic complications.
The
treatment of diabetes is an important health concern and despite a wide
range of available therapies, the epidemic continues. Type 2 diabetes
(T2DM) is a progressive disease caused by insulin resistance and
decreased pancreatic β-cell function. Insulin is produced by the
pancreatic β-cell and mediates cellular glucose uptake and clearance.
Insulin resistance is characterized by the lack of response to the
actions of this hormone which results in decreased cellular clearance of
glucose from the circulation and overproduction of glucose by the
liver.
The
currently available therapies to treat type 2 diabetes augment the
action or delivery of insulin to lower blood glucose. However, despite
therapy, many patients do not achieve control of their type 2 diabetes.
According to the National Health and Nutrition Examination Survey
(NHANES) III, only 36% of type 2 diabetics achieve glycemic control
defined as a A1C<7.0% with current therapies. In an effort to treat
type 2 diabetes, aggressive therapy with multiple pharmacologic agents
may be prescribed. The use of insulin plus oral agents has increased
from approximately 3 to 11% from NHANES II to III.
Thus,
treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major
challenge, particularly in patients who require insulin as the disease
progresses. Various combinations of insulin with oral anti-diabetic
agents (OADs) have been investigated in recent years, and an increasing
number of patients have been placed on these regimens. Poulsen, M. K. et
al., “The combined effect of triple therapy with rosiglitazone,
metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108
(Supp. 6a):23S-32S (2000). Often, these combination therapies become
less effective in controlling hyperglycemia over time, particularly as
weight gain and worsening insulin resistance impair insulin response
pathways.
Hypoglycemia,
weight gain, and subsequent increased insulin resistance are
significant factors that limit optimal titration and effectiveness of
insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or
basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357
(17):1716-1730 (2007)). Weight gain with insulin therapy is
predominantly a consequence of the reduction of glucosuria, and is
thought to be proportional to the correction of glycemia. (Makimattila,
S. et al., “Causes of weight gain during insulin therapy with and
without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42
(4):406-412 (1999)). Insulin drives weight gain when used alone or with
OADs. (Buse, J., supra). In some cases, intensive insulin therapy may
worsen lipid overload and complicate progression of the disease through a
spiral of caloric surplus, hyperinsulinemia, increased lipogenesis,
increased adipocity, increased insulin resistance, beta-cell toxicity,
and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes:
pathogenesis, treatment, and prevention”, JAMA, 299
(10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones
(TZDs) and sulfonylureas intrinsically contribute to weight gain as
glucosuria dissipates with improved glycemic control. Weight gain is
less prominent with metformin, acting through suppression of hepatic
glucose output, or with incretin-based DPP-4 inhibitors. Overall, there
is a pressing need for novel agents that can be safely added to
insulin-dependent therapies to help achieve glycemic targets without
increasing the risks of weight gain or hypoglycemia.
A
novel approach to treating hyperglycemia involves targeting
transporters for glucose reabsorption in the kidney. (Kanai, Y. et al.,
“The human kidney low affinity Na+/glucose cotransporter SGLT2.
Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93
(1):397-404 (1994)). Agents that selectively block the sodium-glucose
cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can
inhibit reabsorption of glucose and induce its elimination through
urinary excretion. (Brown, G. K., “Glucose transporters: structure,
function and consequences of deficiency”, J. Inherit. Metab. Dis., 23
(3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical
models to lower blood glucose independently of insulin. (Han, S. et al.,
“Dapagliflozin, a selective SGLT2 inhibitor, improves glucose
homeostasis in normal and diabetic rats”, Diabetes, 57
(6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel
selective inhibitor of low-affinity sodium glucose cotransporter
(SGLT2), validates the critical role of SGLT2 in renal glucose
reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).
SGLT2
inhibitors inhibitors represent a novel class of agents that are being
developed for the treatment or improvement in glycemic control in
patients with type 2 diabetes. Glucopyranosyl-substituted benzene
derivative are described in the prior art as SGLT2 inhibitors, for
example in
WO 01/27128, WO 03/099836, WO 2005/092877, WO 2006/034489,
WO 2006/064033, WO 2006/117359, WO 2006/117360,
WO 2007/025943, WO 2007/028814, WO 2007/031548,
WO 2007/093610, WO 2007/128749, WO 2008/049923, WO 2008/055870, WO 2008/055940.
The
glucopyranosyl-substituted benzene derivatives are proposed as inducers
of urinary sugar excretion and as medicaments in the treatment of
diabetes.
The
term “canagliflozin” as employed herein refers to canagliflozin,
including hydrates and solvates thereof, and crystalline forms thereof
and has the following structure:
The
compound and methods of its synthesis are described in WO 2005/012326
and WO 2009/035969 for example. Preferred hydrates, solvates and
crystalline forms are described in the patent applications WO
2008/069327 for example.
atigliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:
The compound and methods of its synthesis are described in WO 2004/007517 for example.
ipragliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:
The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
tofogliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:
The compound and methods of its synthesis are described in WO 2007/140191 and WO 2008/013280 for example.
remogliflozin
and prodrugs of remogliflozin, in particular remogliflozin etabonate,
including hydrates and solvates thereof, and crystalline forms thereof.
Methods of its synthesis are described in the patent applications EP
1213296 and EP 1354888 for example.
sergliflozin
and prodrugs of sergliflozin, in particular sergliflozin etabonate,
including hydrates and solvates thereof, and crystalline forms thereof.
Methods for its manufacture are described in the patent applications EP
1344780 and EP 1489089 for example.
luseoghflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:
ertugliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:
and is described for example in WO 2010/023594.
The compound of the formula
is described for example in WO 2008/042688 or WO 2009/014970.
Dapagliflozin
The compound is described for example in WO 03/099836. Crystalline forms are described for example in WO 2008/002824.
Remogliflozin and Remogliflozin Etabonate
The compound is described for example in EP 1354888 A1.
Sergliflozin and Sergliflozin Etabonate
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
1-Chloro-4-(β-D-glucopyranos-1-yl)-2-(4-ethyl-benzyl)-benzene
The compound is described in WO 2006/034489.
(1S)-1,5-anhydro-1-[5-(azulen-2-ylmethyl)-2-hydroxyphenyl]-D-glucitol
The
compound
(4-(Azulen-2-ylmethyl)-2-(β-D-glucopyranos-1-yl)-1-hydroxy-benzene) is
described in WO 2004/013118 and WO 2006/006496. The crystalline choline
salt thereof is described in WO 2007/007628.
(1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-D-glucitol
The compound is described in WO 2004/080990 and WO 2005/012326. A cocrystal with L-proline is described in WO 2007/114475.
Thiophen Derivatives of the Formula (7-1)
wherein
R denotes methoxy or trifluoromethoxy. Such compounds and their method
of production are described in WO 2004/007517, DE 102004063099 and WO
2006/072334.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
The compound is described in WO 2005/012326. A crystalline hemihydrate is described in WO 2008/069327.
Spiroketal Derivatives of the Formula (9-1)
wherein
R denotes methoxy, trifluoromethoxy, ethoxy, ethyl, isopropyl or tert.
butyl. Such compounds are described in WO 2007/140191 and WO
2008/013280.
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Alfa Chemistry employs more than 200 full time staff, of which approximate 80 are Ph.D. and M.S. chemists, specialized in synthetic chemistry, process optimization, and research. Dspiro-PO
ReplyDeleteWow, excellent post. I'd like to draft like this too - taking time and extremely hard work to make a great article. This post has inspired me to write some posts that I am going to write soon on Buy C4-Pharma Combo 450
ReplyDeleteI got here much interesting stuff. The post is great! Thanks for sharing it! Chloromethyl
ReplyDelete