1 ROLAPITANT
2 MAROPITANT
3 LANEPITANT
4 VESTIPITANT
5 CASOPITANT
6 BIRINIPITANT


















............................................................................................................................................................................
2 MAROPITANT



........................................................................
3 LANEPITANT

Lanepitant
LY303870
UNII-17G8FN2E1F
167678-33-3 (diHCl, tri H2O)
170508-05-1 (diHCl)










........................................................................................................
.............................................................................................................
2 MAROPITANT
3 LANEPITANT
4 VESTIPITANT
5 CASOPITANT
6 BIRINIPITANT
1 ROLAPITANT
ROLAPITANT HYDROCHLORIDE
- Rolapitant HCl
- Rolapitant hydrochloride
- Sch 619734
- SCH619734
- UNII-57O5S1QSAQ
(5S ,8S)-8-[[(1R)-1-[3 ,5-
Bis(trifluoromethyl)phenyl] ethoxy] methyl]-8-phenyl-1,7-
diazaspiro[4.5]decan-2-one hydrochloride monohydrate.
Bis(trifluoromethyl)phenyl] ethoxy] methyl]-8-phenyl-1,7-
diazaspiro[4.5]decan-2-one hydrochloride monohydrate.
CAS 914462-92-3
Empirical Formula: C25H26F6N2O2 · HCl · H2O
Molecular Weight: 555
USAN Name: Rolapitant hydrochloride
INN Name: rolapitantum or rolapitant
phase 3
CAS Number: 552292-08-7 (rolapitant free base); 914462-92-3 (rolapitant HCl monohydrdate).
It is in late-stage trials of its drug rolapitant, which showed promising mid-stage results in reducing nausea and vomiting in patients undergoing chemotherapy
Rolapitant hydrochloride is a tachykinin neurokinin 1 (NK1) antagonist in phase III clinical trials at Tesaro for the prevention of chemotherapy-induced nausea and vomiting (CINV). Phase II clinical trials are also under way at OPKO for this indication. At Merck & Co., phase II clinical studies were also under way for the treatment of chronic idiopathic cough and for the prevention of chemotherapy-induced nausea; however, no recent developments have been reported for these indications.
NK1 is a G-protein coupled receptor found in the central and peripheral nervous systems. Substance P is the endogenous ligand for this receptor, whose activation leads to the production of inositol triphosphate. NK1 is believed to be involved in the emetic response.
The drug candidate was originally developed by Schering-Plough (now Merck & Co.), and in 2009 it was licensed to OPKO for the prevention of nausea and vomiting related to cancer chemotherapy and surgery. In 2010, rolapitant was licensed by OPKO to Tesaro on a worldwide basis for the prevention of chemotherapy-induced nausea and vomiting.
Rolapitant is a selective, bioavailable, CNS penetrant neurokinin NK1 receptor antagonist that shows behavioral effects in animals models of emesis. In vitro studies indicate that rolapitant has a high affinity for the human NK1 receptor of 0.66 nM and high selectivity over the human NK2 and NK3 subtypes of >1000-fold. Rolapitant is a functionally competitive antagonist, as measured by calcium efflux, with a calculated Kb of 0.17 nM. (source: Pharmacol Biochem Behav.2012 Mar 31.
Rolapitant is a potent, selective NK1 receptor antagonist that is rapidly absorbed, has a remarkably long half-life (up to180 hours), and appears to have a low potential for drug-drug interactions. A randomized, multicenter, double-blind, dose-ranging study of rolapitant was conducted with placebo and active control groups. Six hundred nineteen adult women undergoing open abdominal surgery were randomly assigned in equal ratios to 1 of 6 study arms: oral rolapitant in 5-mg, 20-mg, 70-mg, or 200-mg doses; IV ondansetron 4 mg; or placebo, stratified by history of PONV or motion sickness. The primary study endpoint was absence of emetic episodes, regardless of use of rescue medication, at 24 hours after extubation.RESULTS: Groups assigned to rolapitant 20-mg, 70-mg, and 200-mg had a higher incidence of no emesis in comparison with placebo at 24 hours after surgery. A linear relationship between rolapitant dose and primary outcome was seen. The probability of an emetic episode was significantly lower in the rolapitant 70-mg and 200-mg groups in comparison with placebo (P ≤ 0.001 based on the log-rank test). No significant differences were noted between rolapitant and the active control (ondansetron) at 24 hours after surgery, but there was a higher incidence of no emesis (regardless of rescue medication use) in the rolapitant 200- and 70-mg groups at 72 and 120 hours, respectively. CONCLUSION: Rolapitant is superior to placebo in reducing emetic episodes after surgery and reduces the incidence of vomiting in a dose-dependent manner. No differences in side effect profile were observed between rolapitant and placebo.
| References |
1: Gan TJ, Gu J, Singla N, Chung F, Pearman MH, Bergese SD, Habib AS, Candiotti KA, Mo Y, Huyck S, Creed MR, Cantillon M; Rolapitant Investigation Group. Rolapitant for the prevention of postoperative nausea and vomiting: a prospective, double-blinded, placebo-controlled randomized trial. Anesth Analg.
2011 Apr;112(4):804-12. Epub 2011 Mar 8. PubMed PMID: 21385988.
2011 Apr;112(4):804-12. Epub 2011 Mar 8. PubMed PMID: 21385988.
2. Reddy GK, Gralla RJ, Hesketh PJ. Novel neurokinin-1 antagonists as antiemetics for the treatment of chemotherapy-induced emesis. Support Cancer Ther. 2006 Apr 1;3(3):140-2. PubMed PMID: 18632487.
3. Drug Data Rep 2003, 25(8): 703
4. A multicenter, randomized, double blind, active-controlled study of the safety and efficacy of rolapitant for the prevention of chemotherapy-induced nausea and vomiting (CINV) in subjects receiving moderately emetogenic chemotherapy (NCT01500226)
ClinicalTrials.gov Web Site 2012, February 06
ClinicalTrials.gov Web Site 2012, February 06
5. Efficacy and safety of rolapitant, a novel NK-1 receptor antagonist, for the prevention of chemotherapy-induced nausea and vomiting in subjects receiving highly emetogenic chemotherapy
48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 9077
48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 9077
6. Proposed international nonproprietary names (Prop. INN): List 97
WHO Drug Inf 2007, 21(2): 160
WHO Drug Inf 2007, 21(2): 160
………………………………
patents
WO 2003051840
WO 2008118328
………………………….
The preparation of diazaspirodecan-2-ones for example, 8-[{1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one, for example, (5S,8S)-8-[{(1R)-1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxy}-methyl]-8-phenyl-1,7-diazaspiro[4.5]decan-2-one (the compound of Formula I) has been described in U.S. Pat. No. 7,049,320 (the ’320 patent), issued May 23, 2006, the disclosure of which is incorporated herein in its entirety by reference.
The compounds described in the ’320 patent are classified as tachykinin compounds, and are antagonists of neuropeptide neurokinin-1 receptors (herein, “NK-1” receptor antagonists). Other NK1 receptor antagonists and their synthesis have been described, for example, those described in Wu et al, Tetrahedron 56, 3043-3051 (2000); Rombouts et al, Tetrahedron Letters 42, 7397-7399 (2001); and Rogiers et al, Tetrahedron 57, 8971-8981 (2001) and in published international application no. WO05/100358, each of which are incorporated herein in their entirety by reference.
“NK-1” receptor antagonists have been shown to be useful therapeutic agents, for example, in the treatment of pain, inflammation, migraine, emesis (vomiting), and nociception. Among many compounds disclosed in the above-mentioned ’320 patent are several novel diazaspirodecan-2-ones, including the compound of Formula I, which are useful in the treatment of nausea and emesis associated with chemotherapy treatments (Chemotherapy-induced nausea and emesis, CINE).
The synthesis method for preparing the compound of Formula I described in the ’320 patent generally follows Scheme I in the provision of 8-[{1-(3,5-Bis-(trifluoromethyl)phenyl)-ethoxyl}-methyl]-8-phenyl-1,7-diaza-spiro[4.5]decan-2-one compounds.
The process for the preparation of the compound of Formula I described in the ’320 patent is carried out in 18 individual steps from commercially available starting materials (see the ’320 patent at col. 43, line 55 to col. 45, line 20; col. 75. line 55 to col. 80, line 21; col. 90 lines 35 to 63; and col. 98, line 1 to col. 99. line 24). In many steps of the process described in the ’320 patent, intermediate compounds must be isolated or isolated and purified before use in a subsequent step, often utilizing column chromatography for this purpose.
……………………………
Examples 72a and 72b
Step 1:
To a solution of crude Compound 53 (19 g) in CH2Cl2 (300 ml) at RT, DIEA (15 ml, 0.087 mol) was added, followed by triphosgene (4.34 g, 0.015 mol). The mixture was stirred at RT for 18 h and was filtered through a pad of silica. Solvents were removed in vacuum to give crude Compound 60 as yellow oil which was used in the next reaction without further purifications.
Step 2:
To the crude Compound 60 in THF (200 ml) at 0° C., LiBH4 (1.26 g, 0.058 mol) was added in small portions. The mixture was stirred at RT for 18 h before quenching with saturated NH4Cl solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (100×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum and purification by column chromatography [hexane-EtOAc, 4:1 (v/v)] gave Compound 61 (12.9 g, 62% overall) as white foam.
Step 3:
Oxalyl chloride (4.2 ml, 0.048 mol) was added to a solution of DMSO (6.8 m[, 0.096) in CH2Cl2 (300 ml) at −78° C. under N2. The mixture was stirred at −78° C. for 15 min before a solution of Compound 61 (8.5 g, 0.012 mol) in CH2Cl2 (100 ml) was added. The mixture was stirred at −78° C. for a further 1 h and Et3N (23.5 ml) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2Cl2 (150 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum gave an aldehyde as yellow oil. To a mixture of NaH (1.44 g, 0.036 mol) in THF at 0° C., methyl diethylphosphonoacetate (6.6 ml, 0.036 mol) was added. The mixture was stirred at 0° C. for 15 min and a solution of aldehyde in THF (100 ml) was added. The cooling bath was removed and the mixture was stirred at RT for 1 h. The reaction was quenched with saturated NH4Cl solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (200 ml×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum and purification by column chromatography [hexane-EtOAc, 4:1 (v/v)] gave an ester as white foam. The ester was dissolved in EtOH (100 ml) and a catalytic amount of palladium (1.28 g, 10% on carbon) was added. The mixture was shaken under H2 (50 psi) for 2 days. Catalytic amount of Pd(OH)2 (20% on carbon) was then added to the mixture and the mixture was again shaken under H2 (50 psi) for 5 h. The mixture was filtered through a pad of Celite and solvents were removed in vacuum to give a white foam. The foam was then dissolved in CH2Cl2 (200 ml) and TFA (8.9 ml, 0.12 mol) was added. The mixture was stirred at RT for 18 h and was cooled at 0° C. before it was neutralized with saturated NaHCO3 solution. Water and EtOAc were added to the mixture. Layers were separated and the aqueous layer was extracted with EtOAc (200 ml×2). The combined organic layers were dried (MgSO4) and filtered. Solvents were removed in vacuum to give a yellow oil. The oil was dissolved in CH3OH (50 ml) and a catalytic amount of K2CO3 (166 mg, 0.0012 mol) was added. The mixture was heated at 60° C. for 2 h. After being cooled to RT, the mixture was filtered through a pad of silica and solvents were removed in vacuum. Purification by column chromatography (EtOAc) gave the mixture of two isomers Example 72a and 72b (2.3 g, 38% overall) as white foam. Separation by HPLC using Chiralcel OD [hexane-isopropanol, 95:5 (v/v)] gave the less polar major isomer Example 72a as white foam. Electrospray MS [M+1]+=501.1. Continuous elution with the same solvent system gave the more polar minor isomer Example 72b as colorless oil.
Electrospray MS [M+1]+=501.1.
………………………..
Example 6 Preparation of Formula I Compound Salt: (5S,8S)-8-({(1R)-1-[3,5-Bis(trifluoromethyl)phenyl]ethoxy}methyl)-8-phenyl-1,7-diazaspiro[4.5]decan-2-one hydrochloride monohydrate
…………………
.................................................................................................................................................................................................
............................................................................................................................................................................
2 MAROPITANT
MAROPITANT
(7R,8S)-N-[(5-tert-Butyl-2-methoxyphenyl)methyl]-7-[di(phenyl)methyl]-1-azabicyclo[2.2.2]octan-8-amine
(2S,3S)-N-[(5-tert-butyl-2-methoxy-phenyl)methyl]-2-(diphenylmethyl)-1-azabicyclo[2.2.2]octan-3-amine
147116-67-4
PRECLINICAL, PFIZER
Maropitant, is described in WO1992021677, US 6,222,038 and US
6,255,230,US 5340826, US 5393762, EP 0769300, WO 2000073304, WO 2005082419, WO 2005082366
.................................................................................................
MAROPITANT CITRATE MONOHYDRATE
359875-09-5,
- Cerenia
- CJ-11,972
- Maropitant citrate
- UNII-LXN6S3999X
Maropitant (trade name Cerenia in the US and other countries), used as maropitantcitrate (USAN), is a neurokinin (NK1) receptor antagonist, which was developed by Zoetisspecifically for the treatment of motion sickness and vomiting in dogs. It was approved by the FDA in 2007 for use in dogs,[1][2] and more recently has also been approved for use in cats.[3]
MORE............
Use of the cryopreserved human hepatocyte sandwich-culture model to measure hepatic metabolism and biliary efflux
1st Int Conf Drug Des Disc (February 4-7, Dubai) 2008, Abst P-140
1st Int Conf Drug Des Disc (February 4-7, Dubai) 2008, Abst P-140
Proposed international nonproprietary names (Prop. INN): List 90
WHO Drug Inf 2004, 18(1): 56
WHO Drug Inf 2004, 18(1): 56
Maropitant, a NK-1 antagonist decreases the sevoflurane MAC during visceral stimulation in dogs
13th World Congr Pain (August 29-September 2, Montreal) 2010, Abst PW 320
13th World Congr Pain (August 29-September 2, Montreal) 2010, Abst PW 320
Identification of metabolites from maropitant using a dual-pressure linear ion trap and mass frontier software
9th Int ISSX Meet (September 4-8, Istanbul) 2010, Abst P343
9th Int ISSX Meet (September 4-8, Istanbul) 2010, Abst P343
Effect of maropitant, a new NK-1 receptor antagonist, on the sevoflurane minimum alveolar concentration during ovarian stimulation in cats
Annu Meet Am Soc Anesthesiol (ASA) (October 15-19, Chicago) 2011, Abst A1585
Annu Meet Am Soc Anesthesiol (ASA) (October 15-19, Chicago) 2011, Abst A1585
| US8183230 | 5-23-2012 | Antimicrobial preservatives to achieve multi-dose formulation using beta-cyclodextrins for liquid dosage forms |
| US2009099364 | 4-17-2009 | Process for preparation of 1-(2s,3s)-2-benzhydryl-n-(5- tert-butyl-2-methoxybenzyl)quinuclidin-3-amine |
| US2007155782 | 7-6-2007 | Nk-1 receptor antagonists anesthesia recovery |
| US2007129328 | 6-8-2007 | Pharmaceutical compositions of neurokinin receptor antagonists and cyclodextrin and methods for improved injection site toleration |
| US2003139443 | 7-25-2003 | Use of tachykinin antagonists, including NK-1 receptor antagonists, to modify unwanted behavior in dogs, cats and horses |
| US6255320 | 7-4-2001 | Polymorphs of a crystalline azo-bicyclo (2,2,2) octan-3-amine citrate and their pharmaceutical compositions |
| US5990125 | 11-24-1999 | NK-1 receptor antagonists for the treatment of cancer |
| EP0790825 | 8-28-1997 | NK-1 RECEPTOR ANTAGONISTS FOR THE TREATMENT OF EYE DISORDERS |
| WO9713514 | 4-18-1997 | NK-1 RECEPTOR ANTAGONISTS FOR PREVENTION OF NEUROGENIC INFLAMMATION IN GENE THERAPY |
| US5576317 | 11-20-1996 | NK-1 receptor antagonists and 5HT3 receptor antagonists for the treatment of |
| WO9614845 | 5-24-1996 | NK-1 RECEPTOR ANTAGONISTS FOR THE TREATMENT OF EYE DISORDERS |
| US5519033 | 5-22-1996 | Azabicyclo derivatives for treatment of urinary incontinence |
| US5393762 | 2-29-1995 | Pharmaceutical agents for treatment of emesis |
| US5340826 | 8-24-1994 | Pharmaceutical agents for treatment of urinary incontinence |
| WO9221677 | 12-11-1992 | bibNUCLIDINE DERIVATIVES |
anhydrous (2S,3S)-N-(methoxy-5-t-butylphenylmethyl-2-diphenylmethyl-1-azobicyclo[2,2,2] octan-3-amine citrate monohydrate salt, its single crystalline polymorphic Form A, and pharmaceutical composition containing them. The invention is also directed to a CNS active NK-1 receptor antagonist for treating emesis in a mammal including humans. Treating is defined here as preventing and treating.

U.S. Pat. No. 5,393,762 and U.S. Ser. No. 08/816,016, both incorporated by reference, describe pharmaceutical compositions and treatment of emesis using NK-1 receptor antagonists. The citrate monohydrate has significantly enhanced stability over other salt forms such as the benzoate which was unstable even at 5° C. The mesylate form is deliquescent.
U.S. Pat. No. 5,393,762 and U.S. Ser. No. 08/816,016, both incorporated by reference, describe pharmaceutical compositions and treatment of emesis using NK-1 receptor antagonists. The citrate monohydrate has significantly enhanced stability over other salt forms such as the benzoate which was unstable even at 5° C. The mesylate form is deliquescent.
synthesis
U.S. 5,807,867, U.S. 6,222,038 and U.S. 6,255,320.
The compound of Formula I, an NK1 receptor antagonist, is effective as an anti-emetic agent for mammals. The compound of Formula I is the subject of U.S. Pat. No. 6,222,038 and U.S. Pat. No. 6,255,320, and the preparation of the compound of Formula I is described therein. U.S. Pat. No. 5,393,762 also describes pharmaceutical compositions and treatment of emesis using NK-1 receptor antagonists. The multiple-use formulation of the compound of Formula I may be parenterally administrated for about five days at the same site for treatment of emesis or other indications. Intravenous or, preferably, subcutaneous administration is desirable for acute use, since retention and absorption of an oral dosage form may be problematic during bouts of emesis. The multiple-use formulation is described in a co-pending U.S. provisional application No. 60/540,897 assigned to and owned by Pfizer. Inc.
The compound of Formula I also improves anesthesia recovery in mammals. A co-pending U.S. provisional application No. 60/540,697 assigned to and owned by Pfizer Inc., describes a method of improving anesthesia recovery by administering a NK-1 antagonist prior to, during or after the administration of general anesthesia.
...........................................
US20090099364



Preparation of (2S,3S)-2-benzhydryl-N-(5-tert-butyl-2-methoxybenzyl) quinuclidin-3-amine citrate monohydrate, Compound of Formula Ia Step C, Scheme II
A solution of (2S,3S)-2-benzhydryl-N-(tert-butyl-2-methoxybenzyl) quinuclidin-3-amine (33.95 kg, 72.4 moles) and anhydrous citric acid (15.3 kg, 79.7 moles) in a mixture of acetone (215 kg) and water (13.6 kg) was heated to 38-42° C. The resultant mixture was then transferred to another reactor via an in-line filter. The transfer line and filter were washed through with acetone (54 kg) and these filtered washings were added to the solution. The resultant mixture was then cooled to 20-25° C. and filtered fart-butyl methyl ether (252 kg) was added portion-wise over a period of approximately 35 minutes. The resultant suspension was then granulated at 20-25° C. for approximately 20 hours. The solid was then collected by filtration on an agitated filter-dryer and the filter cake was washed twice with filtered tert-butyl methyl ether (50 kg each). The resultant solid was then dried at 35° C. under vacuum with agitation to give the title compound (44.4 kg) as a colourless solid. The product was then milted.
1H-NMR (500 MHz, d6-methanol, 30° C.) δ: 7.46 (2H, d), 7.45 (2H, d), 7.37 (4H, m), 7.31 (1H, m), 7.29 (1H, m), 7.24 (1H, dd), 6.95 (1H, d), 6.76 (1H, d), 4.75 (1H, dd), 4.71 (1H, d), 3.76 (1H, m), 3.57 (1H, d), 3.55 (3H, s), 3.37 (1H, m), 3.31 (1H, m), 3.26 (1H, m), 3.24 (1H, d), 3.10 (1H, t), 2.83 (2H, d), 2.75 (2H, d), 2.51 (1H, m), 2.35 (1H, m), 2.11 (1H, m), 2.06 (1H, m), 1.85 (1H, m), 1.29 (9H, s).
13C NMR (125.7 MHz, d6-methanol, 30° C.) δ: 179.4, 175.0, 156.8, 144.0, 141.5, 141.4, 131.1, 130.6, 129.4, 128.9, 128.7, 128.3, 128.2, 127.2, 126.4, 111.0, 74.0, 64.7, 56.1, 54.2, 50.4, 48.5, 48.3, 44.9, 43.8, 34.8, 32.9, 25.3, 22.2, 18.1.
LRMS (ES+): m/z [MH+] 469.
..................................................................................................................
The compound of Formula I also improves anesthesia recovery in mammals. A co-pending U.S. provisional application No. 60/540,697 assigned to and owned by Pfizer Inc., describes a method of improving anesthesia recovery by administering a NK-1 antagonist prior to, during or after the administration of general anesthesia.
...........................................
US20090099364
Preparation of (2S,3S)-2-benzhydryl-N-(5-tert-butyl-2-methoxybenzyl) quinuclidin-3-amine citrate monohydrate, Compound of Formula Ia Step C, Scheme II
A solution of (2S,3S)-2-benzhydryl-N-(tert-butyl-2-methoxybenzyl) quinuclidin-3-amine (33.95 kg, 72.4 moles) and anhydrous citric acid (15.3 kg, 79.7 moles) in a mixture of acetone (215 kg) and water (13.6 kg) was heated to 38-42° C. The resultant mixture was then transferred to another reactor via an in-line filter. The transfer line and filter were washed through with acetone (54 kg) and these filtered washings were added to the solution. The resultant mixture was then cooled to 20-25° C. and filtered fart-butyl methyl ether (252 kg) was added portion-wise over a period of approximately 35 minutes. The resultant suspension was then granulated at 20-25° C. for approximately 20 hours. The solid was then collected by filtration on an agitated filter-dryer and the filter cake was washed twice with filtered tert-butyl methyl ether (50 kg each). The resultant solid was then dried at 35° C. under vacuum with agitation to give the title compound (44.4 kg) as a colourless solid. The product was then milted.
1H-NMR (500 MHz, d6-methanol, 30° C.) δ: 7.46 (2H, d), 7.45 (2H, d), 7.37 (4H, m), 7.31 (1H, m), 7.29 (1H, m), 7.24 (1H, dd), 6.95 (1H, d), 6.76 (1H, d), 4.75 (1H, dd), 4.71 (1H, d), 3.76 (1H, m), 3.57 (1H, d), 3.55 (3H, s), 3.37 (1H, m), 3.31 (1H, m), 3.26 (1H, m), 3.24 (1H, d), 3.10 (1H, t), 2.83 (2H, d), 2.75 (2H, d), 2.51 (1H, m), 2.35 (1H, m), 2.11 (1H, m), 2.06 (1H, m), 1.85 (1H, m), 1.29 (9H, s).
13C NMR (125.7 MHz, d6-methanol, 30° C.) δ: 179.4, 175.0, 156.8, 144.0, 141.5, 141.4, 131.1, 130.6, 129.4, 128.9, 128.7, 128.3, 128.2, 127.2, 126.4, 111.0, 74.0, 64.7, 56.1, 54.2, 50.4, 48.5, 48.3, 44.9, 43.8, 34.8, 32.9, 25.3, 22.2, 18.1.
LRMS (ES+): m/z [MH+] 469.
..................................................................................................................
........................................................................
3 LANEPITANT
LANEPITANT
N-[(2R)-1-[acetyl-[(2-methoxyphenyl)methyl]amino]-3-(1H-indol-3-yl)
propan-2-yl]-2-(4-piperidin-1-ylpiperidin-1-yl)acetamide
propan-2-yl]-2-(4-piperidin-1-ylpiperidin-1-yl)acetamide
- N-[(2R)-1-[Acetyl-[(2-methoxyphenyl)methyl]amino]-3-(1H-indol-3-yl)propan-2-yl]-2-(4-piperidin-1-ylpiperidin-1-yl)acetamid
- (R)- 1-[N-(2-methoxybenzyl)acetylamino]-3-(1H-indol-3-yl)-2-[N-(2-(4-(piperidin-l-yl)piperidin-1-yl)acetyl)amino]propane
- ELI LILLY
170566-84-4 cas no
Molecular Formula: C33H45N5O3 Molecular Weight: 559.7421
167678-33-3 (diHCl, tri H2O)
170508-05-1 (diHCl)
PHASE 2
DESCRIBED IN
Practical and Enantiospecific Synthesis of LY303870
J. Org. Chem., 1995, 60 (21), pp 7033–7036
DOI: 10.1021/jo00126a069
Lanepitant is a chemical compound , as a potential drug for the treatment of pain , including migraine was developed headache.Having been in clinical trials no migraine efficacy was observed, its further clinical development for this indication was from thepharmaceutical company Eli Lilly and Company set. [2] [3] Pharmacologically Lanepitant is a neurokinin - antagonist .
Lanepitant inhibits antagonist via the neurokinin NK 1 receptor mediated effects of substance P . Experimental has Lanepitant anti-inflammatory and pain perception affecting properties. [4] In animal experiments , in which the pathophysiology of migraine has been simulated, the substance inhibits the inflammatory response in the meninges . [5] Animal experiments also submitted a possible efficacy in neuropathic pain [6] and the complex regional pain syndrome [7] [8] near. In clinical studies , however, neither a migraine efficacy has [2] [3] nor an analgesic effect in diabetic neuropathy [9] and osteoarthritis [10] are occupied.

Lanepitan is a chiral chemical compound with a stereogenic center . It is the R - enantiomer , while the biologically active stereoisomer ( eutomer ) Compared with the L -enantiomer, his distomer , has Lanepitant at least 1000-fold higher affinity for the neurokinin NK 1 receptor on. [11]
Tachykinins are a family of peptides which share a common amidated carboxy terminal sequence. Substance P was the first peptide of this family to be isolated, although its purification and the determination of its primary sequence did not occur until the early 1970's.
Between 1983 and 1984 several groups reported the isolation of two novel mammalian tachykinins, now termed neurokinin A (also known as substance K, neuromedin L, and neurokinin α), and neurokinin B (also known as neuromedin K and neurokinin β). See. J.E. Maggio, Peptides. 6 (Supplement 3):237-243 (1985) for a review of these discoveries.
Tachykinins are widely distributed in both the central and peripheral nervous systems, are released from nerves, and exert a variety of biological actions, which, in most cases, depend upon activation of specific receptors expressed on the membrane of target cells. Tachykinins are also produced by a number of non-neural tissues.
The mammalian tachykinins substance P, neurokinin A, and neurokinin B act through three major receptor subtypes, denoted as NK-1, NK-2, and NK-3, respectively. These receptors are present in a variety of organs.
Substance P is believed inter alia to be involved in the neurotransmission of pain sensations, including the pain associated with migraine headaches and with arthritis. These peptides have also been implicated in gastrointestinal disorders and diseases of the gastrointestinal tract such as inflammatory bowel disease. Tachykinins have also been implicated as playing a role in numerous other maladies, as discussed infra.
Tachykinins play a major role in mediating the sensation and transmission of pain or nociception, especially migraine headaches, see. e.g., S.L. Shepheard, et al.. British Journal of Pharmacology. 108:11-20 (1993); S.M. Moussaoui, et al., European Journal of Pharmacology . 238:421-424 (1993); and W.S. Lee, et al.. British Journal of Pharmacology. 112:920-924 (1994). In view of the wide number of clinical maladies associated with an excess of tachykinins, the development of tachykinin receptor antagonists will serve to control these clinical conditions. The earliest tachykinin receptor antagonists were peptide derivatives. These antagonists proved to be of limited pharmaceutical utility because of their metabolic instability.
Recent publications have described novel classes of non- peptidyl tachykinin receptor antagonists which generally have greater oral bioavailability and metabolic stability than the earlier classes of tachykinin receptor antagonists. Examples of such newer non-peptidyl tachykinin receptor antagonists are found in United States Patent 5,491,140, issued February 13, 1996; United States Patent 5,328,927, issued July 12, 1994; United States Patent 5,360,820, issued November 1, 1994; United States Patent 5,344,830, issued September 6, 1994; United States Patent 5,331,089, issued July 19, 1994; European Patent Publication 591,040 Al, published April 6, 1994; Patent Cooperation Treaty publication WO 94/01402, published January 20, 1994; Patent Cooperation Treaty publication WO 94/04494, published March 3, 1994; Patent Cooperation Treaty publication WO 93/011609, published January 21, 1993; Canadian Patent Application 2154116, published January 23, 1996; European Patent Publication 693,489, published January 24, 1996; and Canadian Patent Application 2151116, published December 11, 1995.
United States Patent 5,530,009, issued June 25, 1996, describes a 1,2-diacylaminopropane for use in treating conditions associated with an excess of tachykinins. This patent also teaches processes for preparing this compound.
In essence, this invention provides a class of potent non- peptidyl tachykinin receptor antagonists similar to those of United States Patent 5,530,009. By virtue of their non-peptidyl nature, the compounds of the present invention do not suffer from the shortcomings, in terms of metabolic instability, of known peptide-based tachykinin receptor antagonists.
- ↑ This substance has not yet been classified on their dangerousness either in terms or a reliable and quotable source for this purpose has not been found.
- ↑ a b Goldstein DJ, Open WW, Klein EC, et al. : Lanepitant, antagonist of NK-1 in migraine prevention . In: Cephalalgia . 21, No. 2, March 2001, pp. 102-106. PMID 11,422,091 .
- ↑ a b Goldstein DJ, Wang O, Saper JR, Stoltz R, Silberstein SD, Mathew NT: ineffectiveness of neurokinin-1 antagonist in acute migraine: a crossover study . In: Cephalalgia . 17, No. 7, November 1997, pp. 785-790. PMID 9.39901 million .
- ↑ Iyengar S, Hipskind PA, Gehlert DR, et al. : LY303870, a centrally active neurokinin-1 antagonist with a long duration of action . In: J. Pharmacol. Exp Ther. . 280, No. 2, February 1997, pp. 774-785. PMID 9023291 .
- ↑ LA Phebus, KW Johnson, PW Stengel, Lobb KL, Nixon JA, Hipskind PA: The non-peptide NK-1 receptor antagonist LY303870 Inhibits neurogenic dural inflammation in guinea pigs . In: Life Sci. . 60, No. 18, 1997, pp. 1553-1561. PMID 9126877 .
- ↑ Campbell EA, Gentry CT, Patel S, Panesar MS, Walpole CS, Urban L: Selective neurokinin-1 receptor antagonists are anti-hyperalgesic in a model of neuropathic pain in the guinea-pig . In: Neuroscience . 87, No. 3, December 1998, pp. 527-532. PMID 9758219 .
- ↑ Guo TZ, Offley SC, Boyd EA, Jacobs CR, Kingery WS: Substance P signaling Contributes to the vascular and nociceptive abnormalities observed-in a tibial fracture rat model of complex regional pain syndrome type I in:. Pain . 108, No. 1-2, March 2004, pp. 95-107. doi : 10.1016/j.pain.2003.12.010 . PMID 15,109,512 .
- ↑ Kingery WS, Davies MF, Clark JD: A substance P receptor (NK1) antagonist can reverse vascular and nociceptive abnormalities in a rat model of complex regional pain syndrome type IIin:. Pain . 104, No. 1-2, July 2003, pp. 75-84. PMID 12,855,316 .
- ↑ Goldstein DJ, Wang O, grid BD, Iyengar S: Dose-response study of the analgesic effect of lanepitant in patients with painful diabetic neuropathy . In: Clin Neuropharmacol . 24, No. 1, 2001, pp. 16-22. PMID 11,290,877 .
- ↑ Goldstein DJ, Wang O, Todd LE, grid BD, DeBrota DJ, Iyengar S: Study of the analgesic effect of lanepitant in patients with osteoarthritis pain . In: Clin. Pharmacol. Ther. . 67, No. 4, April 2000, pp. 419-426. doi : 10.1067/mcp.2000.105243 . PMID 10,801,252 .
- ↑ grid BD, Bruns RF, Howbert JJ, et al. : Pharmacological characterization of LY303870: a novel, potent and selective nonpeptide substance P (neurokinin-1) receptor antagonist . In: J.Pharmacol. Exp Ther. . 275, No. 2, November 1995, pp. 737-744. PMID 7473161 .
1-26-2000
Methods of treating or preventing interstitial cystitis
12-15-1999
Methods of treating or preventing sleep apnea
8-5-1999
METHODS OF TREATING OR PREVENTING INTERSTITIAL CYSTITIS METHODS OF TREATING OR PREVENTING INTERSTITIAL CYSTITIS
10-9-1998
METHODS OF TREATING BONE LOSS
9-19-1997
METHODS OF TREATING OR PREVENTING INTERSTITIAL CYSTITIS
9-5-1997
METHODS OF TREATING OR PREVENTING SLEEP APNEA
7-25-1997
METHODS OF TREATING OR PREVENTING PAIN OR NOCICEPTION
9-27-1996
METHODS OF TREATING OR PREVENTING PAIN OR NOCICEPTION
| WO2002085458A2 * | Feb 2, 2002 | Oct 31, 2002 | Hoffmann La Roche | Use of nk-1 receptor antagonists against benign prostatic hyperplasia |
| WO2013004766A1 | Jul 4, 2012 | Jan 10, 2013 | Ferrari Giulio | Nk-1 receptor antagonists for treating corneal neovascularisation |
| US5328927 * | Feb 24, 1993 | Jul 12, 1994 | Merck Sharpe & Dohme, Ltd. | Hetercyclic compounds, processes for their preparation and pharmaceutical compositions containing them |
| US5360820 * | Sep 18, 1992 | Nov 1, 1994 | Glaxo Group Limited | Medical use for tachykinin antagonists |
| US5491140 * | Jun 30, 1994 | Feb 13, 1996 | Eli Lilly And Company | Naphthyl tachykinin receptor antagonists to treat physiological conditions |
| US5530009 * | Jun 5, 1996 | Jun 25, 1996 | Eli Lilly And Company | Bis-piperidinyl non-peptidyl neurokinin receptor antagonists |
| US5554627 * | Oct 27, 1993 | Sep 10, 1996 | Merck, Sharp & Dohme Ltd. | Tachykinin antagonists |
| US5594022 * | Nov 29, 1994 | Jan 14, 1997 | Warner-Lambert Company | Tachykinin antagonists |
| US5652369 * | Apr 6, 1995 | Jul 29, 1997 | Hoffmann-La Roche Inc. | Amino acid derivatives |
| US5328927 * | 24 Feb 1993 | 12 Jul 1994 | Merck Sharpe & Dohme, Ltd. | Hetercyclic compounds, processes for their preparation and pharmaceutical compositions containing them |
| US5344830 * | 10 Dec 1992 | 6 Sep 1994 | Merck & Co., Inc. | N,N-diacylpiperazine tachykinin antagonists |
Preparation 1
Preparation of (R)-3-(lH-indol-3-yl)-N-(2-methoxybenzyl)-2-(N- triphenylmethylamino)propan amide.
In a 50 gallon, glass-hned reactor, L-tryptophan (4.50 kg, 22.0 mol) was added to acetonitrile (30 L, 6.7 vol) at 20 °C. This reactor was vented to a scrubber containing water, intended to scrub ammonia generated during the silylation reaction and HCl generated during the tritylation and esterification reactions. Bis(trimethylsilyl)amine (HMDS, 5.81 L, 27.5 mol, 1.25 eq) was transferred by gravity to the L- tryptophan slurry from a plastic carboy. The carboy was rinsed with acetonitrile (0.5 L). The slurry was heated to 55 °C and stirred until reaction completion. The reaction endpoint was defined as the point at which the slurry has completely gone into solution. The reaction was clear yellow at completion and took about 2 hours.
Trityl chloride (6.45 kg, 23.1 mol, 1.05 eq) was slurried in acetonitrile (30 L, 6.7 vol) and transferred into the reactor at 47 °C, using trapped vacuum at 325 mm Hg. N-methylmorphohne (5.38 L, 48.9 mol, 2.20 eq) was also transferred into the reactor at this time. The reaction slurry was heated and maintained at 55 °C until reaction completion, determined by high performance hquid chromatography analysis. Reaction time was about 2.5 hours.
The reactor was isolated from the scrubber, and cooled to 35- 40 °C. Methyl alcohol (2.29 L, 56.5 mol, 2.55 eq.) was charged to the reactor and the mixture cooled to 25 °C. 2-Chloro-4,6-dimethoxy- 1,3,5- triazine (CDMT, 4.14 kg, 23.61 mol, 1.07 eq) was added to the reactor with acetonitrile (28 L, 6.2 vol) at 25 °C. The reactor was again vented to the scrubber. The reaction slurry was stirred at room temperature until completion. The reaction endpoint is determined by high performance hquid chromatography analysis. Reaction time is approximately 2 hours. The reactor was isolated from the scrubber following the reaction. 2-Methoxybenzylamine (3.11 L, 23.8 mol, 1.08 eq) was charged to the reactor from a plastic carboy by gravity. The slurry thickens with the addition of 2-methoxybenzylamine. The reaction slurry was heated to 35°C and stirred until reaction completion, determined by high performance hquid chromatography analysis. Reaction time was 2.5 hours.
Water (45 kg, 10 vol) was pre-weighed into a separate 50 gallon, glass-lined tank. The water was pressure-transferred into the reaction mixture slurry over about 45 minutes. The resulting yellow- colored slurry was cooled to 0-5 °C over two hours and stirred overnight. The title intermediate was isolated by vertical basket centrifuge isolation using three micron polyethylene multiple filament isolation bag. During the centrifugation, the load speed was generally between 900-1050 rpm, the wash speed was 900-1500 rpm, and the spin speed was 1500-2300 rpm. The title intermediate was then dried by rotary vacuum drying. Yield: 86.4% with isomer purity of 99.6%.
Preparation 2 Reduction of Carbonyl
Preparation of (R)-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)amino]-2-(N- triphenylmethylamino)propane
RED-AL®. [a 3.4 M, solution of sodium bis(2- methoxyethoxy) aluminum hydride in toluene] (535 ml, 1.819 mol), dissolved in anhydrous tetrahydrofuran (400 ml) was slowly added using an addition funnel to a refluxing solution of the acylation product, (R)-3- (lH-indol-3-yl)-N-(2-methoxybenzyl)-2-(N- triphenylmethylamino)propanamide (228.6 g, 0.404 mols) produced supra. in anhydrous tetrahydrofuran (1.0 L) under a nitrogen atmosphere. The reaction mixture became a purple solution. The reaction was quenched after at least 20 hours by the slow addition of excess saturated Rochelle's salt solution (potassium sodium tartrate tetrahydrate). The organic layer was isolated, washed with brine (2X), dried over anhydrous sodium sulfate, filtered, and concentrated to an oil on a rotary evaporator. No further purification was done and the product was used directly in the next step.
Preparation 3 Acylation of Secondary Amine
Preparation of (R)-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)-acetylamino]- 2-(N-triphenylmethylamino)propane
To a stirring solution of (R)-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)amino]-2-(N-triphenylmethylamino)propane (0.404 mol) in anhydrous tetrahydrofuran (1.2 L) under a nitrogen atmosphere at 0°C was added triethylamine (66.5 ml, 0.477 mol) and acetic anhydride (45.0 ml, 0.477 mol). After 4 hours, the mixture was concentrated on a rotary evaporator, redissolved in methylene chloride and ethyl acetate, washed with water (2X) and brine (2X), dried over anhydrous sodium sulfate, filtered, and concentrated to a sohd on a rotary evaporator. The resulting sohd was dissolved in chloroform and loaded onto silica gel 60 (230-400 mesh) and eluted with a 1:1 mixture of ethyl acetate and hexanes. The product was then crystallized from an ethyl acetate/hexanes mixture. The resulting product of (R)-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]-2-(N-triphenylmethylamino)propane was crystallized and isolated over three crops giving 208.97 grams (87% yield) of analytically pure material. Analysis for C40H39N3O2:
Theory: C, 80.91; H, 6.62; N, 7.08. Found: C, 81.00; H, 6.69; N, 6.94. Preparation 4
Deprotection
Preparation of (R)-2-amino-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]prop ane dihy drochloride
A stirring solution of (R)-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]-2-(N-triphenylmethylamino)propane in two volumes of methylene chloride was cooled to between -40°C and -50°C. Anhydrous hydrogen chloride gas was added at such a rate that the temperature of the reaction mixture did not exceed 0°C. The reaction mixture was stirred for 30 minutes to one hour at 0-10°C.
To this reaction mixture was added two volumes of methyl t- butyl ether and the resulting mixture was allowed to stir for 30 minutes to one hour at 0-10°C. The resulting crystalline sohd was removed by filtration and then washed with methyl £ -butyl ether. The reaction product was dried under vacuum at 50°C. (Yield >98%) Analysis for C21H25N3O2 • 2 HCl:
Theory: C, 59.44; H, 6.41; N, 9.90.
Found: C, 60.40; H, 6.60; N, 9.99.
Preparation 5
Preparation of (R)-2-[(2-bromo)acetyl]amino-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]propane
To a stirring solution of (R)-2-amino-3-(lH-indol-3-yl)-l-[N- (2-methoxybenzyl)acetylamino]propane (7.51 g, 21.369 mmol) in anhydrous tetrahydrofuran (100 ml) under a nitrogen atmosphere at 0°C were added diisopropylethylamine (4.1 ml, 23.537 mmol) and bromoacetyl bromide (2.05 ml, 23.530 mmol). After 2 hours, ethyl acetate was added and the reaction mixture washed with water twice, 1.0 N hydrochloric acid (2X), saturated sodium bicarbonate solution (2X), and brine. The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated to a tan foam on a rotary evaporator. In this manner the 2- [(2-bromo)acetyl]amino-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]propane was obtained in quantitative yield. No further purification was necessary.
Preparation 6
Preparation of a polystyrene bound isocyanate resin
To a stirred suspension of 50 grams (61 mmol) aminomethylated polystyrene resin (1.22 mmol/g) in 800 ml toluene was added 193 ml (366 mmol) 1.9 M phosgene in toluene. After stirring the reaction mixture for 10 minutes, 67 ml (482 mmol) triethylamine was added and the reaction mixture was stirred for 18 hours at room temperature. The mixture was filtered and the recovered sohd washed with 10 times with dichloromethane. A hght pink resin mixed with a white sohd was obtained. This sohd mixture was resuspended in 700 ml dichloromethane, stirred for 10 minutes and then filtered and washed well with dichloromethane. The resulting sohd was again suspended, stirred and washed with dichloromethane to provide the desired resin. IR(KBr): 2252 cm"1 (characteristic peak for -N=C=0)
.........................................................................................................................
Synthesis of (R)-2-[N-(2-((4-cyclohexyl)piperazin-l-yl)acetyl)amino]-3-(lH- indol-3-yl)-l-[N-(2-methoxybenzyl)acetylamino]propane
(a) Preparation of 2-(4-(piperidin-l-yl)piperidin-l-yl)acetic acid, potassium salt
4-(Piperidin-l-yl)piperidine (1.20 kg, 7.13 mol) was added to methylene chloride (12.0 L) under a nitrogen atmosphere. Tetrabutylammonium bromide (0.150 kg, 0.47 mol) and sodium hydroxide (1.7 L of a 5 N solution, 8.5 mol) were then added. The reaction mixture was cooled to 10-15°C and methyl bromoacetate (1.17 kg, 7.65 mol) was added and the resulting mixture was stirred for a minimum of 16 hours.
Deionized water (1.2 L) was then added to the mixture and the layers separated. The aqueous layer was back-extracted with methylene chloride (2.4 L). The organic fractions were combined and washed with deionized water (3 x 1.2 L), a saturated sodium bicarbonate solution (1.1 L) and a saturated sodium chloride solution (1.1 L). The organic fraction was then dried over anhydrous magnesium sulfate and concentrated to an oil on a rotary evaporator to -yield 1.613 kg (93.5%) of methyl 2-(4-(piperidin-l-yl)piperidin-l-yl)acetate.
A solution of methyl 2-[4-(piperidin-l-yl)piperidin-l- yl]acetate (2.395 kg, 9.96 mol) in methanol (2.4 L) was added to a solution of potassium hydroxide (0.662 kg, 10.0 mol @ 85% purity) in methanol (10.5 L) under a nitrogen atmosphere. The reaction mixture was heated to 45-50°C for a minimum of 16 hours.
A solvent exchange from methanol to acetone (15.0 L) was performed on the solution on a rotary evaporator. This solution was slowly cooled to room temperature over 16 hours. The resulting solids were filtered, rinsed with acetone (5.0 L) and then dried to yield 2.471 kg (93.8%) of 2-(4-(piperidin-l-yl)piperidin-l-yl)acetic acid, potassium salt. MS 265 (M+1)
(b) Preparation of (R)-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]-2-[N-(2-(4-(piperidin-l-yl)piperidin-l- yl)acetyl)amino]propane
The title compound was. prepared by first admixing (R)-2- amino-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)acetylamino]propane dihydrochloride (50.0 g, 0.118 mol) with 100 ml of methylene chloride under a nitrogen atmosphere.
In a second flask, under a nitrogen atmosphere, 2-(4- (piperidin-l-yl)piperidin-l-yl)acetic acid potassium salt (62.3 g, 0.236 mol) was added to 600 ml of methylene chloride. This mixture was cooled to about -10°C and stirring was continued. To this mixture isobutylchloroformate (23 ml, 0.177 mol) was added dropwise such that the temperature of the 2-(4-(piperidin-l-yl)piperidin-l-yl)acetic acid potassium salt mixture never rose appreciably.
This reaction mixture was stirred at about -10°C for about 1.5 hours at which time the (R)-2-amino-3-(lH-indol-3-yl)-l-[N-(2- methoxybenzyl)acetylamino]propane dihydrochloride/methylene chloride mixture prepared supra was slowly added to the 2-(4-(piperidin- l-yl)piperidin-l-yl)acetic acid potassium salt isobutylchloroformate/methylene chloride solution. The resulting mixture was then stirred for about 1 hour at a temperature between - 15°C and -8°C.
The reaction mixture was removed from the ice bath and allowed to warm to 15-20°C and the reaction was quenched by the addition of 200 ml of water. The pH ofthe solution was adjusted to 2.3-2.7 by the additon of IN sulfuric acid. The layers were separated and the aqueous layer was washed with 100 ml of methylene chloride.
The organic fractions were combined and washed with water (100 ml). The water wash was back extracted with methylene chloride (50 ml) and combined with the aqueous fraction from above. Methylene chloride (500 ml) was added to the combined aqueous layers and the mixture was stirred at room temperature for 15 minutes as basification with 2N sodium hydroxide to a final pH of 9.8 to 10.2 was achieved. The organic and aqueous fractions were separated. The aqueous fraction was washed with methylene chloride and the methylene chloride was added to the organic fraction. The organic fraction was then washed with a mixture of saturated sodium bicarbonate solution (100 ml) and water (50 ml). The bicarbonate wash was separated from the organic fraction and back extracted with methylene chloride (50 ml).
The back extraction was combined with the methylene chloride fraction and the combined fractions were dried over magnesium sulfate. The magnesium sulfate was removed by filtration and the volatiles were removed by vacuum distillation to yield the title product as a foam. (72.5 g, >98% yield). MS 559(M+1)
NMR (DMSO-dβ 3:2 mixture of amide rotamers) δ 1.25-1.70 (m, 10H),
1.77-2.00 (m, 2H), 1.95 (s, 3/5-3H), 2.04 (s, 2/5-3H), 2.10-2.97 (m, 9H), 3.10- 3.65 (m, 3H), 3.72 (s, 2/5-3H), 3.74 (s, 3/5- 3H), 4.26-4.58 (m, 3H), 6.76-7.12 (m, 6H), 7.13-7.35 (m, 2H), 7.42-7.66 (m, 2H), 10.80 (br s, IH). Analysis for C33H45N5O3:
Theory: C, 70.81; H, 8.10; N, 12.51.
Found: C, 70.57; H, 8.05; N, 12.39.
Preparation of 2-(4-(piperidin-l-yl)piperidin-l-yl)acetic acid, potassium salt
4-(Piperidin-l-yl)piperidine (1.20 kg, 7.13 mol) was added to methylene chloride (12.0 L) under a nitrogen atmosphere.
Tetrabutylammonium bromide (0.150 kg, 0.47 mol) and sodium hydroxide (1.7 L of a 5 N solution, 8.5 mol) were then added. The reaction mixture was cooled to 10-15°C and methyl bromoacetate (1.17 kg, 7.65 mol) was added and the resulting mixture was stirred for a minimum of 16 hours.
Deionized water (1.2 L) was then added to the mixture and the layers separated. The aqueous layer was back-extracted with methylene chloride (2.4 L). The organic fractions were combined and washed with deionized water (3 x 1.2 L), a saturated sodium bicarbonate solution (1.1 L) and a saturated sodium chloride solution (1.1 L). The organic fraction was then dried over anhydrous magnesium sulfate and concentrated to an oil on a rotary evaporator to yield 1.613 kg (93.5%) of methyl 2-(4-(piperidin-l-yl)piperidin-l-yl)acetate.
A solution of methyl 2-[4-(piperidin-l-yl)piperidin-l- yl]acetate (2.395 kg, 9.96 mol) in methanol (2.4 L) was added to a solution of potassium hydroxide (0.662 kg, 10.0 mol @ 85% purity) in methanol (10.5 L) under a nitrogen atmosphere. The reaction mixture was heated to 45-50°C for a minimum of 16 hours.
A solvent exchange from methanol to acetone (15.0 L) was performed on the solution on a rotary evaporator. This solution was slowly cooled to room temperature over 16 hours. The resulting solids were filtered, rinsed with acetone (5.0 L) and then dried to yield 2.471 kg (93.8%) of 2-(4-(piperidin-l-yl)piperidin-l-yl)acetic acid, potassium salt. MS 265 (M+l)
dihydrochloride trihydrate
Preparation of (R)-3-( lH-indol-3-yl)- l-[N-(2-methoxybenzyl)acetylamino]- 2-[N-(2-(4-(piperidin-l-yl)piperidin-l-yl)acetyl)amino]propane dihydrochloride trihydrate
3 H20 Under a nitrogen atmosphere 2-(4-(piperidin-l-yl)piperidin- l-yl)acetic acid, potassium salt (0.75 kg, 2.84 mol) was added to methylene chloride (7.5 L). The resulting mixture was cooled to -15 to - 8°C and isobutyl chloroformate (0.29 kg, 2.12 mol) was added at such a rate so as to maintain the temperature of the reaction mixture below - 8°C. After the addition the resulting reaction mixture was stirred for 90 minutes between -15 and -8°C.
The reaction mixture was then cooled to -35°C and solid (R)- 2-amino-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)amino]propane dihydrochloride (0.60 kg, 1.14 mol) was added at such a rate that the reaction temperature was maintained at less than -20°C. After the addition, the reaction mixture was stirred for about one hour with the temperature being maintained between -37°C and -20°C. The reaction was quenched by the addition of deionized water (7.5 L). The reaction mixture was basified to pH 12.8-13.2 by the addition of 5 N sodium hydroxide. The aqueous fraction was removed and retained. Additional deionized water (3.75 L) was added to the organic fraction as was sufficient 5 N sodium hydroxide to re-adjust the pH to 12.8-13.2.
The two aqueous fractions were combined, back-extracted with methylene chloride (1.5 L) and then discarded. The organic fractions were combined and washed with deionized water (4 x 3.5 L). These extracts were combined, back-extracted with methylene chloride (1.5 L), and then discarded. The two organic layers were combined and washed with a saturated sodium chloride solution (3.7 L). The organic fraction was dried over anhydrous magnesium sulfate, filtered, and solvent exchanged from methylene chloride to acetone (3.75 L) on a rotary evaporator. An aqueous solution of hydrochloric acid (0.48 L of 6 N solution, 2.88 mol) and seed crystals (2 g) were added and mixture was stirred for 30-90 minutes. Acetone (13.2 L) was then added and the slurry stirred for one hour. The resulting solid was then filtered, washed with acetone (2 x 1.4 L), and dried to yield 633 g (90%) of (R)-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)acetylamino]-2-[N- (2-(4-(piperidin- 1-yl )piperidin- 1-yl )acetyl)amino]propane dihydrochloride trihydrate.
....................................................................................................................
....................................................................................................................
http://pubs.acs.org/doi/pdf/10.1021/jo00126a069http://pubs.acs.org/doi/pdf/10.1021/jo00126a069
Practical and Enantiospecific Synthesis of LY303870
J. Org. Chem., 1995, 60 (21), pp 7033–7036
DOI: 10.1021/jo00126a069
(R)-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)acetylamino]-2-[N- (2-(4-(piperidin- 1-yl )piperidin- 1-yl )acetyl)amino]propane dihydrochloride trihydrate
(R)-3-(lH-indol-3-yl)-l-[N-(2-methoxybenzyl)acetylamino]-2-[N- (2-(4-(piperidin- 1-yl )piperidin- 1-yl )acetyl)amino]propane dihydrochloride trihydrate
mp 192-196 degC(lossofWATER), 240deg C
1H(300MHz,CDCl3)
1.25-1.70(m, 10H);1.77-2.00(m,3H);1.95,(s,1.8H [rotamerl),2.04,(s,1.2H [rotamerl);2.10-2.97,(m, 9H);
3.10-3.65,(m,3H),
3.72,(s,1.2H[rotamerl);
3.74,(s,1.8Hhotamer]);
4.26-4.58,(m,3H);
6.76-7.12,(m,6H);
7.13-7.35(m,2H),
7.42-7.66(m,2H);
10.80 (broads,1H);
13CNMR(75MHz,DMSO)
169.9,
169.4,
169.3, 156.8, 156.7, 136.1
128.5,127.7,127.5,127.3,127.2,126.9,125.4,124.6,123.2,123.1,
120.08,
120.7,
120.2,
120.0, 118.4, 118.2, 118.1,
111.2,
110.6,
110.5,
110.4, 110.3,
79.1,
61.7, 61.4,
61.3,
55.1,
53.4, 53.3,
53.2,
51.2,49.7,47.9,47.7,47.6,47.5,47.2,43.1,27.8,27.6,27.5,27.4,
27.3,27.1,26.0,25.8,24.5,21.4,21.2;
massspec559au;
IR
1658cm-l;
[alpha D =+15.7deg(c=1,MeOH).
Anal.Calcdfor
C33H53Cl2N506:
C,
57.72,
H,
7.78;
N,
10.20.
Found:
C,
58.00;
H,
7.54;
N,
10.16
..........................................................................................................................................
........................................................................................................
.............................................................................................................

4 VESTIPITANT

VESTIPITANT
(2S)-N-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-2-methylphenyl)-N-methylpiperazine-1-carboxamide
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide
2-(S)-(4-fluoro-2-methylphenyl)piperazine-1-carboxylic acid [1-(R)-(3,5-bis-trifluoromethylphenyl)ethyl]methylamide (vestipitant)
Vestipitant [INN], UNII-S052TOI9BI, DCL001035,
CAS NO 334476-46-9
CAS NO 334476-46-9
Molecular Formula: C23H24F7N3O Molecular Weight: 491.444982
Elemental Analysis: C, 56.21; H, 4.92; F, 27.06; N, 8.55; O, 3.26
Vestipitant, also known as GW597599, is one of the most potent and selective NK(1) receptor antagonists ever discovered, showing appropriate pharmacokinetic properties and in vivo activity. Its actions support the utility of NK(1) receptor blockade in the alleviation of anxiety and, possibly, depression.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potential antiemetic and anxiolytic drug, and as a treatment for tinnitus.
Vestipitant mesylate is a tachykinin NK1 receptor antagonist in phase II clinical trials at GlaxoSmithKline for the treatment of postoperative nausea and vomiting. The drug candidate had been in clinical development at the company for several indications, including the treatment of tinnitus as monotherapy or in combination with paroxetine, the treatment of primary insomnia, the treatment of depression and anxiety and the treatment of chemotherapy-induced nausea and vomiting; however, no recent development has been reported for this research.
Vestipitant has anxiolytic properties and a good safety profile. Vestipitant was investigated for potential effect against chronic tinnitus as a stand-alone treatment and in conjunction with a selective serotonin reuptake inhibitor, paroxetine. No statistically significant treatment benefit effect was detected for tinnitus (intensity, pitch, and distress) VAS scores, arousal-anxiety VAS scores, Tinnitus Handicap Inventory, or tinnitus aggravation scores assessed on Days 1 and 14. However, a statistically significant worsening of tinnitus intensity and distress scores was observed after vestipitant compared with placebo for the mean data collected over the treatment period. No relevant differences in vestipitant plasma concentrations were observed between the subjects given the combination with paroxetine and those receiving vestipitant alone. No specific relationships were observed between tinnitus intensity and vestipitant plasma concentrations.
CONCLUSION: Although well-tolerated vestipitant, alone or in combination with paroxetine, was not effective in ameliorating tinnitus in this patient group.
Elemental Analysis: C, 56.21; H, 4.92; F, 27.06; N, 8.55; O, 3.26
Vestipitant, also known as GW597599, is one of the most potent and selective NK(1) receptor antagonists ever discovered, showing appropriate pharmacokinetic properties and in vivo activity. Its actions support the utility of NK(1) receptor blockade in the alleviation of anxiety and, possibly, depression.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potential antiemetic and anxiolytic drug, and as a treatment for tinnitus.
Vestipitant mesylate is a tachykinin NK1 receptor antagonist in phase II clinical trials at GlaxoSmithKline for the treatment of postoperative nausea and vomiting. The drug candidate had been in clinical development at the company for several indications, including the treatment of tinnitus as monotherapy or in combination with paroxetine, the treatment of primary insomnia, the treatment of depression and anxiety and the treatment of chemotherapy-induced nausea and vomiting; however, no recent development has been reported for this research.
Vestipitant has anxiolytic properties and a good safety profile. Vestipitant was investigated for potential effect against chronic tinnitus as a stand-alone treatment and in conjunction with a selective serotonin reuptake inhibitor, paroxetine. No statistically significant treatment benefit effect was detected for tinnitus (intensity, pitch, and distress) VAS scores, arousal-anxiety VAS scores, Tinnitus Handicap Inventory, or tinnitus aggravation scores assessed on Days 1 and 14. However, a statistically significant worsening of tinnitus intensity and distress scores was observed after vestipitant compared with placebo for the mean data collected over the treatment period. No relevant differences in vestipitant plasma concentrations were observed between the subjects given the combination with paroxetine and those receiving vestipitant alone. No specific relationships were observed between tinnitus intensity and vestipitant plasma concentrations.
CONCLUSION: Although well-tolerated vestipitant, alone or in combination with paroxetine, was not effective in ameliorating tinnitus in this patient group.
Vestipitant is a drug developed by GlaxoSmithKline which acts as a selective antagonist for the NK1 receptor. It is under development as a potentialantiemetic and anxiolytic drug,[1][2] and as a treatment for tinnitus.[3]
- Reddy, GK; Gralla, RJ; Hesketh, PJ (2006). "Novel neurokinin-1 antagonists as antiemetics for the treatment of chemotherapy-induced emesis". Supportive cancer therapy 3 (3): 140–2.doi:10.3816/SCT.2006.n.011. PMID 18632487.
- Brocco, M; Dekeyne, A; Mannoury La Cour, C; Touzard, M; Girardon, S; Veiga, S; De Nanteuil, G; Dejong, TR et al. (2008). "Cellular and behavioural profile of the novel, selective neurokinin1 receptor antagonist, vestipitant: a comparison to other agents". European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology 18 (10): 729–50.doi:10.1016/j.euroneuro.2008.06.002. PMID 18657401.
- ClinicalTrials.gov NCT00394056 Vestipitant Or Vestipitant/Paroxetine Combination In Subjects With Tinnitus And Hearing Loss
.........................
vestipitant
.........................
VESTIPITANT MESYLATE
CAS: 334476-64-1 of MESYLATE
- GW597588B
- UNII-OWR424W90Q
D06293, 334476-64-1
Journal of Thermal Analysis and Calorimetry, 2010 , vol. 102, 1 pg. 297 - 303
..........
INTRODUCTION

International patent application number WO2001/25219 describes piperazine derivatives. One such compound described therein is 2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide (otherwise known as vestipitant) and it has the following chemical structure (I).

WO2001/25219 also describes the methanesulphonate salt of the compound (I).
The compound (I) and its pharmaceutically acceptable salts may be prepared by the processes described in International patent applications WO2001/25219 and WO2007/048642, which are incorporated herein by reference. Specifically, Examples 37 and 36 of WO2001/25219 describe the synthesis of the compound (I) as free base and as methanesulphonate salt respectively. Hydrochloride and acetate salts of the compound(I) are described in the Examples
38 and 18 respectively. Example 1 of WO2007/048642 discloses a process for preparing an intermediate in the synthesis of the compound(I).
...........................
Synthetic Process of Vestipitant
The following synthetic route was reported by Giuseppe Guercio et al from GlaxoSmithKline:
Org. Process Res. Dev., 2009, 13 (6), pp 1100–1110
DOI: 10.1021/op9002032
The initial chemical development synthetic route, derived from the one used by medicinal chemistry, involved several hazardous reagents, gave low yields and produced high levels of waste. Through a targeted process of research and development, application of novel techniques and extensive route scouting, a new synthetic route for GW597599 was developed. This paper reports the optimisation work of the third and last stage in the chemical synthesis of GW597599 and the development of a pilot-plant-suitable process for the manufacturing of optically pure arylpiperazine derivative 1. In particular, the process eliminated the use of triphosgene in the synthesis of an intermediate carbamoyl chloride, substantially enhancing safety, overall yield, and throughput.


1H NMR (600 MHz, DMSO-d6) δ 1.48 (d, J = 6.9 Hz, 3H); 2.31 (s, 3H); 2.39 (s, 3H); 2.74 (s, 3H); 2.95 (t, J = 12.2 Hz, 1H); 3.00−3.06 (m, 1H); 3.27 (dd, J = 12.5, 2.3 Hz, 1H); 3.28−3.35 (m, 1H); 3.38−3.43 (m, 1H); 3.47 (dt, J = 13.3, 3.1 Hz, 1H); 4.49 (dd, J = 11.8, 3.3 Hz, 1H); 5.35 (q, J = 6.5 Hz, 1H); 6.84 (td, J = 8.4, 2.6 Hz, 1H); 7.01 (dd, J = 10.2, 2.7 Hz, 1H); 7.29 (dd, J = 8.5, 6.0 Hz, 1H); 7.71 (bs, 2H); 8.02 (bs, 1H); 8.71 (bs, 1H); 9.02 (bs, 1H).
ES+: m/z 492 [MH − CH3SO3H]+, 341, 221; ES−: m/z 586 [M − H]−; 95 [CH3SO3]−.
13C NMR (150 MHz, DMSO-d6) δ 16.37, 18.81, 30.54, 39.79, 42.41, 45.70, 46.58, 52.41, 53.42, 112.48, 116.55, 121.02, 123.19 (d), 127.19, 127.44 (d), 130.34 (d), 134.00, 138.56, 144.79, 160.89, 163.2.
IR (Nujol mull, cm−1): 1653 (str. C═O), 1600 (str. C═C aromatic) (cm−1).
HPLC column type Betabasic C18; mobile phase A: buffer ammonium hydrogen carbonate 5 mM pH = 10/methanol 40/60% v/v and B buffer ammonium hydrogen carbonate 5 mM pH = 10/methanol 10/90% v/v; gradient: 0 min 100% A to 20 min 100% B. flow 1 mL/min; column temperature 40 °C; detector UV DAD @210 nm. Retention times 1: 13 min, purity >98%.
HPLC column type Chiralpack AD; mobile phase n-hexane/ethanol 86/14% v/v + 0.2% v/v purified water; flow 1 mL/min; column temperature 25 °C; detector UV DAD @210 nm. Retention time1: 4.56 min and opposite enantiomer 4.15 min, other diastereomers 5.20 and 14.2 min, respectively.
............................
SYNTHESIS
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-l- carboxylic acid [l-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide
Preparation 1
(S)-2-(4-fluoro-2-methylphenyl)piperazine dihydrochloride
A suspension of (S)-3-(4-fluoro-2-methylphenyl)piperazin-2-one (S)-2-hydroxy-2- phenylacetate (14.0Kg; contains 16%w/w EtOAc hence 11.8 kg corrected for solvent) and tetrabutylammoniunn bromide (TBAB, 236g) in THF (94L) was warmed to 40°C to obtain a clear solution that was cooled to 30°C and then added to a slurry of sodium borohydride (powder grade, 5.5kg) in THF (41L) at 20°C, followed by THF (5.6L). The mixture was warmed to 35°C and then boron trifluoride-THF complex (36.6kg) was added over 90min, followed by THF (1L). The mixture was stirred for 6h and then IMS (47L) added over 3 hours. The mixture was distilled to ca. 94L, diluted with IMS (47L) and further distilled to 94L. The slurry was cooled to 25°C, filtered and the solids washed with IMS (2x35L). The combined filtrates were heated to 70°C and hydrogen chloride (5-6N in isopropanol, 15kg) added over 72min. The resulting slurry was heated at reflux for 3h, cooled to 20°C over 2h and then held at this temperature for 2h. The suspension was filtered, washed with IMS (3x24L) and the solids dried under vacuum at 45-50°C to give the title compound (6.87kg) as a white powder.
*H NMR NMR (D20) δ (ppm) 7.44 (dd, 1H), 7.03-7.00 (m, 2H), 4.89 (dd, 1H), 3.82-3.51 (m, 6H), 3.35 (s, 3H).
Preparation 2
(S)-tert-butyl3-(4-fluoro-2-methylphenyl)piperazine-l-carboxylate
hydrochloride
Triethylamine (5.5kg) was added to a slurry of (S)-2-(4-fluoro-2-methylphenyl)piperazine dihydrochloride (6.60kg, 94.6% assay) in EtOAc (38L) and was rinsed in with EtOAc (1L). The slurry was stirred at 40°C for 120 minutes and was then cooled to 20°C. 79.2%w/w Di-fe/ -butyl dicarbonate in EtOAc solution (6.29kg) was added over 60 minutes and was rinsed in with EtOAc (1L). The slurry was stirred for 15 minutes. Further 79.2%w/w di-fe/ -butyl dicarbonate in EtOAc solution (0.19kg) and EtOAc (1L) was added and the slurry was stirred for 43 minutes. EtOAc (5L), 79.2%w/w di-fe/ -butyl dicarbonate in EtOAc solution (0.25kg) and EtOAc (1L) were added and the slurry was then stirred for 15 minutes to complete the reaction. Water (18.7L) was added to dissolve all solids present and the lower aqueous layer was separated. The organic layer was washed with water (18.7L). The solution was distilled under reduced pressure to a total volume of 25L. Fresh EtOAc (37L) was added and the solution was distilled under reduced pressure to a total volume of 25L. EtOAc (49L) was added and the temperature was adjusted to 15°C. A slurry of the title compound (31.2g) in EtOAc (310ml) was added followed by 5.5M hydrogen chloride in isopropanol solution (0.412kg) rinsed in with EtOAc (1L). The mixture was stirred for 60 minutes to give a slurry. 5.5M Hydrogen chloride in isopropanol solution (3.6kg) was added portionwise over 55 minutes and was rinsed in with EtOAc (1L). The resultant slurry was stirred for 30 minutes at 15°C. The slurry was filtered and the solid was washed with EtOAc (2 x 16.8kg). The solid was dried under vacuum at 40°C to give the title compound (6.84kg) as a white solid.
*H NMR (500 MHz, DMSO-o^) δ ppm 9.89 (brs, 2 H), 7.88 (dd, 1 H), 7.13 - 7.20 (m, 2 H), 4.43 (d, 1 H), 4.07 (d, 1 H), 3.96 (d, 1 H), 3.30 - 3.38 (m, 2 H), 3.21 (m, 2 H), 2.39 (s, 3 H), 1.42 (s, 9 H). Preparation 3
(R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-methylethanamine
To a suspension of (R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-nnethylethanannine (S)-2- hydroxysuccinate (9Kg) in EtOAc (27L), 13% w/w aqueous sodium carbonate solution (27L) was added. The mixture was stirred for 30 minutes at 25°C to ensure complete dissolution. The layers were separated and the organic phase was washed with water (27L). EtOAc (36L) was added and the solution concentrated in vacuo to 18L. Further EtOAc (49Kg) was added and the solution concentrated in vacuo to 18L to give a colourless 33.4% w/w solution of the title compound in EtOAc (17.9Kg).
*H NMR for title compound (500 MHz, DMSO-i¼) δ ppm 8.01 (s, 2 H), 7.90 (s, 1 H), 3.79 (q, 7=6.56 Hz, 1 H), 2.35 (br s, 1 H), 2.10 (s, 3 H), 1.25 (d, 7=6.56 Hz, 3 H)
H NMR for EtOAc peaks (500 MHz, DMSO-i¼) δ ppm 4.02 (q, 7=7.17 Hz, 2 H), 1.98 (s, 3 H), 1.17 (t, 7=7.10 Hz, 3 H)
NMR shows a ratio of 1:6.1 the title compound: EtOAc.
Preparation 4
(S)-N-((R)-l-(3,5-bis(trifluoromethyl)phenyl)ethyl)-2-(4-fluoro-2- methylphenyl)-N-methylpiperazine-l-carboxamide methanesulfonate (Crystalline Form 1)
To a 33.4% w/w solution of (R)-l-(3,5-bis(trifluoromethyl)phenyl)-N-methylethanamine in EtOAc (14.70Kg) was added EtOAc (22L). The solution was vacuum purged three times with carbon dioxide gas and stirred under a flow of C02 at 20°C for 1 hour. Triethylamine (2.40Kg) was added followed by EtOAc (1.35Kg) and the solution stirred for 50 minutes under a flow of C02. Chlorotrimethylsilane (2.50Kg) was added over 30 minutes keeping the internal temperature below 25°C followed by EtOAc (1.35Kg) and the suspension stirred under a flow of C02 at 20°C for 30 minutes. Pyridine (2.85Kg) was added followed by EtOAc (2.70Kg). Thionyl chloride (3.25Kg) was added over 20 minutes followed by EtOAc (2.70Kg) and the suspension heated to 25°C for 6 hours. The reaction was cooled to 10°C and quenched with 28% w/w aqueous malic acid solution (14.30Kg). The layers were separated at 20°C and the organic phase washed with 14% w/w aqueous malic acid solution (13.50Kg), water (12.70Kg) and 20% w/w aqueous potassium phosphate dibasic solution (22.40Kg). EtOAc (4.50Kg) was added and the solution concentrated in vacuo to 15L. Further EtOAc (15L) was added and the solution concentrated in vacuo to 15L.
To the concentrated solution, EtOAc (5L) was added followed by (S)-tert-butyl 3-(4-fluoro-2- methylphenyl)piperazine-l-carboxylate hydrochloride (5.00Kg) and EtOAc (2.50Kg). Tributylamine (7.00Kg) was added and the suspension heated to reflux for 1 hour. The reaction was cooled to 30°C and EtOAc (27.20Kg) followed by water (15.00Kg) were added. The layers were separated, diethylamine (l.lOKg) was added to the organic phase and the solution heated to 40°C for 1 hour. The reaction was cooled to 30°C and washed with 0.5M sulfuric acid (25.90Kg), 0.5M sulfuric acid (15.45Kg) and water (15.00Kg).
To the organic phase, methanesulfonic acid (5.85Kg) was added and the solution heated to 40°C for 1 hour. The reaction was cooled to 10°C then 13%w/w aqueous ammonia solution (23.75Kg) was added over 30 minutes keeping the internal temperature below 35°C. The layers were separated at 30°C and the organic phase was washed with 1% w/w aqueous ammonia solution (15.15Kg) and water (15.00Kg). EtOAc (4.50Kg) was added to the organic phase and the solution was concentrated in vacuo to 15L. EtOAc (40L) was added and the solution concentrated in vacuo to 15L
Further EtOAc (10L) was added followed by methanesulfonic acid (1.20Kg) and (S)-N-((R)- l-(3,5-bis(trifluoromethyl)phenyl)ethyl)-2-(4-fluoro-2-methylphenyl)-N-methylpiperazine-l- carboxamide methansulfonate (25g) in isooctane (0.25Kg) and the suspension was stirred at 20°C for 70 minutes. Isooctane (50L) was added over 90 minutes and the reaction stirred for 1 hour. The suspension was filtered and washed with 2: 1 isooctane/EtOAc (12.5L) three times. The solid was co- milled to give the title compound (6.31Kg) as a white solid.
*H NMR (400 MHz, DMSO-i¾) δ ppm 8.96 (br. s., 2 H), 8.00 (s, 1 H), 7.71 (s, 2 H), 7.29 (dd,
7=8.56, 6.11 Hz, 1 H), 6.99 (dd, 7=10.27, 2.69 Hz, 1 H), 6.83 (td, 7=8.56, 2.45 Hz, 1 H), 5.35 (q, 7=6.60 Hz, 1 H), 4.52 (dd, 7=11.74, 3.18 Hz, 1 H), 3.52-3.22 (m, 4 H), 3.12-2.92 (m, 2 H), 2.74 (s, 3 H), 2.39 (s, 3 H), 2.37 (s, 3 H), 1.49 (d, 7=7.09 Hz, 3 H)
ES+: m/z 492 [MH - CH3S03H]+
Melt onset is 171°C obtained by Differential Scanning Calorimetry (DSC).
........................
SYNTHESIS
Example 36 2-(SM4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid M -(R)-
(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide methansulphonate
To a suspension of intermediate 81 (4.9Kg) in AcOEt (137.2L), triethylamine (5.63L) was added. The mixture was cooled to 0°C then a solution of diterbuthyl dicarbonate (3.134Kg) in AcOEt (24.5L) was added in 35 min, maintaining the temperature between 0 and 5°C. The suspension was stirred at 0°C for 15 min, at 20/25°C for 1 hr, then washed with water (3 x 39.2L), concentrated to 24.5L and then added to a solution of triphosgene (1.97Kg) in AcOEt (24.5L) cooled to 0°C. Triethylamine (3.28L) was then added in 40 min, maintaining the temperature between 0 and 8°C. The suspension was stirred for 1 h and 45 min at 20/25°C and 30 min at 70Cand then the solution of intermediate 82 diluted with AcOEt (49L) and triethylamine (2.6L) was added in 30 min. The mixture was refluxed for 15 hrs.
The reaction mixture, cooled at 20/25°C was treated with aqueous solution of NaOH 10%v/v (36.75L). Organic phase was washed with HCI 4%v/v (46.55L) and NaCI 11 ,5%p/p (4 x 24.5L) then concentrated to 14.7L. and diluted with Ciclohexane (39.2L). The mixture was filtered through a silica pad (4.9Kg) that was washed twice with a mixture of CH/AcOEt 85/15 (2 x 49L). To the Eluted phases (14.7L) cooled at 20/25°C, methyl tertbutyl ether (49L) and methansulphonic acid (4.067L) were added. The mixture was washed with NaOH 10%v/v (31.85L) then with water (4 x 31.85L). Organic phase was concentrated to 9.8L, methyl tertbutyl ether (49L) was added and the solution filtered through a δmicron filter then concentrated to 9.8L. At 20/25°C MTBE (29.4L) and metansulphonic acid (1.098L) were added. The suspension was refluxed for 10 min, stirred at 20/25°C for 10hrs and 2 hrs at O°C.Then the precipitate was filtered, washed with methyl tertbutyl ether (4.9L) dried under vacuum at 20/25°C for 24 hrs to obtain the title compound (5.519Kg.) as white solid.
1H-NMR (DMSO) δ (ppm) 8.99 (bm, 1 H); 8.66 (bm, 1 H); 8.00 (bs, 1 H) 7.69 (bs, 2H); 7.27 (dd, 1 H); 7.00 (dd, 1 H); 6.83 (m, 1 H); 5.32 (q, 1 H) 4.47 (dd, 1 H); 3.50-3.20 (m, 4H); 2.96 (m, 2H); 2.72 (s, 3H); 2.37 (s, 3H) 2.28 (s, 3H); 1.46 (d, 3H). ES+: m/z 492 [MH - CH3SO3H]+ ES": m/z 586 [M - H]"; 95 [CH3SO3]"
Example 37
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid Ii -(R)- (3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide
To a solution of intermediate 40a (15.6g) in anhydrous THF (94ml), at 0°C, under N2, BH3THF 1 M/THF (154ml) was added. The solution was heated at reflux for 3 hr. HCI 37% (54ml) was slowly added maintaining the reaction mixture in an ice-bath and the reaction mixture was stirred at rt for 1 hr. Water was then added (125 ml) and solid NaHCO3 (62.4g) was added portionwise until a pH of 6.5.The aqueous phase was extracted with Et O (4x160 ml) and the combined organic extracts were dried over
Na2SO , the solids were filtered and evaporated to leave a colourless oil which was purified by flash chromatography (silica gel, EtOAc/Methanol 7/3). The obtained product was suspended in Et2O (220ml) and washed with NaHCO3 sat. (2x36ml). The combined organic phases were dried (Na2SO ) and evaporated to give the title compound as white foam (8.7g,). 1H-NMR (CDCI3) δ (ppm) 7.78 (s, 1 H); 7.60 (s, 2H); 7.28 (m, 1 H); 6.85 (dd, 1 H); 6.79 (td, 1 H); 5.53 (q, 1 H); 4.43 (dd, 1 H); 2.9-3.5 (m, 5H); 2.78 (m, 1 H), 2.71 (s, 3H); 2.43 (s, 3H); 1.47 (d, 3H).
Intermediate 40
2-(S)-(4-Fluoro-2-methyl-phenyl)-3-oxo-piperazine-1 -carboxylic acid ri-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide ( 40a ) 2-(S)-(4-Fluoro-2-methyl-phenyl)-3-oxo-piperazine-1 -carboxylic acid ri-(S)-(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide.(-40b) To a solution of intermediate 39 (12.1g) in anhydrous DCM (270 mL), TEA (16.4 mL) was added. The solution was cooled down to 0°C and a solution of triphosgene (7.3 g) in anh. DCM (60 mL) was added drop-wise over 40 min. The reaction mixture was stirred at 0°C for 4 hr and was brought back to r.t. DIPEA (20.2 mL) was then added, followed by a solution of [1-(3,5- bis-trifluoromethyl-phenyl)-ethyl]-methyl-amine (23.6 g) in acetonitrile (300 mL) and an additional amount of acetonitrile (300 mL). The reaction mixture was warmed up to 95°C (oil bath T°C) without a water condenser to evaporate the DCM. When the internal temperature had reached 70°C, the flask was equipped with a water condenser, and the reaction mixture was heated at 70°C for an additional 2 hr (4 hr total). It was then brought back to r.t. and the solvent was evaporated. The residue was partitioned between DCM / 2% HCI and the phases were separated. The aqueous layer was extracted with DCM (1x) and the combined organic extracts were dried. The solids were filtered and the solvent evaporated to give a crude mixture of title compounds which were purified by flash chromatography (AcOEt/CH 8:2) to obtain the title compounds 40a (8.8 g) and 40 b (9.0 g) as white foams.
NMR (1H, DMSO-de): δ 8.16 (s, 1 H), 7.98 (s, 2H), 7.19 (dd, 1 H), 6.97 (dd, 1 H), 6.87 (td, 1 H), 5.34 (s, 1 H), 5.14 (q, 1 H), 3.45-3.2 (m, 4H), 2.53 (s, 3H), 2.27 (s, 3H), 1.56 (d, 3H).
Intermediate 40b: NMR (1H, DMSO-d6): δ 8.16 (s, 1 H), 7.95 (s, 2H), 7.19 (dd, 1 H), 6.98 (dd, 1 H), 6.90 (td, 1 H), 5.29 (q, 1 H), 5.28 (s, 1 H), 3.45-3.15 (m, 4H), 2.66 (s, 3H), 2.27 (s, 3H), 1.52 (d, 3H).
Intermediate 81 (S)-3-(4-Fluoro-2-methyl-phenyl)-piperazine dihydrochloride
To a solution of intermediate 39 (60.35g) in dry THF (180ml), at 0-3°C, under N2, BH3 THF 1 M/THF (1220mL) was added dropwise. The solution was refluxed for 4 hours then cooled to 0-3°C and methanol (240mL) was added. The reaction mixture was heated to room temperature then it was concentrated to dryness. The residue was redissolved in methanol (603.5mL), excess HCI 1 N in Et2O (1207mL) was added and the mixture was refluxed for 2 hours then cooled at 3°C for 4 hours. The suspension was filtered to obtain a white solid that was washed with Et2O (60.35mL) and dried to yield the title compound (72.02q)
1H-NMR (DMSO) δ (ppm) 11.0-9.5 (b, 4H); 7.99-7.19 (dd-m, 3H); 4.96 (dd, 1 H); 3.65-3.15 (m, 6H); 2.42 (s, 3H).
..................
HYDROCHLORIDE SALT
Example 38
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid |i -(R)-
(3,5-bis-trifluoromethyl-phenyl)-ethvπ-methyl-amide hydrochloride Example 37 (0.1 g) was dissolved in Ethyl Ether (0.8ml) at room temperature, then 1 M HCI solution in Ethyl Ether (0.6ml) was added. The suspension was stirred at 3°C for 3 hour, then filtered and washed with Ethyl Ether (1 ml) to afford the title compound ( 0.015g ) as a white solid. 1H-NMR (DMSO) δ (ppm) 9.31 (bm, 1 H); 9.11 (bm, 1 H); 8.02 (bs, 1 H); 7.72 (bs, 2H); 7.28 (dd, 1 H); 7.00 (dd, 1 H); 6.84 (m, 1 H); 5.34 (q, 1 H); 4.54 (dd, 1 H); 3.50-3.20 (m, 4H); 3.08 (m, 1 H); 2.93 (m, 1 H); 2.73 (s, 3H); 2.38 (s, 3H); 1.48 (d, 3H).
ACETATE SALT
Example 18
2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine-1 -carboxylic acid M -(R)-
(3.5-bis-trifluoromethyl-phenyl)-ethvn-methyl-amide acetate salt
To a solution of intermediate 40a (8.8 g) in dry THF (33 mL) under N2 BH3.THF (1 M solution in THF - 87 mL) was added and the reaction mixture was stirred at reflux for 3 hr, then cooled to r.t. and HCI (37%, 30 mL) was added drop-wise maintaining the reaction mixture in an ice-bath. The reaction mixture was stirred at r.t. for 1 hr. Water was then added (70 mL) and solid NaHCO3 (35.2 g) was added portion-wise until a pH of 6.5. The THF was evaporated and the aqueous phase was extracted with Et2θ (3 x 88 mL). The combined organic phases were dried, and evaporated to leave a colourless oil (7.37 g).
This crude oil was purified by flash chromatography (AcOEt/MeOH 7:3). The product obtained was suspended in Et2θ (125 mL) and washed with NaHCO3 sat. (2 x 20 mL). The clear combined organic phases were dried and evaporated to obtain the 2-(S)-(4-Fluoro-2-methyl-phenyl)-piperazine- 1 -carboxylic acid [1 -(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl- amide as white foam (5.27 g). This material (5.27 g) was dissolved in Et2θ (79 mL) and acetic acid (613 μL) was added drop-wise. The mixture was stirred at r.t. for 1 h and then at 0°C for 1h. The suspension was filtered to give the title compound (4.366 g) as a white solid. NMR (1H, DMSO-de): δ (ppm) 7.98 (s, 1 H), 7.70 (s, 2H), 7.87 (m, 1 H), 6.91 (m, 1 H), 6.77 (m, 1 H), 5.29 (q, 1 H), 4.23 (dd, 1 H), 3.2-2.6 (m, 6H), 2.68 (s, 3H), 2.3 (s, 3H), 1.89 (s, 3H), 1.48 (d, 3H). MS (m/z): 492 [M-CH3COO]+.
[ ]D = - 120.4°C Solvent (CHCI3); Source: Na; Cell volume [mL]: 1 ; Cell pathlength [dm]: 1 ; Cell temperature [°C]: 20; Wavelength [nm]: 589
'''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''
EXAMPLE 1 N-[1-(R) 3,5-bis-trifluoromethyl phenyl)-ethyl]-N-methyl carbamoyl chloride
[1-(R) 3,5-bis-trifluoromethyl phenyl)-ethyl]methyl amine L(−)maleate (13.5 g; 33.33 mmol) was suspended in ethyl acetate (39.9 ml) and ethanol (0.1 ml); aqueous sodium carbonate 13% (40 ml) was added and the mixture was stirred at a temperature 20-25° C. until a clear solution was formed. The water phase was discarded and the organic phase was washed with water (40 ml). Fresh ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added, the solution was concentrated to 40 ml, a second amount of fresh ethyl acetate (49.87 ml) and ethanol (0.13 ml) was added and the solution was concentrated to 40 ml. Fresh ethyl acetate (109.7 ml) and ethanol (0.3 ml) were added under CO2 flow. A cycle of vacuum and CO2 in the vessel was applied, then CO2 was maintained for 10 minutes. Then, a neat Et3N (6.1 ml; 46.34 mmol) was added and the reaction mixture was stirred at a temperature 20-25° c. for 30 minutes. Trimethylmethylsilylchloride (6.4 ml; 40.42 mmol) was added in 30 minutes (exothermic step) and the reaction mixture was stirred for further 30 minutes at room temperature. Pyridine (5.4 ml; 66.66 mmol) was added, then SOCl2 (3.6 ml; 40.42 mmol) was added in 10 minutes. The reaction mixture was stirred at room temperature for 10 hours under CO2 atmosphere. 13% w/w aqueous racemic malic acid (60 ml) was added and the mixture was stirred for 15 minutes; the water phase was discarded then the organic phase was washed with water (60 ml); the water phase was discarded then the organic phase was washed with sodium carbonate 13% w/w (60 ml). Finally, the water phase was discarded and ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added and the solution was concentrated to 50 ml; further ethyl acetate (49.87 ml) and ethanol (0.13 ml) were added and the solution was concentrated to dryness to give the title compound as a pale yellow (10.41 gr; 31.33 mmol 94% yield)
NMR-(d6-DMSO) δ (ppm)
8.04 δ (br s, 1H), 7.97 δ (br s, 2H), 5.52 δ (q, 1H), 2.97 δ (s, 3H), 1.66 δ (d, 3H)
EXAMPLE 2 (2R)-2-(4-fluoro-2-methylphenyl)-4-oxo-1-piperidinyl carbonyl; chloride
(2R)-2-(4-fluoro-2-methylphenyl)-4-oxo-1-piperidine L(−) mandelate (2 g; 5.57 mmol) was suspended in ethyl acetate (8 ml); aqueous sodium carbonate 13% w/w (10 ml) was added and the mixture was stirred at a temperature 20-25° C. until a clear solution was formed.
The water phase was discarded and the organic phase was washed with aqueous sodium chloride 10% w/w (4 ml). Fresh ethyl acetate (8 ml) were added, the solution was concentrated to 6 ml, a second amount of fresh ethyl acetate (8 ml) was added and the solution was concentrated to 6 ml.
Fresh ethyl acetate (2 ml) and neat Et3N (1.94 ml; 13.92 mmol) were added under CO2 flow at 0° C. The mixture was stirred for 10 minutes, then Trimethylmethylsilylchloride (1.42 ml; 11.14 mmol) was added in 5 minutes (exothermic step) and the reaction mixture was stirred for further 30 minutes at 0° C. Pyridine (0.58 ml; 7.24 mmol) was added, then SOCl2 (0.53 ml; 7.24 mmol) was added in 5 minutes. The reaction mixture was stirred at 0° C. for 1 h, then at a temperature 20-25° C. for 5 hours under CO2 atmosphere. Water (20 ml) was added was added; the water phase was discarded then the organic phase was washed with sodium carbonate 13% w/w (20 ml); the water phase was discarded then the organic phase was dried on sodium sulphate. The organic phase was filtered and concentrated to dryness to give the title compound as a pale yellow (1.5 gr; 5.57 mmol 100% yield)
HPLC Rt: 2.33 min; MS: [H+] 270
......................
J. Med. Chem., 2009, 52 (10), pp 3238–3247
DOI: 10.1021/jm900023b

a(a) (i) Mg, I2, THF, T = 70 °C, 2 h; (ii) LiBr, Cu2Br2, THF, room temp 1 h; (iii) CH3OCOCOCl, room temp, 2 h; (b) ethylenediamine, toluene, reflux, 6 h; (c) H2 (1 atm), 10% Pd/C, MeOH, 16 h; (d) (i) S-(+)-mandelic acid orR-(−)-mandelic acid, AcOEt, T = 3−5 °C, 2 h; (ii) filtration of the salt, then crystallization in AcOEt; (iii) 0.73 M NaOH; (e) (i) triphosgene, Et3N, CH2Cl2, T = 0 °C, 4 h; (ii) 1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-N-methylamine, N(i-Pr)2Et, CH3CN, T = 70 °C, 2 h; (f) (i) 1 M BH3·THF, THF, reflux, 3 h; (ii) Et2O, AcOH.
.................
patents
WO 2012175434
WO 2008046882
WO 2004091624
WO 2004067093
...................
| WO1993005791A1 | Sep 18, 1992 | Apr 1, 1993 | Univ Pennsylvania | Prevention of hemolysis |
| WO2001025219A2 | Oct 5, 2000 | Apr 12, 2001 | Giuseppe Alvaro | Piperazine compounds |
| WO2004091624A1 * | Apr 16, 2004 | Oct 28, 2004 | Renzo Carletti | Combinations comprising paroxetine and 2- (s) - (4-fluoro-2-methyl-phenyl) -piperazine-1-carboxylic acid [1- (r)- (3,5-bis-trifluoro-2-methyl-phenyl) -ethyl]-methyl amide for treatment of depression and/or anxiety |
| WO2005082419A1 | Jan 6, 2005 | Sep 9, 2005 | Wayne Alan Boettner | Pharmaceutical compositions of neurokinin receptor antagonists and cyclodextrin and methods for improved injection site toleration |
| WO2007048642A1 | Oct 26, 2006 | May 3, 2007 | Ilaria Bientinesi | Process for preparing n, n-substituted carbamoyl halides |
| EP1897542A1 | Sep 7, 2006 | Mar 12, 2008 | Sanofi-Aventis | Aqueous formulation comprising an antitumor agent |
.......................................
1. Organic Process Research and Development, 2009 , vol. 13, 6 pg. 1100 - 1110
2. Magnetic Resonance in Chemistry, 2010 , vol. 48, 7 pg. 523 - 530
3. Org. Process Res. Dev., 2009, 13 (3), pp 489–493.
4. Synthesis of the NK1 receptor antagonist GW597599. Part 1: Development of a scalable route to a key chirally pure arylpiperazine
Org Process Res Dev 2008, 12(6): 1188.............
Org Process Res Dev 2008, 12(6): 1188.............
5.Journal of Thermal Analysis and Calorimetry, 2010 , vol. 102, 1 pg. 297 - 303
Di Fabio R, Alvaro G, Griffante C, Pizzi DA, Donati D, Mattioli M, Cimarosti Z, Guercio G, Marchioro C, Provera S, Zonzini L, Montanari D, Melotto S, Gerrard PA, Trist DG, Ratti E, Corsi M.
J Med Chem. 2011 Feb 24;54(4):1071-9. doi: 10.1021/jm1013264. Epub 2011 Jan 13.
Provera S, Guercio G, Turco L, Curcuruto O, Alvaro G, Rossi T, Marchioro C.
Magn Reson Chem. 2010 Jul;48(7):523-30. doi: 10.1002/mrc.2611.
Provera S, Martini L, Guercio G, Turco L, Costa L, Marchioro C.
J Pharm Biomed Anal. 2010 Nov 2;53(3):389-95. doi: 10.1016/j.jpba.2010.04.027. Epub 2010 Apr 29.
Sabbatini FM, Di Fabio R, Griffante C, Pentassuglia G, Zonzini L, Melotto S, Alvaro G, Capelli AM, Pippo L, Perdona' E, St Denis Y, Costa S, Corsi M.
Bioorg Med Chem Lett. 2010 Jan 15;20(2):623-7. doi: 10.1016/j.bmcl.2009.11.078. Epub 2009 Nov 20.
Di Fabio R, Griffante C, Alvaro G, Pentassuglia G, Pizzi DA, Donati D, Rossi T, Guercio G, Mattioli M, Cimarosti Z, Marchioro C, Provera S, Zonzini L, Montanari D, Melotto S, Gerrard PA, Trist DG, Ratti E, Corsi M.
J Med Chem. 2009 May 28;52(10):3238-47. doi: 10.1021/jm900023b.
...............................................................
5 CASOPITANT

CASOPITANT
Tachykinin NK1 Antagonists
(2S,4S)-4-(4-Acetyl-1-piperazinyl)-N-[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl]-2-(4-fluoro-2-methylphenyl)-N-methyl-1-piperidinecarboxamide
4-(S)-(4-Acetyl-piperazin-1-yl)-2-
(R)-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid,
[1-(R)-(3,5-bis-trifluoromethyl- phenyl)-ethyl]-methylamide
414910-30-8 MESYLATE
414910-27-3 (free base)
414910-27-3 (free base)
679769
GW-679769
GW-679769B
| MF C30H35F7N4O2.CH4O3S MESYLATE | |
| Molecular Weight | 712.719 |
Casopitant (trade names Rezonic (US), Zunrisa (EU)) is an neurokinin 1 (NK1) receptor antagonist undergoing research for the treatment of chemotherapy-induced nausea and vomiting (CINV).[1] It is currently under development by GlaxoSmithKline (GSK).
In July 2008, the company filed a marketing authorisation application with the European Medicines Agency. The application was withdrawn in September 2009 because GSK decided that further safety assessment was necessary.[2] Casopitant
mesylate, a tachykinin NK1 receptor antagonist, had been filed for
approval in the U.S. and the E.U. by GlaxoSmithKline for the prophylaxis
of chemotherapy-induced nausea/vomiting.
In 2009 the company
discontinued the development of the drug candidate for this indication.
An MAA had also been filed for the treatment of postoperative nausea and
vomiting, and in 2009 the application was withdrawn by the company.
Additional
phase II clinical trials were ongoing at GlaxoSmithKline for the
treatment of depression, anxiety, sleep disorders, fibromyalgia and
overactive bladder, however, no recent developments have been reported
for these indications.
- Lohr L (2008). "Chemotherapy-induced nausea and vomiting". Cancer J 14 (2): 85–93.doi:10.1097/PPO.0b013e31816a0f07. PMID 18391612.
- "GlaxoSmithKline withdraws its marketing authorisation application for Zunrisa". London: EMEA. 13 October 2009. Retrieved 21 December 2009
- Casopitant mesilate
Drugs Fut 2008, 33(9): 737 - WO 2002032867
- WO 2008046882
- Development of a control strategy for a defluorinated analogue in the manufacturing process of casopitant mesylate
Org Process Res Dev 2010, 14(4): 832 NMR FREE BASE, MESYLATE - WO 2006061233
- WO 2004091616
- US20040014770 ENTRY 1B MP MESYLATE 243
- Tetrahedron, 2010 , vol. 66, 26 p. 4769 - 4774 NMR FREE BASE
- Journal of Medicinal Chemistry, 2011 , vol. 54, 4 p. 1071 - 1079 NMR MESYLATE
| WO2006061233A1 * | Dec 7, 2005 | Jun 15, 2006 | Glaxo Group Ltd | The use of medicament 4-(s)-(4-acetyl-piperazin-1-yl)-2-(r)-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid, [1-(r)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamide |
| WO2001044200A2 * | Dec 14, 2000 | Jun 21, 2001 | David J Blythin | Selective neurokinin antagonists |
| WO2002010141A1 * | Jul 25, 2001 | Feb 7, 2002 | Michael Kirk Ahlijanian | Imidazole derivatives |
| WO2002032867A1 * | Oct 12, 2001 | Apr 25, 2002 | Giuseppe Alvaro | Chemical compounds |
| US20020123491 * | Dec 14, 2000 | Sep 5, 2002 | Neng-Yang Shih | Selective neurokinin antagonists |
| US20030064980 * | Jun 6, 2002 | Apr 3, 2003 | Neng-Yang Shih | Selective neurokinin antagonists |
| US20030144270 * | Nov 12, 2002 | Jul 31, 2003 | Schering Corporation | NK1 antagonists |

Casopitant (Rezonic,
Zunrisa, casopitant mesylate, GW-679769, 679769, CAS #414910-27-3),
4-(4-Acetyl-piperazin-1-yl)-2-(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic
acid [1-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methyl-amide, is a NK-1
receptor antagonist.
Casopitant is under investigation for the
treatment of emesis, nausea, drug-induced nausea, chemotherapy-induced
nausea and vomiting, post-operative nausea and vomiting, sleep
disorders, anxiety disorders, depressive disorders, overactive bladder,
and myalgia (Drug Report for Casopitant, Thomson Investigational Drug Database (Sep. 15, 2008); Reddy et al., Supportive Cancer Therapy 2006, 3(3), 140-142; and WO 2006/061233).
Casopitant has
also shown promise in treating disorders of the central nervous system,
tinitis, and sexual dysfunction (WO 2006/061233).
compound may be
of value in the treatment of Sexual dysfunctions including Sexual
Desire Disorders such as Hypoactive Sexual Desire Disorder and Sexual
Aversion Disorder sexual arousal disorders such as Female Sexual Arousal
Disorder and Male Erectile Disorder orgasmic disorders such as Female
Orgasmic Disorder, Male Orgasmic Disorder and Premature Ejaculation
sexual pain disorder such as Dyspareunia and Vaginismus, Sexual
Dysfunction Not Otherwise Specified; paraphilias such as Exhibitionism,
Fetishism, Frotteurism, Pedophilia, Sexual Masochism, Sexual Sadism
Transvestic Fetishism, Voyeurism and Paraphilia Not Otherwise Specified
gender identity disorders such as Gender Identity Disorder in Children
and Gender Identity Disorder in Adolescents or Adults and Sexual
Disorder Not Otherwise Specified.

Casopitant is
subject to CYP3A4-mediated oxidative metabolism at the 3-carbon of the
piperazine ring to form a hydroxylated metabolite which may be further
oxidized to the corresponding 3-oxo metabolite (Minthorn et al, Drug Metab. Disp., 2008,
36(9), 1846-1852). Adverse effects associated
with casopitantadministration include: neutropenia, nausea, hiccups,
headache, constipation, dizziness, pruritis, alopecia, and fatigue.
Overactive
bladder is a term for a syndrome that encompasses urinary frequency,
with or without urge incontinence, generally but not necessarily
combined with pollacisuria and nocturia. Overactive bladder is also
characterised by involuntary detrusor contractions which are either
triggered by provocation or occur spontaneously. If the detrusor
hyperactivity observed is based on neurological causes (e. g.
Parkinson's disease, apoplexy, some forms of multiple sclerosis, spinal
cord injury or the cross section of the bone marrow) it is known as
neurogenic detrusor hyperactivity. If no clear cause can be detected
this is known as idiopathic detrusor hyperactivity. In addition,
detrusor hyperactivity may be associated with anatomical changes in the
lower urinary tract, for example, in patients with bladder outlet
obstruction (an enlargement of the prostate gland in males)
International
patent application WO 02/32867 describes novel piperidine derivatives. A
0 particular preferred compound described therein is
4-(S)-(4-Acetyl-piperazin-1-yl)-2-(R)-
(4-fluoro-2-methyl-phenyl)-piperidine-1-carboxylic acid
...........................................................................
CASOPITANT MESYLATE
EXAMPLE 4
[0330]
4-(S)-(4-Acetyl-piperazin-1-yl)-2-(R)-(4-fluoro-2-methyl-phenyl)-piperidine-1-Carboxylic
Acid, [1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamide
Methanesulphonate
[0331] A solution of intermediate 4a (7.7 g) in
acetonitrile (177 mL) was added to a solution of 1-acetyl-piperazine
(3.9 g) in acetonitrile (17.7 mL) followed by sodium
triacetoxyborohydride (6.4 g) under a nitrogen atmosphere. The reaction
mixture was stirred at room temperature for 24 hours and then quenched
with a saturated sodium hydrogen carbonate (23.1 mL) and water (61.6
mL). The resulting solution was concentrated in vacuo, then AcOEt (208
mL) was added; the layers were separated and the aqueous layer was
back-extracted with further AcOEt (2×77 mL). The collected organic
phases were washed with brine (2×118 mL), dried and concentrated in
vacuo to give the crude mixture of syn and anti diastereomers (nearly
1:1) as a white foam (9.5 g).
[0332] A solution of this
intermediate in THF (85.4 mL) was added to a solution of methansulfonic
acid (0.890 mL) in THF (6.1 mL) at r.t. After seeding, the desired syn
diastereomer started to precipitate. The resulting suspension was
stirred for 3 hours at 0° C. and then filtered under a nitrogen
atmosphere. The resulting cake was washed with cold THF (15.4 mL) and
dried in vacuo at +20° C. for 48 hours to give the title compound as a
white solid (4.44 g).
[0333] NMR (d6-DMSO): δ (ppm)
9.52 (bs, 1H); 7.99 (bs, 1H); 7.68 (bs, 2H); 7.23 (m, 1H); 6.95 (dd,
1H); 6.82 (m, 1H); 5.31 (q, 1H); 4.45 (bd, 1H); 4.20 (dd, 1H); 3.99 (bd,
1H); 3.65-3.25 (bm, 5H); 3.17 (m, 1H); 2.96 (m, 1H); 2.88-2.79 (m+m,
2H); 2.73 (s, 3H); 2.36 (s, 3H); 2.30 (s, 3H); 2.13-2.09 (bd+bd, 2H);
2.01 (s, 3H); 1.89-1.73 (m+m, 2H); 1.46 (d, 3H).
[0334] m.p 243.0° C.
[0335] The compound is isolated in a crystalline form.
intermediate 4a is needed for above syn, ignore 4b
[0168] Intermediate 4
[0169]
2-(R)-(4-Fluoro-2-methyl-phenyl)-4-oxo-piperidine-1-Carboxylic Acid,
[1-(R)-3,5-bis-trifluoromethyl-phenyl)ethyl]-Methylamide (4a) and
2-(S)-(4-Fluoro-2-methyl-phenyl)-4-oxo-piperidine-1-Carboxylic Acid,
[1-(R)-3,5-bis-trifluoromethyl-phenyl)-ethyl]-Methylamide (4b) Method A
[0170]
A solution of triphosgene (147 mg) dissolved in dry DCM (5 mL) was
added drop-wise to a solution of intermediate 2 (250 mg) and DIPEA (860
μL) in dry DCM (15 mL) previously cooled to 0° C. under a nitrogen
atmosphere. After 2 hours,
[1-(R)-3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride
(503 mg) and DIPEA (320 μL) in dry acetonitrile (20 mL) were added and
the mixture was heated to 70° C. for 16 hours. Further
[1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride
(170 mg) and DIPEA (100 μL) were added and the mixture was stirred at
70° C. for further 4 hours. Next, the mixture was allowed to cool to
r.t., taken up with AcOEt (30 mL), washed with a 1N hydrochloric acid
cold solution (3×15 mL) and brine (2×10 mL). The organic layer was dried
and concentrated in vacuo to a residue, which was purified by flash
chromatography (CH/AcOEt 8:2) to give:
[0171] 1. intermediate 4a (230 mg) as a white foam,
[0172] 2. intermediate 4b (231 mg) as a white foam. ................ignore
[0173] Intermediate 4a
[0174] NMR (d6-DMSO):
δ (ppm) 7.98 (bs, 1H); 7.77 (bs, 2H); 7.24 (dd, 1H); 6.97 (dd, 1H);
6.89 (m, 1H); 5.24 (t, 1H); 5.14 (q, 1H); 3.61 (m, 1H); 3.55 (m, 1H);
2.71 (m, 2H); 2.56 (s, 3H); 2.50 (m, 2H); 2.26 (s, 3H); 1.57 (d, 3H).
[0175] Intermediate 4b
[0176] NMR (d6-DMSO):
δ (ppm) 7.96 (bs, 1H); 7.75 (bs, 2H); 7.24 (dd, 1H); 6.98 (dd, 1H);
6.93 (dt, 1H); 5.29 (q, 1H); 5.24 (t, 1H); 3.56 (m, 1H); 3.48 (m, 1H);
2.70 (s, 3H); 2.50 (m, 4H); 2.26 (s, 3H); 1.54 (d, 3H). ........ ignore
[0177] Intermediate 4a
[0178] Method B
[0179]
A saturated sodium hydrogen carbonate solution (324 mL) was added to a
solution of intermediate 9 (21.6 g) in AcOEt (324 mL) and the resulting
mixture was vigorously stirred for 15 minutes. The aqueous layer was
back-extracted with further AcOEt (216 mL) and the combined organic
extracts were dried and concentrated in vacuo to give intermediate 8 as a
yellow oil, which was treated with TEA (19 mL) and AcOEt (114 mL). The
solution obtained was added drop-wise over 40 minutes to a solution of
triphosgene (8 g) in AcOEt (64 mL) previously cooled to 0° C. under a
nitrogen atmosphere, maintaining the temperature between 0° C. and 8° C.
[0180]
After stirring for 1 hours at 0° C. and for 3 hours at 20° C.,
[1-(R)-(3,5-bis-trifluoromethyl-phenyl)-ethyl]-methylamine hydrochloride
(29.7 g), AcOEt (190 mL) and TEA (38 mL) were added to the reaction
mixture which was then heated to reflux for 16 hours.
[0181] The
solution was washed with 10% sodium hydroxide solution (180 mL), 1%
hydrochloric acid solution (4×150 mL), water (3×180 mL) and brine (180
mL). The organic layer was dried and concentrated in vacuo to a residue,
which was purified through a silica pad (CH/AcOEt 9:1) to give the
title compound (21.5 g) as a brown thick oil.
[0182] NMR (d6-DMSO):
6 (ppm) 7.97-7.77 (bs+bs, 3H); 7.24 (dd, 1H); 6.97 (dd, 1H); 6.88 (td,
1H); 5.24 (m, 1H); 5.14 (q, 1H); 3.58 (m, 2H); 2.7 (m, 2H); 2.56 (s,
3H); 2.49 (m, 2H); 2.26 (s, 3H); 1.57 (d, 3H).
intermediate 2
[0152] Intermediate 2
[0153] 2-(4-Fluoro-2-methyl-phenyl)-piperidine-4-one
[0154] Method A
[0155]
2-Methyl-4-fluoro-benzaldehyde (4 g) was added to a solution of
4-aminobutan-2-one ethylene acetal (3.8 g) in dry benzene (50 mL) and
the solution was stirred at r.t. under a nitrogen atmosphere. After 1
hour the mixture was heated at reflux for 16 hours and then allowed to
cool to r.t. This solution was slowly added to a refluxing solution of
p-toluensulphonic acid (10.6 g) in dry benzene (50 mL) previously
refluxed for 1 hour with a Dean-Stark apparatus. After 3.5 hours the
crude solution was cooled and made basic with a saturated potassium
carbonate solution and taken up with AcOEt (50 mL). The aqueous phase
was extracted with AcOEt (3×50 mL) and Et2O (2×50 mL). The organic layer
was dried and concentrated in vacuo to a yellow thick oil as residue
(7.23 g). A portion of the crude mixture (3 g) was dissolved in a 6N
hydrochloric acid solution (20 mL) and stirred at 60° C. for 16 hours.
The solution was basified with solid potassium carbonate and extracted
with DCM (5×50 mL). The combined organic phases were washed with brine
(50 mL), dried and concentrated in vacuo to give the title compound (2.5
g) as a thick yellow oil.
[0156] Method B
[0157]
L-selectride (1M solution in dry THF, 210 mL) was added drop-wise, over
80 minutes, to a solution of intermediate 1 (50 g) in dry THF (1065 mL)
previously cooled to −72° C. under a nitrogen atmosphere. After 45
minutes, 2% sodium hydrogen carbonate solution (994 mL) was added
drop-wise and the solution was extracted with AcOEt (3×994 mL). The
combined organic phases were washed with water (284 mL) and brine (568
mL). The organic phase was dried and concentrated in vacuo to get
1-benzyloxycarbonyl-2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one as a
pale yellow thick oil (94 g) which was used as a crude.
[0158]
This material (94 g) was dissolved in AcOEt (710 mL), then 10% Pd/C
(30.5 g) was added under a nitrogen atmosphere. The slurry was
hydrogenated at 1 atmosphere for 30 minutes. The mixture was filtered
through Celite and the organic phase was concentrated in vacuo to give
the crude 2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one as a yellow oil.
This material was dissolved in AcOEt (518 mL) at r.t. and racemic
camphorsulphonic acid (48.3 g) was added. The mixture was stirred at r.t
for 18 hours, then the solid was filtered off, washed with AcOEt (2×50
mL) and dried in vacuo for 18 hours to give
2-(4-fluoro-2-methyl-phenyl)-piperidine-4-one, 10-camphorsulfonic acid
salt as a pale yellow solid (68.5 g). (M.p.: 167-169° C.-NMR (d6-DMSO):
6 (ppm) 9.43 (bs, 1H); 9.23 (bs, 1H); 7.66 (dd, 1H); 7.19 (m, 2H); 4.97
(bd, 1H); 3.6 (m, 2H); 2.87 (m, 3H); 2.66 (m, 1H); 2.53 (m, 2H); 2.37
(s+d, 4H); 2.22 (m, 1H); 1.93 (t, 1H); 1.8 (m, 2H); 1.26 (m, 2H); 1.03
(s, 3H); 0.73 (s, 3H).
[0159] This material (68.5 g) was suspended
in AcOEt (480 mL) and stirred with a saturated sodium hydrogen
carbonate (274 mL). The organic layer was separated and washed with
further water (274 mL). The organic phase was dried and concentrated in
vacuo to give the title compound (31 g) as a yellow-orange oil.
[0160] NMR (d6-DMSO):
6 (ppm) 7.49 (dd, 1H); 7.00 (m, 2H); 3.97 (dd, 1H); 3.27 (m, 1H); 2.82
(dt, 1H); 2.72 (bm, 1H); 2.47 (m, 1H); 2.40 (m, 1H); 2.29 (s, 3H); 2.25
(dt, 1H); 2.18 (m, 1H).
[0161] MS (ES/+): m/z=208 [MH]+.
intermediate 9
[0220] Intermediate 9
[0221] 2-(R)-(4-Fluoro-2-methyl-phenyl)-piperidin-4-one Mandelic Acid.
[0222]
A solution of L-(+)-mandelic acid (22.6 g) in AcOEt (308 mL) was added
to a solution of intermediate 2 (31 g) in AcOEt (308 mL). Then
isopropanol (616 mL) was added and the solution was concentrated in
vacuo to 274 mL. The solution was then cooled to 0° C. and further cold
isopropanol (96 mL) was added. The thick precipitate was stirred under
nitrogen for 5 hours at 0° C., then filtered and washed with cold Et2O
(250 mL) to give the title compound as a pale yellow solid (20.3 g).
[0223] M.p.: 82-85° C.
[0224] NMR (d6-DMSO):
δ (ppm) 7.51 (dd, 1H); 7.40 (m, 2H); 7.32 (m, 2H); 7.26 (m, 1H); 7.0
(m, 2H); 4.95 (s, 1H); 4.04 (dd, 1H); 3.31 (m, 1H); 2.88 (m, 1H);
2.49-2.2 (m, 4H); 2.29 (s, 3H).
[0225] Chiral HPLC: HP 1100 HPLC
system; column Chiralcel OD-H, 25 cm×4.6 mm; mobile phase:
n-hexane/isopropanol 95:5+1% diethylamine; flow: 1.3 ml/min; detection:
240/215 nm; retention time 12.07 minutes.
......................
NMR
mesylate
Org. Process Res. Dev., 2010, 14 (6), pp 1337–1346
DOI: 10.1021/op100150b

1H NMR (600 MHz, DMSO-d6):
9.57 (br s, 1H), 7.99 (br s, 1H), 7.68 (br s, 2H), 7.23 (m, 1H), 6.95
(dd, 1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99
(m, 1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96
(m, 1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30
(s, 3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73
(m, 1H), 1.46 (d, 3H), MS: m/z 617 [MH]+, as free base.
...............
Org. Process Res. Dev., 2010, 14 (6), pp 1407–1419
DOI: 10.1021/op100209c
(2R,4S)-4-(4-Acetyl-1-piperazinyl)-N-{(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-1-piperidinecarboxamide Methanesulfonate Salt (Casopitant Mesylate 1)

A solution of casopitant 2 (0.86 wt) was diluted with EtOAc (overall solution of 2
in EtOAc was 4 L) and acetone (4.5 L) and was heated to the required
temperature (from 39 °C). Thereafter, neat methanesulfonic acid (0.12 L,
1.64 mol) was charged, followed by a slurry of 2 (0.005 kg) in
EtOAc (0.05 L) as seed. The obtained suspension was stirred for 1 h
followed by the addition of 3 L of isooctane in the required time (1 h).
The slurry was cooled to 20 °C in 2 h and aged 3 h. The suspension was
filtered and the solid washed with EtOAc (3 × 4 L). The white solid was
dried overnight under vacuum at 40 °C to give the desired casopitant
mesylate 1 (0.94 kg).
1H NMR (600 MHz, DMSO-d6)
δ 9.57 (br s, 1H), 7.99 (br s, 1H), 7.68 (br s, 2H), 7.23 (m, 1H), 6.95
(dd, 1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99
(m, 1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96
(m, 1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30
(s, 3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73
(m, 1H), 1.46 (d, 3H). MS: m/z 617 [MH]+, as free base.
.............
Org. Process Res. Dev., 2010, 14 (4), pp 805–814
DOI: 10.1021/op1000622
NMR CASOPITANT FREE BASE
(2R,4S)-4-(4-Acetyl-1-piperazinyl)-N-{(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-1-piperidinecarboxamide (Casopitant 2)
HCOOH (0.49 L, 13 mol) was added to a cooled suspension of NaBH(OAc)3 (0.82 kg, 3.87 mol) in CH3CN (4 L), keeping the internal temperature between 10−15 °C; then the lines were washed with more CH3CN (1 L), and the mixture was stirred for 40 min.
1-Acetylpiperazine (0.7 kg, 5.46 mol) was added neat over the solution of piperidone-urea 3, and the mixture was diluted with CH3CN (3 L). The resulting mixture was added over the previous suspension; fresh CH3CN
(4 L) was used to wash the line. The reaction mixture was stirred at 15
°C for 12 h. The solvent was evaporated under reduced pressure to 4 L.
The resulting suspension was diluted with fresh EtOAc (4 L), and then washed with ammonia [21% w/w solution (4 L,  11.25 M in NH3)], Na2CO3
[15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic
layer was washed with water (4 L). The organic phase was then
concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the
solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3
[15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic
layer was washed with water (4 L). The organic phase was then
concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the
solution was concentrated to 2.5 L to give a solution of casopitant 2.
11.25 M in NH3)], Na2CO3
[15% w/w solution (4 L)]. More EtOAc (4 L) was added, and the organic
layer was washed with water (4 L). The organic phase was then
concentrated to 2.5 L; again fresh EtOAc (4 L) was added, and the
solution was concentrated to 2.5 L to give a solution of casopitant 2.
1H NMR (600 MHz, DMSO-d6):
δ 7.99 (s, 1H), 7.68 (s, 2H), 7.18 (dd, 1H), 6.90 (dd, 1H), 6.76 (td,
1H), 5.33 (q, 1H), 4.14 (dd, 1H), 3.38 (m, 5H), 2.71 (s, 3H), 2.72 (m,
1H), 2.54 (m, 1H), 2.47 (m, 2H), 2.41 (m, 2H), 2.34 (s, 3H), 1.95 (s,
3H), 1.85 (m, 1H), 1.77 (m, 1H), 1.62 (dq, 1H), 1.47 (d, 3H), 1.40 (q,
1H).
.....................................
J. Med. Chem., 2011, 54 (4), pp 1071–1079
DOI: 10.1021/jm1013264
(2R,4S)-1′-acetyl-N-{(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4,4′-bipiperidine-1-carboxamide methanesulfonate salt 16a (casopitant)
nmr mesylate
1H NMR (600 MHz, DMSO-d6):
9.57 (bs, 1H), 7.99 (bs, 1H), 7.68 (bs, 2H), 7.23 (m, 1H), 6.95 (dd,
1H), 6.82 (m, 1H), 5.31 (q, 1H), 4.45 (m, 1H), 4.20 (dd, 1H), 3.99 (m,
1H), 3.56 (m, 1H), 3.47 (m, 3H), 3.37 (m, 1H), 3.15 (m, 1H), 2.96 (m,
1H), 2.87 (m, 1H), 2.80 (t, 1H), 2.74 (s, 3H), 2.36 (s, 3H), 2.30 (s,
3H), 2.13 (m, 1H), 2.08 (m, 1H), 2.10 (s, 3H), 1.87 (m, 1H), 1.73 (m,
1H), 1.46 (d, 3H). MS: m/z 617 [MH]+, as free base.
syn of intermediates

a(a)
(i) 2-Bromo-5-fluorotoluene, Mg, THF, 60−70 °C; (ii) 4-methoxypyridine,
benzyl chloroformate, THF, −20 °C, then Grignard’s reagent, −20 °C, 1
h; (b) (i) tris(triphenylphosphine)rhodium(I) chloride, 2-propanol, H2 (p = 5 atm), 60 °C, 5 h; (ii) Pd/C 5%, H2 (p = 4 atm), 20 °C, 5 h; (iii) (R,S)-10-camphorsulfonic acid, toluene; (c) CH2Cl2, H2O, 8% NaHCO3 (aq); l-(+)-mandelic acid, 2-propanol, heptanes; (d) MeNH2, EtOH, NaBH4, 25 °C, 1.5 h; (e) (i) ethyl acetate, NaHCO3 (aq. sat. soln), 5; (ii) triphosgene, triethylamine, ethyl acetate, then 5, 20 °C, 2 h; (f) R′RNH, CH3CN, NaBH(OAc)3, room temp, 24 h.

...........................................................
6 BIRINIPITANT
BIRINAPANT, Apoptosis inhibitor
(2S,2′S)-N,N’-((2S,2′S)-((3S,3′S,5R,5′R)-5,5′-((6,6′-difluoro-1H,1′H-[2,2'-biindole]-3,3′-diyl)bis(methylene))bis(3-hydroxypyrrolidine-5,1-diyl))bis(1-oxobutane-2,1-diyl))bis(2-(methylamino)propanamide)
1260251-31-7 cas no
US20110003877,WO 2013049350 A1
| Molecular Weight: | 806.94 |
| Birinapant Formula: | C42H56F2N8O6 |
Birinapant, also known as TL32711, is a synthetic small molecule and peptido mimetic of second mitochondrial-derived activator of caspases (SMAC) and inhibitor of IAP (Inhibitor of Apoptosis Protein) family proteins, with potential antineoplastic activity. As a SMAC mimetic and IAP antagonist, TL32711 binds to and inhibits the activity of IAPs, such as X chromosome-linked IAP (XIAP) and cellular IAPs 1 and 2. Since IAPs shield cancer cells from the apoptosis process, this agent may restore and promote the induction of apoptosis through apoptotic signaling pathways in cancer cells. IAPs are overexpressed by many cancer cell types and suppress apoptosis by binding and inhibiting active caspases-3, -7 and -9 via their baculoviral lAP repeat (BIR) domains
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014

 Guangyao Yu, Yijun Deng, Gurpreet Singh Kapoor, Condon, Susan Rippin, Martin Graham, Angeline Mufalli, Jennifer Burns, Martin Seipel, Eric Neiman, Thomas Haimowitz, Christopher Benetatos, Yasuhiro Mitsuuchi, Srinivas Chunduru Not pictured: Divya Goel")
(From left) Guangyao Yu, Yijun Deng, Gurpreet Singh Kapoor, Condon, Susan Rippin, Martin Graham, Angeline Mufalli, Jennifer Burns, Martin Seipel, Eric Neiman, Thomas Haimowitz, Christopher Benetatos, Yasuhiro Mitsuuchi, Srinivas Chunduru. Not pictured: Divya Goel
Credit: Courtesy of TetraLogic
It’s often said that two heads are better than one. For Malvern, Pa.-basedTetraLogic Pharmaceuticals, it was combining two copies of a molecule into one drug candidate that did the trick. Stephen M. Condon, vice president for chemistry at TetraLogic, told the story of that candidate, birinapant, in New Orleans.
Birinapant was designed to reinstate cancer cells’ ability to die. Many cancers that are resistant to conventional chemotherapy drugs have defects in the cell death pathway known as apoptosis. The human body uses apoptosis every day to clear away abnormal or unwanted cells.
Apoptosis is a tightly regulated process, Condon explains, with a network of proteins that activate or block the process. TetraLogic’s target is a family of proteins called the Inhibitor of Apoptosis proteins. As their name suggests, these proteins block apoptosis. They interfere with protease enzymes that carry out cellular dismantling.
TetraLogic’s aim is to lift that blockade to restart apoptosis in tumors. Many tumors have excesses of the apoptosis inhibitor proteins relative to normal cells, so targeting this process has the potential to be less toxic to normal cells compared with conventional chemotherapy.
It turns out nature has a way of negating the inhibitor proteins’ actions—a protein known as Smac. TetraLogic’s founders demonstrated that only a tiny portion of Smac is necessary to keep the inhibitor proteins at bay—the four amino acids at the protein’s N-terminus. “Once you can get a protein down to a tetrapeptide,” about the size of a small-molecule drug, “you start getting a lot of interest from the pharma community,” Condon told C&EN.
Peptides fall apart in the body too quickly to be drugs, so Condon’s team worked with molecular mimics of the Smac tetrapeptide. Their biggest advance was realizing that combining two copies of their tetrapeptide mimics into one molecule made their compounds highly effective at reinstating apoptosis in cancer cell lines. Many proteins in the apoptosis pathway function as dimers, so using these so-called bivalent mimics against them makes sense, Condon said.
However, several of the bivalent compounds were associated with pronounced body weight loss in mice. Condon’s team eventually learned that replacing a branched side chain on their peptide mimics and adding a hydroxyl group to a proline residue improved the tolerability for the animals without impacting the antitumor effect. With that, birinapant was born.
In mice, birinapant shrank tumors. The compound has been in clinical trials since 2009, both on its own and in combination with other chemotherapy drugs such as irinotecan and gemcitabine. On the basis of other biochemical work on the apoptosis pathway, TetraLogic thinks these drugs could act in synergy with birinapant to treat cancer, Condon said.
Birinapant is currently in Phase II clinical trials in patients with acute myeloid leukemia, pancreatic cancer, or ovarian cancer. Although these trials don’t have a control group, the emerging data are promising, TetraLogic chief executive officer John M. Gill told C&EN. (Early-stage cancer clinical trials are commonly run without placebo groups.) The birinapant trials show preliminary evidence both that the drug is having the desired effect and that this effect is associated with signs of clinical activity. Given these results, the company plans to launch randomized Phase II studies early in 2014.








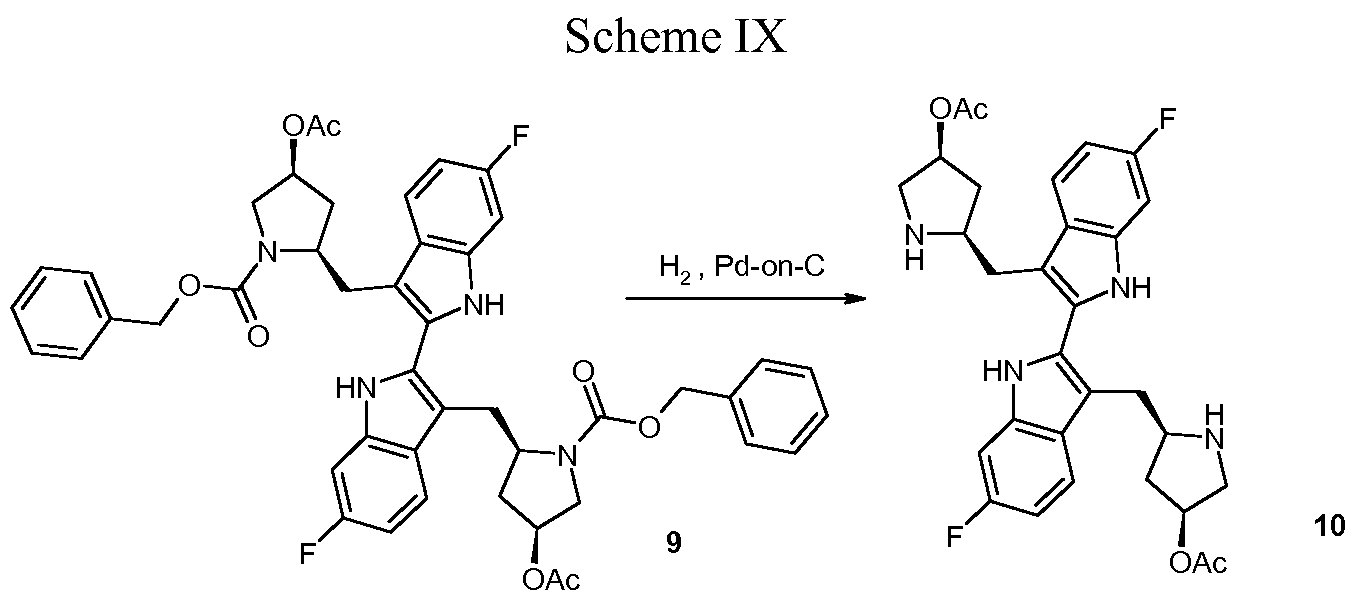


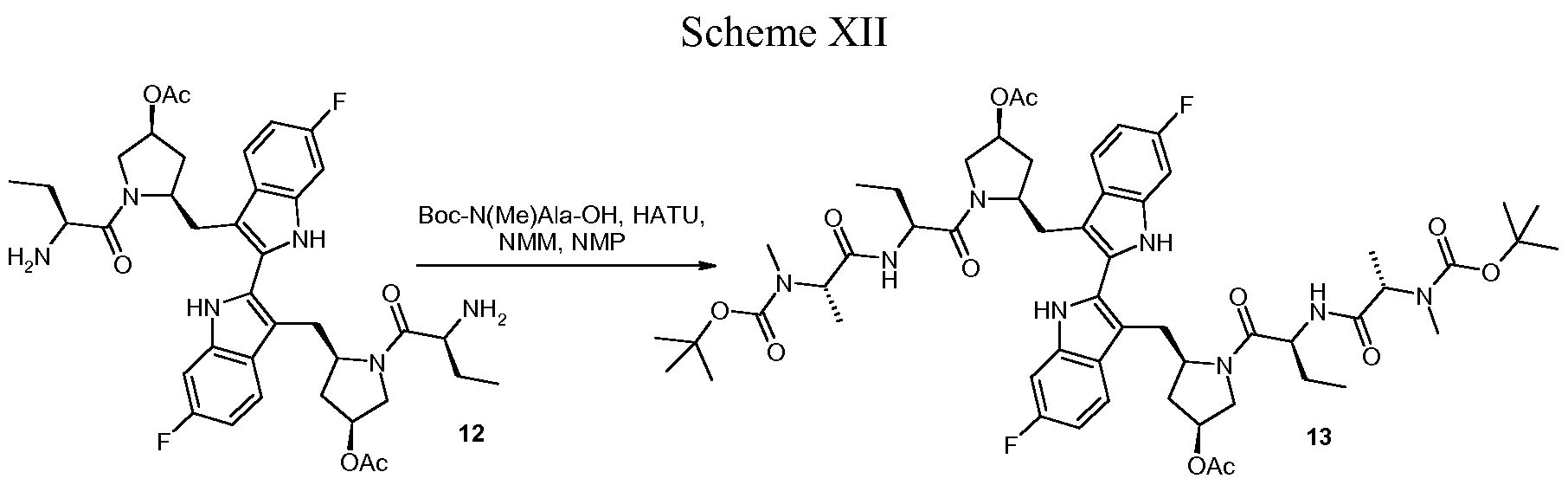


spectral data
1H NMR (300 MHz, CDC13): 511.74 (s, 2H), 8.27 (d, J= 8.7 Hz, 2H), 7.71 (dd, J= 5.4, 8.4 Hz, 2H), 7.55 (dd, J =2.4, 9.6 Hz, 2H), 6.88 (ddd, J= 2.4, 9.3, 9.3 Hz, 2H), 4.62-4.78 (m, 4H), 4.43 (dd, J= 9.3, 9.9 Hz, 2H), 4.03 (dd, J= 4.8, 11.4 Hz, 2H), 3.80 (d, J = 11.4 Hz, 2H), 3.66 (dd, J= 2.7, 14.4 Hz, 2H), 3.53 (dd, J = 11.4, 14.4 Hz, 2H), 3.11 (q, J = 6.9 Hz, 2H), 2.56 (s, 6H), 2.45 (m, 2H), 2.19 (d, J= 14.4 Hz, 2H), 1.76-2.10 (m, 6H), 1.59 (br s, 2H), 1.39 (d, J= 6.9 Hz, 6H), 1.22-1.38 (m, 2H), 1.07 (t, J = 7.2 Hz, 6H) ppm;
13C NMR (75 MHz, d6- DMSO): 5175.2, 172.8, 161.6, 158.5, 137.3, 137.2, 128.4, 128.3, 126.4, 120.8, 120.6, 109.4, 108.7, 108.4, 98.4, 98.0, 70.8, 60.2, 59.9, 56.6, 51.8, 36.4, 35.3, 28.3, 25.6, 20.0, 10.6 ppm.
Mass spectrum (ESI), m/z 807.5 [(M)+; calcd for C42H56F2N806: 806.9].

Aprepitant (MK-0869L-754,030), lUPAC name 5-([(2R,3S)-2-((R)-1 -[3,5- bis(trifluoromethyl)phenyl]ethoxy)-3-(4-fluorophenyl)morpholino]methyl)-1 /-/-1 ,2,4- triazol-3(2H)-one,

as described and claimed in the following US patents: US5,719,147, US 5,538,982, US 6,048,859, US 6,096,742 and US 6,235,735, the contents of which are incorporated herein by reference in their entirety. Also described in: Hale JJ et al, J Med Chem 1998; 41 (23) 4607-14; as well as pro-drugs thereof, such as:
Fosaprepitant (L-758,298, Emend) lUPAC name [3-{[(2R,3S)-2-[(1 R)-1 -[3,5- bis(trifluoromethyl)phenyl] ethoxy]-3-(4-fluorophenyl)morpholin-4-yl]methyl}-5-oxo- 2H- 1 ,2,4-triazol-1 -yl]phosphonic acid

e.g. in the form of a salt such as the dimeglumine salt as described and claimed at least in US 5,691 ,336, the contents of which are incorporated herein by reference in its entirety; b. ZD4974 as described in WO02026724 and WO01077089, the contents of which are incorporated herein by reference in its entirety:

The following compound, described in WO01077069 and WO00059873, the contents of which are incorporated herein by reference in entirety:

d. The following compound described in DE19519245, the contents of which are incorporated herein by reference in its entirety:

e. The following compound described in WO9732865, the contents of which are incorporated herein by reference in its entirety:

f. The following compound described in EP1295599, the contents of which are incorporated herein by reference in its entirety:

g. CGP49823 described in WO9626183 and WO9610562, the contents of which are incorporated herein by reference in their entirety:

h. The following compound as described in WO9514017, the contents of which are incorporated herein by reference in its entirety:

LY303870, Lanepitant, described in WO9907681 , the contents of which incorporated herein by reference in its entirety:

j. LI 686017, described in WO03091226, the contents of which are incorporated
herein by reference in its entirety:

k. FK888, as described in Hagiwara D et al, J Med Chem 1994; 37: 2090-9 and
WO9222569, WO93141 13, WO9321215, EP655442 and WO9637488, the contents of which are incorporated herein by reference in their entirety:

I. The following compound, described in WO9222569, WO93141 13, WO9321215, EP655442 and WO9637488, the contents of which are incorporated herein by reference in their entirety:

m. The following compound, described in WO9222569, WO93141 13, WO9321215, EP655442 and WO9637488, the contents of which are incorporated herein by reference in their entirety:

n. The following compound, described in WO00053572, the contents of which are incorporated herein by reference in its entirety:

o. Netupitant, described in WO020008232, the contents of which are incorporated herein by reference in its entirety:

p. Befetupitant, described in WO020008232, the contents of which are incorporated herein by reference in its entirety:

q. The following compound, described in WO202062784 and WO020008232, the
contents of which are incorporated herein by reference in their entirety:

r. R1 16031 , described in WO9724356 and WO0716440, the contents of which are incorporated herein by reference in their entirety:

s. The following compound, described in EP522808, the contents of which are incorporated herein by reference in its entirety:

t. The following compound:

KA731 , described in WO9831704, the contents of which are incorporated herein by reference in its entirety:

x. NKP608, described in WO04024714, the contents of which are incorporated herein by reference in its entirety:

y. CP96,345 described in Lowe JA et al. 1992; 35:2591 -600, and in WO92021677, the contents of which are incorporated herein by reference in their entirety;

z. The following compound, described in Lowe JA et al. J Med Chem 1994; 37:2831 – 40, and in WO92021677, the contents of which are incorporated herein by reference in their entirety;

aa.CP99,994, described in Desai MC et al. J Med Chem 1992; 35:491 1 -3, the contents of which are incorporated herein by reference in its entirety;

bb.CP-122,721 described in Rosen TJ et al. Bioorg Med Chem Lett 1998; 8:281 -4, the contents of which are incorporated herein by reference in its entirety:

cc. CJ-17,493, described in WO9925714, the contents of which are incorporated herein by reference in its entirety:

dd.Ezlopitant, CJ-1 1 ,974 described in WO1992021677 the contents of which are
incorporated herein by reference in its entirety:

ee.Maropitant, CJ-1 1 ,972, described in WO1992021677, US 6,222,038 and US
6,255,230, the contents of which are incorporated herein by reference in their entirety:

ff. RP77580 described in EP429366, the contents of which are incorporated herein reference in its entirety:

gg.Dapitant, RPR100893, described in WO9321 154, the contents of which are incorporated herein by reference in its entirety:

hh.The following compound, described in EP512901 , the contents of which are incorporated herein by reference in its entirety:

ii. Nolpitantium, SR140333 described in EP512901 , the contents of which are incorporated herein by reference in its entirety:

jj. The following compound, described in WO9526338, the contents of which are incorporated herein by reference in its entirety:

kk. SSR240600, described in WO00068292, the contents of which are incorporated herein by reference in its entirety:

II. SCH388714 described in WO06065654, the contents of which are incorporated herein by reference in its entirety:

mm. The following compound described in Paliwal S et al, Bioorg Med Chem Lett 2008; 18:4168-71 , the contents of which are incorporated herein by reference in its entirety:

nn.Rolapitant, described in WO03051840, the contents of which are incorporated
herein by reference in its entirety:

oo. The following compound, dexcribed in EP566069, the contents of which are
incorporated herein by reference in its entirety:

pp.TAK-637, described in JP10259184, the contents of which are incorporated herein by reference in its entirety:

qq.The following compound described in JP2004002334, the contents of which are incorporated herein by reference in its entirety:

rr. The following compound described in JP2007277231 and JP2008239618, the contents of which are incorporated herein by reference in their entirety:

. The following compound described in JP2007277231 and JP2008239618, the contents of which are incorporated herein by reference in their entirety:

tt. The following compound described in WO9317032 and WO951 1686, the contents of which are incorporated herein by reference in their entirety:

The following compound described in WO9630367 and WO01025233, the contents of which are incorporated herein by reference in their entirety:

vv. HSP1 17 described in WO9630367 and WO01025233, the contents of which are incorporated herein by reference in their entirety:

ww. The following compound, described in Set S, et al. Bioorg Med Chem ILKett 2005; 15:1479-84 and WO03062245, the contents of which are incorporated herein by reference in their entirety:

xx. The following compound, described in Seto S, et al. Bioorg Med Chem Lett 2005;
15:1479-84 and WO03062245, the contents of which are incorporated herein by reference in their entirety:

yy. The following compound, KRP-103, described in WO03062245 and WO05019225, the contents of which are incorporated herein by reference in their entirety:

zz. The following compound described in WO06106727, the contents of which are incorporated herein by reference in its entirety:

aaa. The following compound, described in WO07074491 , the contents of which are incorporated herein by reference in its entirety:

bbb. SLV317, described in US20020065276, the contents of which are
incorporated herein by reference in its entirety:

ROLAPITANT IS

Palonosetron hydrochloride has recently emerged as a highly efficacious anti-nauseant and anti-emetic agent. See PCT publications WO 2004/045615 and 2004/073714 from Helsinn Healthcare. Palonosetron hydrochloride is sold in the United States as a sterile injectable liquid under the ALOXI® brand, in sterile unit dose vials containing 0.075 or 0.25 mg. of palonosetron hydrochloride. Palonosetron hydrochloride also is also sold as an orally administered soft-gel dosage form containing 0.5 mg. of palonosetron hydrochloride. The official chemical name for palonosetron hydrochloride is (3aS)-2-[(S)-l-Azabicyclo [2.2.2]oct-3-yl]- 2,3,3a,4,5,6-hexahydro-l-oxo-lHberiz[Je] isoquinoline hydrochloride (CAS No. 119904-90-4); its empirical formula is C19H24N2OHCI, and its molecular weight is 332.87. The compound is represented by the following chemical structure:

Methods of synthesizing palonosetron are described in U.S. Patent Nos. 5,202,333 and
5,510,486. Pharmaceutically acceptably dosage forms are described in PCT publications WO 2004/067005 and WO 2008/049552 from Helsinn Healthcare.
NKi antagonists have also recently emerged as a tool for combating nausea and vomiting from emetogenic medical procedures. Most recently, aprepitant was approved by the Food and Drug Administration ("FDA") for use in combination with other anti-emetic agents for the prevention of nausea and vomiting from moderately and highly emetogenic chemotherapy. However, it quickly became apparent that aprepitant' s effect was limited principally to vomiting - not nausea - and that aprepitant did not provide as much benefit during the acute phase of CINV. When tested against nausea in humans, aprepitant was unable to induce a significant reduction in the incidence or severity of nausea following moderately or highly emetogenic chemotherapy when compared to a 5-HT3 antagonist alone. See FDA Approved Labeling for Emend®. Thus, while aprepitant is approved by FDA for the prevention of nausea and vomiting in humans, this indication is somewhat misleading because aprepitant did not reduce nausea in the clinical trials preformed for aprepitant more than nausea controlled by the other components of the anti-emetic regimen. In addition, the results reported in Grunberg et ah, SUPPORT CANCER CARE (2009) 17:589-594, from a combined treatment of aprepitant and palonosetron, were far from promising.
Merck & Co. markets aprepitant, as EMEND® in the United States. The product is approved in a capsule dosage form, and is marketed for the prevention of CINV (acute and delayed) in combination with other anti-emetic agents such as ondansetron and metoclopramide. The product reportedly has a terminal half-life of from 9 to 13 hours. While aprepitant has demonstrated some effect against nausea, its effects have been inconsistent. Casopitant is another NKi antagonist that has been tested against nausea and vomiting in humans. A clinical study of casopitant is discussed in Therapeutics and Clinical Risk Management 2009:5 pp375- 384 to Ruhlmann et al. and Drug Metabolism and Disposition, vol.37, No. 8, 2009, pp.1635- 1645 to Pellegatti et al. As reported by Ruhlmann et al. in THERAPEUTICS AND CLINICAL RISK MANAGEMENT, 2009:5 375-384, casopitant had no statistically significant effect against nausea when administered in response to moderately emetogenic chemotherapy, and even induced nausea as a side effect. Casopitant has the formula (2R,4S)-4-(4-acetytlpiperazin-l-yl)-N-{(lR)- 1 - [3,5-bis(trifluoromethyl)phenyl]ethyl } -2-(4-fluoro-2-methylphenyl)-N-methylpiperidine- 1 - carboxamide, and the below chemical structure:

DR ANTHONY CRASTO
THANKS AND REGARD'S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I
was paralysed in dec2007, Posts dedicated to my family, my
organisation Glenmark, Your readership keeps me going and brings smiles
to my family
No comments:
Post a Comment