1 LOSARTAN
2 IRBESARTAN
3 VALSARTAN
4 CANDESARTAN
5 EPROSARTAN
6 OLMESARTAN
7 AMG 925
8 Allisartan isopropoxil
9
10
11
10
11
more
1 LOSARTAN
LOSARTAN
CAS 114798-26-4
DuP-753
E-3340
L-158086
MK-0954
MK-954
Ex-89 (free acid)
E-3340
L-158086
MK-0954
MK-954
Ex-89 (free acid)
launched 1994, merck
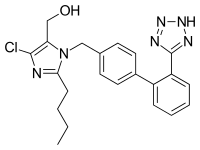
COZAAR (losartan potassium, cas 124750-99-8) is an angiotensin II receptor (type AT1)antagonist. Losartan potassium, a nonpeptide molecule, is chemically described as 2-butyl-4-chloro-1-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]imidazole-5-methanol monopotassium salt. Its empirical formula is C22H22ClKN6O, and its structural formula is:
 |
Losartan potassium is a white to off-white free-flowing crystalline powder with a molecular weight of 461.01. It is freely soluble in water, soluble in alcohols, and slightly soluble in common organic solvents, such as acetonitrile and methyl ethyl ketone. Oxidation of the 5-hydroxymethyl group on the imidazole ring results in the active metabolite of losartan.
COZAAR is available as tablets for oral administration containing either 25 mg, 50 mg or 100 mg of losartan potassium and the following inactive ingredients: microcrystalline cellulose, lactose hydrous, pregelatinized starch, magnesium stearate, hydroxypropyl cellulose, hypromellose, and titanium dioxide.
COZAAR 25 mg, 50 mg and 100 mg tablets contain potassium in the following amounts: 2.12 mg (0.054 mEq), 4.24 mg (0.108 mEq) and 8.48 mg (0.216 mEq), respectively. COZAAR 25 mg, COZAAR 50 mg, and COZAAR 100 mg may also contain carnauba wax.
LOSARTAN POTASSIUM
|
Losartan (rINN) /loʊˈsɑrtən/ is an angiotensin II receptor antagonist drug used mainly to treat high blood pressure (hypertension). Losartan was the first angiotensin II antagonist to be marketed. Losartan potassium is marketed by Merck & Co. Inc. under the trade nameCozaar. Losartan is available in generic form.
As with all angiotensin II type 1 receptor (AT1) antagonists, losartan is indicated for the treatment of hypertension. It may also delay progression of diabetic nephropathy, and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes, hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).
Although clinical evidence shows calcium channel blockers and thiazide-type diuretics are preferred first-line treatments for most patients (from both efficacy and cost points of view), an angiotensin II receptor antagonist such as losartan is recommended as first-line treatment in patients under the age of 55 who cannot tolerate an ACE inhibitor.The LIFE study demonstrated losartan was significantly superior to atenolol in the primary prevention of adverse cardiovascular events (myocardial infarction or stroke), with a significant reduction in cardiovascular morbidity and mortality for a comparable reduction in blood pressure. A study hints that losartan has a beneficial effect on mitochondria by reversing age related dysfunction in maintaining normal blood pressure and cellular energy usage. The maximal effects on blood pressure usually occur within 3–6 weeks upon starting losartan.
Losartan is also available as hydrochlorothiazide/losartan, a combination drug with a low dose thiazide diuretic to achieve an additive antihypertensive effect.
- Activation of AT1 receptors in the outer membrane of vascular smooth muscle cells of the heart and arteries causes those tissues to constrict. Blocking of vasoconstriction mediated by AT1 receptors has been found to be beneficial to patients with hypertension.
- [0003]AT1 receptors are activated by an octa-peptide, angiotensin II. Angiotensin II helps to maintain constant blood pressure despite fluctuations in a person’s state of hydration, sodium intake and other physiological variables. Angiotensin II also performs the regulatory tasks of inhibiting excretion of sodium by the kidneys, inhibiting norephedrin reuptake and stimulating aldosterone biosynthesis.
- [0004]Inhibiting angiotensin II binding to AT1 receptors with an AT1 receptor antagonist disrupts the vasoconstriction mediated by AT1 receptors that contributes to hypertension.
- [0005]In the early 1970s, it was discovered that certain oligopeptides competitively inhibited angiotensin receptors (at that time the existence of two receptor subtypes, AT1 and AT2, was unknown). This discovery spurred interest in development of therapeutic oligopeptides with increased potency, but interest in peptide analogs waned due in part to their poor oral bioavailability.
- [0006]In 1982, Furukawa. Kishimoto and Nishikawa of Taketa Chemical Indus. discovered a class of non-peptide-containing imidazoles that also inhibited the vasoconstriction effect of angiotensin II. See U.S. Patents Nos. 4,340,598 and 4,355,040. Later, U.S. Patent No. 5,138,069 was obtained by Carini, Denucia and Pancras of E.I. DuPont de Nemours on another class of imidazoles, which encompasses the compound losartan. In 1995, losartan (CA Index: 2-butyl-4-chloro-1-[[2'-(1H-tetrazol-5-yl) [1,1'-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol) (formula I):
 became the first nonpeptide AT1 antagonist approved by the U.S. Food and Drug Administration for clinical use. Losartan can be administered orally as its monopotassium salt. Losartan potassium is available by prescription in tablet form as a sole active ingredient (Cozaar®: Merck) and as a co-active ingredient with hydrochlorothiazide (Hyzaar®: Merck).
became the first nonpeptide AT1 antagonist approved by the U.S. Food and Drug Administration for clinical use. Losartan can be administered orally as its monopotassium salt. Losartan potassium is available by prescription in tablet form as a sole active ingredient (Cozaar®: Merck) and as a co-active ingredient with hydrochlorothiazide (Hyzaar®: Merck). - [0007]Losartan has been prepared by a variety of synthetic pathways. In several of these synthetic pathways, the penultimate product is 2-butyl-4-chloro-1-[[2'-(2-triphenylmethyl-2H-tetrazol-5-yl) [1,1'-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol (“trityl losartan”). Trityl losartan is an intermediate in processes described in U.S. Patents Nos. 5,138,069; 5,962,500 and 5,206,374.
- [0008]In a process described in Example 316 of U.S. Patent No. 5,138,069, the tetrazole ring of losartan is formed by reacting 1-[(2'-cyanobiphenyl-4-yl)methyl]-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction gives a trimethylstannyl substituted tetrazole compound directly. The trimethylstannyl group is cleaved from the product by reacting with trityl chloride. This reaction results in attachment of the trityl group to the tetrazole ring. In the last step, the trityl group is cleaved with acid to give losartan (Scheme 1).

- [0009]In the last step, trityl losartan was suspended in methanol and cooled to ~10°C. 3.4 N Hydrochloric acid was added to the slurry. After a period of time, the pH of the reaction mixture was raised to 13 with 10 N NaOH. Methanol was then distilled off while makeup water was added. After distillation, additional water and toluene were added. The toluene phase was separated and the aqueous phase was extracted once more with toluene. Ethyl acetate and acetic acid were then added to the aqueous phase. Losartan was recovered from the aqueous phase as a solid and further purified by slurrying in ethyl acetate. Losartan was obtained in 88.5% yield and 98.8% purity as determined by HPLC. This process is also described in U.S. Patents Nos. 5,128,355 and 5,155,188.
- [0010]U.S. Patent No. 5,962,500, Examples 3-5, describe a process for preparing losartan in which the tetrazole ring of losartan is present in the starting material, 5-phenyltetrazole. The ’500 patent process, depicted in Scheme 2, is convergent and uses a Suzuki coupling reaction (Miyaura, N.; Suzuki, A. Chem. Rev., 1995, 95, 2457) in the convergent step. On one branch of the synthesis, 5-phenyltetrazole is converted into the boronic acid coupling partner for the Suzuki reaction by ortho metalation with n-butyl lithium, followed by reaction with trisopropylborate. The tetrazole ring is protected from reacting with the strong allcyl lithium base with a trityl group. The trityl group is conventionally attached by reacting the tetrazole with trityl chloride in the presence of a non-nucleophilic base. On the other branch of the convergent synthesis, 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner.

- [0011]The direct product of Suzuki coupling is trityl losartan. In the next and last step, the tetrazole ring of trityl losartan is deprotected with 4N H2SO4 in THF. In that step, the acidic solution was aged overnight at 20 to 25°C. The solution was then extracted with isopropyl acetate and residual organic solvent was removed from the aqueous phase under vacuum. The solution was then carried forward to from the potassium salt without intermediate isolation of losartan. This process is also described in U.S.Patents Nos, 5,206,374, Example 21, and 5,310,928, Example 21.
- [0012]Larsen, R.D et al. [J. Org. Chem. (1994), 59, 6391-6394] discloses a similar convergent synthesis of lasartan, whereby the trityl lasartan, generated by Suzuki coupling, is deprotected using 0.7 M H2SO4 in a 50 : 50 mixture of acetonitrile /water.
- [0013]U.S. Patent No. 5,206,374 Examples 1 and 4-8 describe another process for making Iosartan that also involves a Suzuki coupling reaction. However, unlike the ’500 patent process, the ’374 patent process is not convergent. The ’374 patent process is depicted in Scheme 3.

- [0014]In the ’374 patent process, as in the `500 patent process, the tetrazole ring of 5-phenyltetrazole is protected with a trityl group before orthometallation of the phenyl moiety with n-butyl lithium in preparation for making the boronic acid Suzuki coupling partner. In the Suzuki coupling step, the boronic acid is reacted with 4-bromotoluene. The methyl group attached to one of the phenyl rings of the Suzuki product is then halogenated with N-bromosuccinamide and the benzylic bromine atom of that product is displaced with 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde. Reduction of the aldehyde group with sodium borohydride yields trityl losartan. The tetrazole group of trityl losartan was deprotected with 12% aqueous HCl in THF. After 12 hours, the pH of the reaction mixture was raised to 12.5 with 30% NaOH. The THF was then distilled off while make-up water was added to the mixture. After distillation, the mixture was cooled and the triphenyl methanol byproduct of deprotection, which had precipitated, was removed by filtration. The filtrate and rinsate, with which it was combined, were extracted with toluene. Then, ethyl acetate was added and 36% HCI was added until the pH of the reaction mixture was lowered to 3.8. The mixture was cooled, causing losartan to precipitate from the solution. Losartan was obtained in 83% theoretical yield starting from trityl losartan.
EP 253310 discloses a process, wherein 2-n-butyl-4-chloro-1H-imidazolyl-5-methanol (III) is coupled with 5-(4′-bromomethyl-1,1′-biphenyl-2-yl)-2-triphenylmethyl-2H-tetrazole (IV) in N,N-dimethylformamide as solvent in presence of sodium methoxide as the base to furnish trityl losartan. The other bases that have been claimed are sodium hydride, alkali metal carbonates such as sodium carbonate and potassium carbonate and amine bases such as triethyl amine and pyridine.

The coupling reaction results in a mixture of trityl losartan and its regio isomer (V). These are separated by column chromatography.
U.S. Pat. Nos. 5,130,439 and 5,310,928 disclose a method for coupling (IV) and (VI) in N,N-dimethylacetamide solvent in the presence of anhydrous potassium carbonate as base. The imidazole aldehyde (VI) gives predominantly the desired regio isomer (VII). The intermediate VII is then reduced with sodium borohydride to furnish the trityl losartan. The product is isolated by extraction into toluene from aqueous N,N-dimethylacetamide, concentration of the toluene solution and crystallization using ethyl acetate or ethanol as solvent. The synthesis steps are depicted as follows.

In a process published in J. Med. Chem. (1991), 34, 2525-2547, Losartan is prepared by coupling (III) and (IV) in N,N-dimethylformamide in the presence of sodium methoxide. The desired compound is isolated after vacuum distillation of solvent followed by extractive work-up. The resultant product mixture is purified by chronmatography.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 describe a process for the preparation of losartan, wherein the tetrazole ring of losartan is formed by reacting 1-((2′-cyanobiphenyl-4-yl)methyl)-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction results in trimethylstannyl substituted tetrazole compound, which is then reacted with trityl chloride and sodium hydroxide.

The trityl losartan thus formed is treated with 3.4N hydrochloric acid in methanol at about 10° C. to give losartan.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 also disclose another process for making trityl losartan, where in the coupling between IV and VI is carried out in a biphasic solvent system comprising of chlorinated solvent and water. The reaction is carried out at room temperature in presence of sodium hydroxide as the base and aliquat 336 as the phase transfer catalyst. The resulting intermediate VII is then reduced in situ with sodium borohydride to furnish trityl losartan.

U.S. Pat. No. 5,206,374, 5,310,928 and 5,962,500 disclose another process for preparing losartan in which 5-phenyltetrazole (X) is converted into the boronic acid coupling partner (XII) for the Suzuki reaction by tritylation of phenyltetrazole with trityl chloride in presence of a non-nucleophilic base, ortho metalation with n-butyl lithium, followed by reaction with triisopropylborate. 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde (VI) is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner (XIII). The product of Suzuki coupling is trityl losartan. This process is published in J. Org. Chem. (1994), 59, 6391-6394.


European patents EP 470,794 and EP 470,795 describe a method for the manufacture of biphenyl carbonitriles (XVI). These patents also describe a method of preparation of trityl losartan by coupling of intermediates (III) and (IV) employing the procedure described in EP 253,310.

Losartan potassium exhibits polymorphism. Several polymorphic forms have been prepared and characterized. The following paragraphs briefly describe various polymorphs.
U.S. Pat. No. 5,608,075 discloses the polymorphic forms of losartan, wherein the trityl losartan is deprotected with H2SO4 in 50:50 acetonitrile:water and the free acid is treated with KOH solution. The aqueous solution containing losartan potassium is added slowly to a refluxing azeotropic mixture of cyclohexane/iso propanol and the ternary azeotrope cyclohexane/iso propanol/water is distilled till the water content of the pot is less than 0.05%. The white crystalline solid thus obtained is polymorphic form-I, which is characterized by DSC, XRD and IR. Polymorphic form-II is prepared by heating form-I in a DSC cell. This process is also described in U.S. Pat. No. 5,859,258.
U.S. Pat. No. 6,710,183 discloses the synthesis of losartan potassium starting from trityl losartan, wherein trityl losartan is reacted in an alcohol of formula R—OH (where R is C1 to C4 straight chain alkyl group) with 0.1 to 1 equivalent KOH. Losartan potassium thus formed is isolated after crystallizing out by changing the solvent to an aprotic or weakly protic solvent. The alcohol used is preferably methanol and the protic dipolar solvent used for the crystallization of the final product is preferably acetonitrile or straight or branched chain or cyclic aliphatic hydrocarbons.
EP 1294712 (WO 02/094816) discloses the process to manufacture losartan potassium form-I, wherein trityl losartan or losartan is suspended in a solvent and KOH is added to obtain a clear solution, which is then concentrated under reduced pressure to remove most of the solvent. An anti solvent is added to crystallize losartan potassium. The solvents to prepare losartan potassium include methanol, ethanol, and butanol but preferably the salt formation is carried out in methanol. Anti solvent is selected from common solvents such as ethyl acetate, acetonitrile, toluene and acetone, but the preferred anti solvent is acetone.
US application 2004/0006237 (WO 03/048135) relates to novel amorphous and novel crystalline forms III, IV, V of losartan potassium and the processes for their preparation. The patent also discloses novel processes for preparing losartan potassium forms I and II. The preparation of amorphous losartan includes the step of dissolving losartan potassium in a solvent to form a solution and distilling the solvent form the solution to dryness. Losartan form III (hydrated) is obtained by exposing losartan potassium amorphous or form I to an atmosphere having high relative humidity. Losartan potassium form IV is obtained by treating a saturated solution of losartan potassium in ethanol with methylene chloride. Losartan form V is obtained by treating a saturated solution of losartan potassium in ethanol with hexane. Losartan potassium form II is obtained by adding a saturated solution of losartan potassium in ethanol to xylene to form a mixture and evaporating ethanol from the mixture. Losartan form I is obtained by treating a saturated solution of losartan potassium in ethanol or iso propanol, with less soluble solvent like ethyl acetate, toluene, acetone, methyl ethyl ketone, methylene chloride, acetonitrile, dimethyl carbonate or hexane.
US application 2004/0034077 (WO 03/093262) discloses a process for preparing losartan and losartan potassium, wherein trityl losartan is treated with an acid in a diluent comprising a ketone. Especially preferred liquid ketones are acetone, methyl ethyl ketone and methyl isobutyl ketone, and acetone being the most preferred. Acids, which have been found suitable, include hydrochloric acid, sulphuric acid, acetic acid, trifluoroacetic acid, hydrobromic acid and formic acid. After the trityl losartan has been substantially converted to losartan, reaction mixture is basified. Preferred bases are alkali metal hydroxides and alkoxides. After addition of the base, the liquid ketone is evaporated under vacuum. After separation of triaryl methyl alcohol the residue is acidified to yield losartan. Free losartan is suspended in an alcohol and treated with a solution of potassium ions. Finally losartan potassium is precipitated from the alcohol. The alcohol is selected from the group consisting of isopropyl alcohol, butyl alcohol and isobutyl alcohol. The potassium ion solution is prepared by dissolving potassium iso propoxide, potassium butoxide and potassium iso butoxide or potassium hydroxide in the diluent.
US application 2004/0097568 discloses a process for preparing form III of losartan potassium, wherein trityl losartan is treated with aqueous solution of potassium hydroxide in methanol to obtain losartan potassium. The solvent is evaporated under vacuum and traces of water are removed as an azeotrope with toluene. Methanol and carbon are added to the resulting mixture. The carbon is filtered and the methanol is distilled. The resulting mixture is cooled to 20-25° C. to obtain crystalline form III losartan potassium.
US 5,138,069 and
WO 93/10106. The advantages provided by pharmaceutical products in the crystalline form in terms of easiness of processes for the preparation of related medicaments are well known. Crystalline compounds are in fact known to be more suited to the formulation of galenic forms, thanks both to their flowability in the form of powders or granulates, and to the surface properties of the crystals which promote adhesion, for example during the preparation of tablets. Furthermore, the solubility of crystalline compounds in aqueous solutions, in particular in the gastric juices, can also be significantly different than that of the corresponding amorphous compounds. There is therefore the need to discriminate between the crystalline and the amorphous forms of biologically active compounds, so as to fulfil the various pharmaceutical requirements.
A number of crystalline and amorphous forms of losartan potassium are known from
WO 95/17396 and
WO 03/048135. According to
WO 95/17396, crystalline losartan potassium is prepared by salification of acid losartan with an alkali hydroxide. The losartan potassium aqueous solution is then added to a isopropanol-cyclohexane azeotropic mixture under reflux. Water is then removed by azeotropic distillation of the resulting water-isopropanol-cyclohexane ternary mixture, which boils at 64°C. When the solution is anhydrous, the head temperature raises to 69°C and losartan potassium crystallizes.
US 5,859,258 discloses another crystallization process which comprises dissolution of losartan potassium in isopropanol-water, distillation of the binary azeotrope to an approx. 2.6% water content, precipitation by addition of a losartan potassium suspension in cyclohexane, subsequent distillation of the ternary azeotrope to a water content ranging from 0.02 to 0.11 %, and finally drying crystalline losartan potassium under vacuum at a temperature of approx. 45-50°C.
…………………

……………………
Marcus Baumann , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK
Corresponding author email
Editor-in-Chief: J. Clayden
Beilstein J. Org. Chem. 2011, 7, 442–495.
The imidazole ring of losartan, an antihypertensive and angiotensin II blocker is formed in a condensation reaction between valeroamidine 160 and dihydroxyacetone [50]. It was found that direct chlorination of the imidazole 162also forms the dichlorination product 164 (as shown in Scheme 33) with formaldehyde as a by-product which proved difficult to suppress and made purification of the reaction mixture problematic. Hence, a sequence involving silyl protection, chlorination and deprotection was established which gave the desired product in 90% overall yield (Scheme 33).
![[1860-5397-7-57-i33]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i33.png?max-width=550&background=FFFFFF)
Scheme 33: Synthesis of functionalised imidazoles towards losartan.
Alternatively, glycine can be reacted with methyl pentanimidate 169 to form the corresponding amidine 171 in high yield. Cyclisation, followed by a Vilsmeier-type reaction then furnishes the key chloroimidazolyl building block 172in good yield (Scheme 34) [51].
![[1860-5397-7-57-i34]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i34.png?max-width=550&background=FFFFFF)
Scheme 34: Direct synthesis of the chlorinated imidazole in losartan.
- 50———Shi, Y.-J.; Frey, L. F.; Tschaen, D. M.; Verhoeven, T. R. Synth. Commun. 1993, 23, 2623–2630.doi:10.1080/00397919308012598
- 51—-Griffiths, G. J.; Hauck, M. B.; Imwinkelried, R.; Kohr, J.; Roten, C. A.; Stucky, G. C.; Gosteli, J. J. Org. Chem. 1999,64, 8084–8089. doi:10.1021/jo9824910
- 52–Zhong, Y.-L.; Lee, J.; Reamer, R. A.; Askin, D. Org. Lett. 2004, 6, 929–931. doi:10.1021/ol036423y
……………………..
NMR
NMR: (1H, DMSO, 300 mHz): δ 0.80 (3H, t, J=10. CH3), 1.25 (2H, sext, J=10. CH3CH2), 1.45 (2H, quin, J=10. CH3CH2CH2), 2.45-2.55 (2H, m, CH3CH2CH2CH2), 4.25 (2H, d, J= 3, CH2OH), 5.15-5.25 (3H, m, CH2Ar and OH), 6.88 (d, 2H, J=12, ArH), 7.08 (d, 2H, J=12, ArH), 7.23-7.36 (3H, m, ArH), 7.50-7.55 (1H, ArH).
SEACOND SET
IR v max (KBR): 3201.01, 1580.73, 1460.18, 764.81, 540.09
1H NMR (MeOD) δ, 0.87 (t, 3H), 1.33 (sext, 2H), 1.53 (quint, 2H), 2.56 (t, 2H), 4.43 (s, 2H), 5.24 (s, 2H), 6.89-7.53 (m, 8H).
13C NMR (MeOD) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.3 (M+1).
……………………………..
Melting point: 179-180.2
IR, v max (KBR): 3376.27, 1579.77, 1468.86, 762.88, 556.4
1H NMR (CDCl3) δ, 0.87 (t, 3H), 1.31 (sext, 2H), 1.54 (quint, 2H), 2.57 (t, 2H), 4.45 (s, 2H), 5.30 (s, 2H), 7.01-7.68 (m, 8H).
13C NMR (CDCl3) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.5 (M+1).
……………………………..
ADDITIONAL WRITEUP FOR READERS, NUMBERINGS ARE ALL NEW
Losartan and its potassium salt, having the formulae (1) & (2) respectively are angiotensin-II receptor (Type AT1) antagonists.

In adults Losartan is currently indicated for the treatment of hypertension (in hypertensive patients with left ventricular hypertrophy, it is also indicated to reduce the risk of stroke).
Losartan Potassium having the formula 2 and its principle active metabolite block the vasoconstrictor and aldosterone. Secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor found in many tissues (e.g., vasicular smooth muscle, adrenal gland) otherwise called as angiotensin receptor blockers (ARBs).
The present invention relates to a short, simple and practical process for the preparation of Losartan 1 which belongs to a novel class of tetrazole-imidazole compounds.
There are many processes recorded in literature. The latest prior art information for the preparation of Losartan is the disclosure made in the patent application of Novartis in their PCT WO 2005/014602 dated 17 Feb. 2005.
The process described in the application comprises the reaction of 4′-(Bromomethyl)-2-cyanobiphenyl (BromoOTBN) of the formula 3 with 2-n-butyl-4-chloro-5-formyl imidazole (BCFI) of the formula of 4 in the presence of Potassium carbonate and acetonitrile to give ‘cyano aldehyde’ of the formula 5. The Cyano aldehyde of the formula 5 is reduced with sodium borohydride to get ‘cyano alcohol’ of the formula 6. The Cyano alcohol is reacted with diethyl aluminium azide in the presence of triethyl aluminium to give Losartan of the formula 1.
The reaction scheme of the process is shown in the Scheme 1

Even though the process is simple, handling of triethyl aluminium used needs special attention like very anhydrous conditions, reactions are to be performed under nitrogen or argon and transferring of triethyl aluminium from the containers needs anhydrous systems. The neat liquid and dense solutions of triethyl aluminium are known to ignite very easily at room temperature in presence of air (Pyrophoric). So handling of both triethyl aluminium and diethyl aluminium needs special attention like anhydrous conditions, nitrogen atmosphere etc.,
In EP 0578125A1 of Takeda Chemical Industries dated 12 Jan. 1994, yet another method for the preparation of Losartan has been disclosed in which Trioctadecyl or Trioctyl tin azide has been used as a tetrazole-forming agent. This method also uses the Cyano alcohol of the formula (6). The process comprises reacting the cyano alcohol of the formula (6) with tri-n-octyl tin azide in presence of toluene to give tri-n-octyl tetrazole derivative, which was treated with nitrous acid to give Losartan of the formula (1) in 94.7% yield. The process is shown in the reaction scheme 2

Even though the yields are better (94.7%) in this process again handling of tri-n-octyl tin azide is involved.
Dupont/Merck in their patents and papers always described that trityl Losartan of the formula 7 is detritylated to get Losartan 1 For example they described in J. Med. Chem., 1991, 34, 2525-2547, the preparation of Losartan of the formula 1, from trityl Losartan of the formula 7 using mineral acids such as Hydrochloric acid and sulfuric acid in 93% yield. The reaction scheme of the process is shown in the scheme 3

In this paper ‘Aldehyde Tetrazole’ of the formula 8 is isolated from trityl tetrazole aldehyde of the formula 21 and were further used for preparing derivatives of aldehyde such as benzene sulfonyl hydrazones of the formula 9 but not for Losartan. This process is shown in the scheme 4

In J. Org. Chem 1994, 59, 6391-6394 again by Merck team reported Trityl Losartan and Losartan synthesis by coupling of boronic acid derivative 11 with 3-(4-bromobenzyl) derivative of BCBMI of the formula 10. The formed trityl Losartan of the formula 7 is converted to Losartan of the formula 1 with acid. The whole process is described in Scheme 5

The Compound of the formula 10 is prepared from the reaction of BCFI of the formula 4 with p-bromo benzyl bromide of the formula 12 in potassium carbonate and Dimethyl formamide followed by reduction with sodium borohydride (NaBH4). The details are given in the Scheme 6

The Compound of the formula 11 is prepared from 5-phenyl tetrazole of the formula 14 by reacting with trityl chloride to get N-trityl-5-phenyl tetrazole of the formula 13, which on reaction with butyl lithium and triisopropyl borate followed by hydrolysis to give compound of the formula 11. This process is shown in the Scheme 7

In one of the first patent filed by Dupont/Merck (date of filing 9 Jul. 1987, priority 11 January 1986 EP0253310) reported a procedure for the preparation of Losartan. Bromo OTBN of the formula 3 is reacted with BCHMI of the formula 15 in the presence of a base to give cyano alcohol of the formula 6, and its regioisomer of the formula 14. Separation of the isomer needs column chromatography. The cyano alcohol 6 is reacted with sodium/ammonium azide in DMF for 13 days to get Losartan 1 in 21% yield. The process is shown in the Scheme 8

The drawbacks of the above process are
- 1). Separation of the regioisomer using column chromatography which is industrially not feasible for the preparation of large scale (ton) material/product
- 2). The tetrazole formation takes 13 days with 21% yield, which is unproductive.
- 3). Dupont/Merck uses BCHMI 15 as the starting material for preparing cyano alcohol of the formula 6. BCHMI 15 is an expensive intermediate compared to BCFI 4, and also the formation of unwanted regio isomer 14 is higher. The process is schematically described in scheme 8. Even though the process looks simple it has two problems.
First: Cyano alcohol is produced as a mixture of regioisomers and needs column chromatography for purification.
Second: Tetrazole formation. This takes 13 days with 21% yield, which limits commercialization of the process.
In U.S. Pat. No. 4,820,843 and U.S. Pat. No. 4,879,186, Dupont prepares Losartan by reaction of BCFI of the formula 4 and N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 in the presence of base, followed by reduction with sodium borohydride to give Trityl Losartan of the formula 7, which is treated with mineral acid to give Losartan 1.
The process is shown in scheme 9

In U.S. Pat. No. 4,874,867 of Dupont/Merck, a process for the preparation of N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 is described by the reaction of OTBN of the formula 20 with trimethyl tin azide to give the compound 17, which is treated with Hydrochloric acid to give tetrazole derivative of OTBN of the formula 18. The tetrazole derivative of OTBN of the formula 18 is protected with trityl chloride to give compound of the formula 19, followed by bromination with N-bromosuccinimide to give N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16.
The process is shown in the scheme 10.

In all the above papers and patents by Dupont/Merck, the process yields in many steps are good 75-95% and in some steps are less to moderate 21-49%. The drawbacks, or the problems in all these processes is, the number of unit operations.
For example:
- 1). In J. Med. Chem 1991, 34, 2525-2547 the number of steps are six (6) to prepare Losartan of the formula 1 from the readily available intermediates.
- 2). In J. Org. Chem 1994, 59, 6391-6394 the number of steps are nine (9) to prepare Losartan of the formula 1 from the readily available intermediates.
- 3). In EP 0253310 patent the number of operations are two (2) but the problem is time & yields i.e., 13 days and poor yield (21%), also the uneconomical column chromatographic separation of regioisomer.
- 4). In U.S. Pat. Nos. 4,820,843 and 4,879,186 the number of steps are six (6).
- 5). In U.S. Pat. No. 4,874,867 the number of steps are seven (7).
………………………..
INTERMEDIATES
(1-(2′-Cyano biphenyl-4-methyl)-2-butyl-4-chloro-5-formyl imidazole) of the formula 5.

Melting point: 107-108° C.
HPLC Purity: >98%
IR. v max (KBR): 2218 (—CN), 1662.40 (—CHO)
1H NMR (CDCl3) δ, 0.91 (t, 3H), 1.38 (sext, 2H), 1.73 (quint, 2H), 2.67 (t, 2H), 5.61 (s, 2H), 7.16-7.77 (m, 8H), 9.77 (s, 1H).
13C NMR (CDCl3) δ, 13.51, 22.18, 26.33, 29.04, 47.74, 110.05, 118.36, 124.11, 126.59, 127.65, 129.16, 129.81, 132.76, 133.61, 136.01, 137.69, 142.96, 144.33, 154.46, 177.73

2 IRBESARTAN
IRBESARTAN
IRBESARTAN, SR 47436, BMS-186295
Avapro® (Bristol-Myers Squibb) and Karvea®
(Sanofi-Winthrop)
(Sanofi-Winthrop)
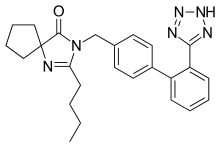
2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one
138402-11-6 CAS NO
U.S. Patents 5,270,317 and 5,352,788, 6,162,922
The compound prepared according to US 5270317 is polymorph A
- Irbesartan is known by following chemical names:
- (a) 2-Butyl-3-[[2'-(1H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (b) 2-Butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (c) 2-n-butyl-4-spirocyclopentane-1-[(2'-(tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one.
- The structural formula of Irbesartan is represented below.
 Irbesartan
Irbesartan - The synthesis of irbesartan is first disclosed in US5270317 (equivalentEP0454511 ) and subsequently, several other patents disclose the synthesis of irbesartan by different methods. Basically the synthesis of this molecule involves two common intermediates namely spiroimidazole and substituted 4′-bromomethylbiphenyl.
- US 5270317 describes preparation of irbesartan wherein 1-[(2'-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin -5-one which is reacted with tributyltin azide in xylene at reflux temperature for 66 hours to give a product which is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol/THF mixture using 4N hydrochloric acid to get irbesartan.
- US5629331 describes a process for the preparation of irbesartan from 1-[(2'-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
Irbesartan (INN) /ɜrbəˈsɑrtən/ is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under thetrade names Aprovel, Karvea, and Avapro.
It is marketed in Brazil by Sanofi-Aventis under the trade name Aprovel .
As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
Irbesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Irbesartan/hydrochlorothiazide combination preparations are marketed under similar trade names to irbesartan preparations, including Irda, CoIrda, CoAprovel, Karvezide,Avalide and Avapro HCT.
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to placebo.[3]

BMS annual sales approx $1.3bn. Sanofi-aventis annual sales approx $2.1bn. In the United States, a generic version is available. Patent expired March 2012.
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. (2001). “Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes”. N Engl J Med 345 (12): 851–60. doi:10.1056/NEJMoa011303.PMID 11565517.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A (December 2008). “Irbesartan in patients with heart failure and preserved ejection fraction”. N. Engl. J. Med. 359 (23): 2456–67.doi:10.1056/NEJMoa0805450. PMID 19001508.
4……….C. A. Bernhart, P. M. Perreaut, B. P. Ferrari, Y. A. Muneaux,
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5…. K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6… C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5…. K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6… C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
Irbesartan of formula (I).

The chemical name of Irbesartan is 2-Butyl-3-[[2'-(lH-tetrazol-5-yl)[l,l'-biphenyl]-4- yl]methyl]-l,3-diazaspiro[4,4]non-l-en-4-one and formula is C2SH2SN6O and molecular weight is 428.53. The current pharmaceutical product containing this drug is being sold by Sanofi Synthelabo using the tradename AVAPRO, in the form of tablets. Irbesartan is useful in the treatment of diabetic neuropathy, heart failure therapy and hypertension. Irbesartan is angiotension II type I (AΙIi)-receptor antagonist. Angiotension II is the principal pressor agent of the rennin-angiotension system and also stimulates aldosterone synthesis and secretion by adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system and smooth muscle cell growth. Irbesartan blocks the vasoconstrictor and aldosterone- secreting effects of angiotension II by selectively binding to the ATi angiotension II receptor. U.S. Pat. Nos. 5,270,317 and 5,559,233 describes a process for the preparation of N- substituted heterocyclic derivatives which involves reacting a heterocyclic compound of the formula

with a (biphenyl-4-yl)methyl derivative of the formula

wherein R1, R2, R3, R4, R5, and t, z and Hal have the meanings given in said U.S. Pat. No.
5,270,317, in the presence of an inert solvent such as DMF, DMSO or THF, with a basic reagent, for example KOH, a metal alcoholate, a metal hydride, calcium carbonate or triethylamine. The products of the reaction were purified by chromatography.
U.S. Pat. Nos. 5,352,788, and 5,559,233, and WO 91/14679 also describe identical alkylation of the nitrogen atom of the heterocyclic compound with the halo-biphenyl compound using the same inert solvent and the same basic reagents.
- US5629331 describes a process for the preparation of irbesartan from 1-[(2'-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
- WO 2005/051943 A1 describes a process for the preparing irbesartan wherein 1-[(2'-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with tributyltin chloride, sodium azide and TBAB in toluene at reflux temperature for 20 hours. Product is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol and formic acid to get irbesartan.
- WO 2006/023889 describes a method for preparing irbesartan, wherein 1-(2′-cyanobiphenyl-4-yl)methyl)-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with sodium azide and triethylamine hydrochloride in N-methyl-2-pyrrolidone to give irbesartan.
- WO 2005/113518 describes a process for preparing irbesartan wherein cyano irbesartan in xylene, is reacted with tributyltin chloride and sodium azide at reflux temperature till reaction is completed followed by aqueous work-up and recrystallization to give irbesartaN
- The process involving use of zinc salt for the transformation of nitrile to tetrazole is a safe and efficient process as reported in JOC (2001) 66, 7945-50. The use of zinc salt for transforming nitrile to tetrazole has also been published in WO9637481 and US5502191
Also Canadian Patent No. 2050769 describes the alkylation of the nitrogen atom of the heterocycle of the formula
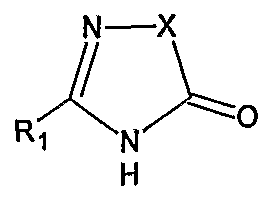
with a compound of the formula

wherein X, R1, Z1 and Z6 have the meanings given therein, in the presence of N,N- dimethylformamide and a basic reagent, such as alkali metal hydrides for example sodium or potassium hydride.
All of the above identified patents describe alkylation in solvents, such as N5N- dimethylformamide or DMSO, etc. in the presence of a basic reagent, for example, a metal hydride or a metal alcoholate etc. The strong bases, such as metal hydride or a metal alcoholate require anhydrous reaction conditions. Since N,N-dimethylformamide is used as a solvent, its removal requires high temperature concentration by distillation, which can result in degradation of the final product. The product intermediate is also purified by chromatography which is commercially not feasible and cumbersome on large scale. Another process given in Canadian Patent No. 2050769 provides synthetic scheme as herein given below.

This process comprises the steps of protecting carboxylic group present on cyclopentane ring which is deprotected in consecutive step by vigourous hydrogenation condition in autoclave which is operationally difficult at a large scale.
US Patent No. 2004242894 also discloses the process of preparation of lrbesartan from 4- bromomethyl biphenyl 2′-(lH-tetrazol (2-triphenylmethyl) 5-yl) and Ethyl ester of 1- Valeramido cyclopentanecarboxylic acid in toluene in presence of base and PTC, and then hydrolyzing the protecting group. However this requires chromatographic purification.
This patent also discloses the process of preparation of tetrazolyl protected lrbesartan using 2,6 lutidine and oxalylchloride in toluene. However in this process the yield is as low as 30%.
US Patent No. 2004192713 discloses the process of preparation of lrbesartan by condensing the two intermediates via Suzuki coupling reaction. The reaction scheme is as given herein below.

However, this process has several disadvantages such as use of the reagents like butyl lithium and triisobutyl borate at low temp such as -20 to -30°C under Argon atmosphere condition which is difficult to maintain at commercial scale.
WO2005113518 discloses the process of preparation of Irbesartan by condensing n- pentanoyl cycloleucine (V) with 2-(4-aminomethyl phenyl) benzonitrile (VI) using dicyclocarbodiimide (DCC) and 1 -hydroxy benzotriazole as catalyst to give an open chain intermediate of formula (VIII) which is then cyclized in the presence of an acid, preferably trifluoro acetic acid to give cyano derivative of formula (VII) and which in turn is converted to Irbesartan by treating it with tributyl tin chloride and sodium azide.

In this application further describes another process comprising the steps of reacting 2- butyl-l,3-diazasρiro[4,4]non-l-en-4-one monohydrochloride (A) with 4-bromobenzyl bromide (B) in presence of base and solvent to give 3-[4-bromobenzyl]-2-butyl-l,3- diazaspiro[4,4]non-l-en-4-one (C) which is condensed with 2-[2'-(triphenylmethyl-2'H- tetrazol-5'-yl)phenyl boronic acid in the presence of tetrakis triphenyl phosphine palladium and base to give lrbesartan (I). However these processes suffer with several disadvantages such as it uses trifluoroacetic acid for the cyclization step which is highly corrosive material. The process requires an additional step of activation by DCC. This step not only increases number of steps but also create problem in handling DCC at an industrial scale as it is highly prone to hazard which makes the process least preferred on a large scale production of lrbesartan. Further it uses phenyl boronic acid derivative and triphenyl phosphine complex which are harmful for the skin and eye tissue and also harmful for respiratory system. Tetrakis triphenyl phosphine palladium is also a costly material which increases overall cost for the production of lrbesartan. Moreover the yield is as low as 22%. All the above patents/applications are incorporated herein as reference. In summary, prior art relating to the process for the preparation of lrbesartan suffers with several drawbacks such as i) It requires chromatographic purification of intermediates at various stages. ii) It requires specific autoclave conditions for a deprotection of protecting group. iii) It requires maintaining low temperature conditions such as -300C and requires special handling care and air and moisture tight condition with the reagents such as butyl lithium and triisobutyl borate. iv) It uses hazardous and highly corrosive reagents, v) It suffers low yield problem. vi) All the process is having more number of reaction steps.
- Irbesartan is described in Bernhart et al., U.S. Patent No. 5,270,317
- Irbesartan, is a potent, long-acting angiotensin II receptor antagonist which is particularly useful in the treatment of cardiovascular ailments such as hypertension and heart failure. Its chemical name is2-n-butyl-4-spirocyclopentane-1-[(2'-(tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one.
Irbesartan is an antihypertensive agent known from EP 454511. From EP 708103, which discloses their X-ray spectra, two polymorphs are known where form A can be produced form a solvent system containing less than 10% of water, while Form B from a system with more than 10% of water. The specific morphological variant of form A can be prepared having properties as disclosed in EP 1089994. Additional form has been disclosed in WO 04089938. Amorphous irbesartan is known from WO 03050110. It is said that Irbesartan produced as taught in EP 454511 is a fluffy material with relatively low bulk and tap densities and undesirable flow characteristics, which consequently has unadvantageous electrostatic properties, among them a high chargeability as measured by tribugeneration between -30 and -40 nanocoulomb/g (10’9As/g). Alternativelyirbesartan could be prepared by complex process using sonifications and/or temperature oscillations according to EP 1089994 to exhibit a chargeability as measured by tribugeneration between -0 and -10 nanocoulomb/g.
According to EP 454511 a solid composition in form of tablets is prepared by mixing the active ingredient with a vehicle such as gelatine, starch, lactose, magnesium stearate, talc, gum Arabic or the like and can be optionally coated. The compositions containing from 20% to 70% by weight of irbesartan are known from EP 747050.
WO 04/007482 teaches the acidification to pH 2 – 3,5 of trityl irbesartan, which is sufficient to remove the protecting group, but not to convert into an acid addition salt; WO 04/065383 is likewise silent on hydrohalide acid addition salts. WO
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
……………………………………………
…………………

Example 1Preparation of Compounds of formula IVa and IVb:

- A jacketed 1,000 mL 3-neck flask was charged with 4′-methylbiphenyl-2-carbonitrile (Compound 1, 100.0 g) and CH2CI2 (500 mL) under nitrogen. To a 500 mL Erlenmeyer flask with magnetic stirrer, sodium bromate (NaBrO3; 31.2 g) was dissolved in water (170 mL). The NaBrO3 solution was transferred to the 1,000 mL flask and the reaction mixture was cooled to about 5 °C or less. Aqueous HBr solution (48 %, 105.0 g) was added to the 1,000 mL flask and the resulting reaction mixture was recycled though a UV lamp reactor. The reaction mixture was kept at 0-20 °C and the recycling was continued until the reaction was deemed complete by HPLC. Optionally, additional sodium bromate and hydrogen bromide may be added. The relative amounts of Compound 2 and Compound 3 were about 80-90% and about 10-20% respectively. Aqueous sodium metabisulfite solution (2.0 g of in 10 mL water) was added to the reaction mixture. Allow the phases to settle and the methylene chloride phase was washed with water and used in the next step without further purification.
Example 2Preparation of Compound II:

- A 1L 3-neck flask was charged with Compound V (134.0 g), MTBAC (5.0 g) and CH2Cl2 (170 mL) and cool to -5 to 5 °C. An aqueous solution of KOH (182.6 g in 212 mL water) was added slowly to the 1L flask and the reaction temperature was kept at ≤ 5 °C. The methylene chloride solution of Compound IVa and Compound IVb from Example 1 was added to the reaction mixture slowly, while maintaining the temperature at 0-10 °C. Diethyl phosphite (39.66g) was added drop wise at 0-10 °C. Check the reaction mixture for completion of the reduction reaction, and additional diethyl phosphite may be added.
- The reaction mixture was allowed to warm to ambient (20-30 °C) and agitated until the reaction was deemed complete by HPLC. Water (150 mL) was added and the phases were separated. The organic layer was extracted with water (230 mL) and polish filtered.
- The methylene chloride (which contained the crude Compound II) was distilled off and exchanged with about 400 mL of methyl tert-butyl ether (MTBE) (optionally, the MTBE recycled from washing below can be used here). Upon cooling, crystallization occurred (optionally seeds were added) and after further cooling to below 25°C, crystals of Compound II were isolated, washed with MTBE and dried in vacuum at a temperature of less than 60°C. HPLC retention time: 18.126 min. Typically, the yield was about 85 to about 88%. Alternatively, IPA could be used as the crystallization and washing solvent
- Optionally, the solvent (i.e., MTBE or IPA) used to wash the crystals of Compound II above can be recycled and used to crystallize the crude Compound II in the next batch. Since the washed solvent contains Compound II as well as impurities, it was surprisingly found that the washed solvent can be recovered and used again in crystallizing the crude compound of formula II in the next batch without sacrificing its purity while increasing its yield.
Example 3Preparation of Compound I:

- A reactor was charged with Compound II (1 kg), triethylamine chlorhydrate (0.713 kg), sodium azide (0.337 kg) and N-methyl pyrrolidinone (2.07 kg), and the reaction mixture was heated to about 122°C under stirring. After completion of the reaction as determined by HPLC, the reaction mixture was cooled to about 45°C, and an aqueous solution of sodium hydroxide (35%, 5.99 kg) and water (3.0 kg) were added, the resulting mixture was stirred at a temperature between about 20 and about 40°C for about 0.5 hours. The aqueous phase was discarded and the organic phase was treated with toluene (1.73 kg) and water (5.0 kg), and stirred for about 0.5 hours at about 20 – about 30°C. The toluene phase was discarded and the aqueous phase was washed with ethyl acetate (1.8 kg) and treated with aqueous HCl until pH was adjusted to about 4.8 – about 5.2. Precipitation occurred and the resulting suspension was stirred for about 1 hour at about 20 – about 25°C. The precipitation was collected and washed with water three times (1.0 kg x 3). The crude wet product was recrystallized using a mixture of iso-propanol (0.393 kg) and water (4.5 kg). HPLC retention time: 11.725 min. The yield for Compound I was about 87%.
…………………………………………….
SPECTRAL DATA
The ESI mass spectrum of irbesartan showed a protonated molecular ion peak at m/z 429.3 confirming the molecular weight 428. The fragmentation pattern of parent ion 429.3 showed the fragment ions at m/z 385.9, 235.1, 207, 195.4, 192.1, 180.2 and 84
The FT-IR spectrum exhibited a characteristic stretching absorption band at 1732 cm-1 for the carbonyl group of amide functionality. The presence of this band at higher frequency was due to the ring stretching due to five member ring system. Another band at 1614cm-1 was due to C=N stretching vibrations
1H and 13C- NMR were recorded using DMSO-d6 as a solvent. In 1H-NMR the signal due to tetrazole NH proton was not detected may probably due to the tautomerism.
SEE
DP 1 IS IMPURITY
………………………………………….
NMR
1H-NMR (DMSO d6): δppm 0.78 (t, 3H); 1.17-1.30 (sex, 2H); 1.40-1.50 (quent, 2H); 1.64-1.66 (m, 2H); 1.80-1.82 (m, 6H); 2.22-2.29 (t, 2H); 4.67 (s, 2H); 7.07 (s, 4H); 7.50- 7.68 (m, 4H) M+: 429.6
,…………………..
m.p:181-182oC,
IR (KBr, cm-1) 1732 (C=O), 1616 (C=N); 1H NMR (DMSO-d6): δ 7.95–7.32 (m, 8 H), 4.80 –4.60 (s, 2 H), 3.60– 3.00 (br s, 1 H), 2.40– 2.20 (t, 2 H , J = 6.04 Hz), 2.00– 1.60 (m, 8 H),1.60–1.45 (quint, 2 H), 1.40– 1.20 (sext, 2 H), 0.91–0.70 (t, 3H, J = 7.41 Hz);
13C-NMR (DMSOd6): δ 186.5, 162.0,155.9, 141.9, 139.2, 137.2. 131.9, 131.4, 130.1, 128.7, 127.1, 124.3, 76.7, 43.1,
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
MS: m/z= 429 [M+1];
Anal. Calcd for C25H28N6O : C, 70.07; H,
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
………………………………………………..
1H NMR in DMSO-D6 : 7.68 (d. 2H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.08 (s, 4 H, Ar-H), 4.68(s, 2H, -CH2), 2.69(t,2H,-CH2),2.18(m,2H,-CH2),1.83(m,2H,-CH2),1.81 (t, 2H, -CH2), 1.65 (t, 2H, -CH2), 1.45 (m, 2 H, -CH2), 1.24(m , 2H, -CH2), 0.77 (t, 3H, -CH3),
IR (KBR): 3061 (Aromatic C-H stretching), 2960 (Aliphatic C-H stretching), 3443 (N-H stretching), 1733 (C=0 stretching), 1617(CN stretching), 1337.99(CN stretching), 1407(N=N stretching) cm“1.
……………………….
HPLC condition:
Column: Alltima C18 (Alltech 88050) 15.0cm in length x 4.6mm in internal diameter and 5 micron particle size;
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
The chromatographic purity of
the compounds was analyzed using Agilent 1200 series HPLC instrument under the following conditions:
Column : Symmetry C18, 4.6 × 75 mm, 3.5 µm
Mobile phase : Eluent A: Deionized water, Eluent B: HPLC grade Methanol
Chromatographic Conditions
a. Column temperature : Ambient
b. Sample compartment : Ambient
c. Detector : 225 nm
d. Injection volume : 10 µL
e. Run time : 45 minutes
f. Flow rate :1.0 mL/min
g. Injector :Auto sampler with variable volume injector
h. Diluent : HPLC grade Acetonitrile
............................................................................................................
3 VALSARTAN
VALSARTAN
Greening the Valsartan Synthesis: Scale-up of Key Suzuki–Miyaura Coupling over SiliaCat DPP-Pd
† SiliCycle Inc., 2500 Parc-Technologique Blvd, Quebec City, Quebec, Canada G1P 4S6
‡ Istituto per lo Studio dei Materiali Nanostrutturati, CNR, via U. La Malfa 153, 90146 Palermo, Italy
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op400118f
Publication Date (Web): June 17, 2013

The study of the scale-up of the heterogeneous Suzuki-Miyaura coupling reaction in batch conditions between 2-chlorobenzonitrile and 4-tolylboronic acid, a key step in valsartansynthesis, to produce 4′-methyl-2-biphenylcarbonitrile over the SiliaCat DPP-Pd catalyst in ethanol under reflux allows to identify the optimal reaction conditions.
The catalyst, regardless of limited Pd leaching, is not reusable, and the method can be effectively applied to the high yield synthesis of several coupling products, opening the route to efficient continuous coupling syntheses.
ABOUT VALSARTAN
Valsartan (Angiotan or Diovan) is an angiotensin II receptor antagonist (more commonly called an “ARB”, or angiotensin receptor blocker), with particularly high affinity for the type I (AT1) angiotensin receptor. By blocking the action of angiotensin, valsartan dilates blood vessels and reduces blood pressure.[1] In the U.S., valsartan is indicated for treatment ofhigh blood pressure, congestive heart failure (CHF), or post-myocardial infarction (MI).[2] In 2005, Valsartan was prescribed more than 12 million times in the United States[citation needed] and global sales were approximately $6.1 billion in 2010.[3] The patents for valsartan and valsartan/hydrochlorothiazide expired in September 2012.[4][5]
A study released in 2010, based on 819,491 cases in U.S. Department of Veterans Affairs database from 2002 to 2006, demonstrated a significant reduction in the incidence and progression of Alzheimer’s disease and dementia.[6] An earlier study released by theJournal of Clinical Investigation in 2007 found some efficacy in the use of valsartan in the treatment and prevention of Alzheimer’s disease (in a mouse model).[7]
Valsartan, also known as (S)—N-(1-Carboxy-2-methyl-prop-1-yl)-N-pentanoyl-N-[2′-(1H-tetrazol-5-yl)bi phenyl-4-ylmethyl]-amine, has the following structure:

and is marketed as the free acid under the name DIOVAN. DIOVAN is prescribed as oral tablets in dosages of 40 mg, 80 mg, 160 mg and 320 mg of valsartan.
Valsartan and/or its intermediates are disclosed in various references, including: U.S. Pat. Nos. 5,399,578, 5,965,592, 5,260,325, 6,271,375, WO 02/006253, WO 01/082858, WO 99/67231, WO 97/30036, Peter Bühlmayer, et. al., Bioorgan. & Med. Chem. Let., 4(1) 29–34 (1994), Th. Moenius, et. al., J. Labelled Cpd. Radiopharm., 43(13) 1245–1252 (2000), and Qingzhong Jia, et. al., Zhongguo Yiyao Gongye Zazhi, 32(9) 385–387 (2001).
Valsartan is an orally active specific angiotensin II antagonist acting on the AT1 receptor subtype. Valsartan is prescribed for the treatment of hypertension. U.S. Pat. No. 6,395,728 is directed to use of valsartan for treatment of diabetes related hypertension. U.S. Pat. Nos. 6,465,502 and 6,485,745 are directed to treatment of lung cancer with valsartan. U.S. Pat. No. 6,294,197 is directed to solid oral dosage forms of valsartan.
The synthesis of valsartan is discussed, inter alia, in U.S. Pat. No. 5,399,578. In the synthesis disclosed therein, the final synthetic step (exclusive of work-up and purification) involves the reaction of a cyano group on the biphenyl ring with an azide, for example, tributyl tin azide. The reaction scheme of the ’578 patent is as follows:

Peter Bühlmayer, et. al., Bioorgan. & Med. Chem. Let., 4(1) 29–34 (1994)
In Moenius, et. al., J. Labelled Cpd. Radiopharm., 43(13) 1245–1252 (2000), various schemes for synthesis of valsartan are provided, with one being:

Another paper, Qingzhong Jia, et. al., Zhongguo Yiyao Gongye Zazhi, 32(9) 385–387 (2001), discloses a synthesis scheme for valsartan as follows:

There is a need in the art for an improved synthetic process for the preparation of valsartan and precursors of valsartan.
DOSE
Oral tablets, containing 40 mg (scored), 80 mg, 160 mg, or 320 mg of valsartan. Usual dosage ranges from 40–320 mg daily.
In some markets available as a hard gelatin capsule, containing 40 mg, 80 mg, or 160 mg of valsartan.
Diovan HCT contains a combination of valsartan and hydrochlorothiazide but, unlike Diovan, is only indicated for hypertension, not for CHF or post-MI. Diovan HCT is available in oral tablets, containing (valsartan/HCTZ mg) 80/12.5, 160/12.5, 160/25, 320/12.5, and 320/25.
Whether angiotensin receptor blockers may or may not increase the risk of myocardial infarction (heart attack) was announced in BMJ[8] and was debated in 2006 in the medical journal of the American Heart Association.[9][10] To date[when?], there is no consensus on whether ARBs have a tendency to increase MI, but there is also no substantive evidence to indicate that ARBs are able to reduce MI.
In the VALUE trial, the angiotensin II receptor blocker valsartan produced a statistically significant 19% (p=0.02) relative increase in the prespecified secondary end point of myocardial infarction (fatal and non-fatal) compared with amlodipine.[11]
The CHARM-alternative trial showed a significant +52% (p=0.025) increase in myocardial infarction with candesartan (versus placebo) despite a reduction in blood pressure.[12]
Indeed, as a consequence of AT1 blockade, ARBs increase Angiotensin II levels several-fold above baseline by uncoupling a negative-feedback loop. Increased levels of circulating Angiotensin II result in unopposed stimulation of the AT2 receptors, which are, in addition upregulated. Unfortunately, recent data suggest that AT2 receptor stimulation may be less beneficial than previously proposed and may even be harmful under certain circumstances through mediation of growth promotion, fibrosis, and hypertrophy, as well as proatherogenic and proinflammatory effects.[13][14][15]
In patients with impaired glucose tolerance, valsartan may decrease the incidence of developing diabetes mellitus type 2.[16] However, the absolute risk reduction is small (less than 1 percent per year) and diet, exercise or other drugs, may be more protective. In the same study, no reduction in the rate of cardiovascular events (including death) was shown.
There is a case report of a stillbirth in which valsartan is implicated.[18]In the US, UK and Australia, valsartan is marketed by Novartis under the trade name Diovan. In Pakistan, it is marketed by Efroze under the trade name Angiotan. In India, it is marketed by Cipla under the trade name Valtan and by Torrent Pharmaceuticals under the trade name Valzaar. In Egypt and in France, it is marketed by Novartis under the name of Tareg. In Ukraine, it is marketed by Фарма Старт under the trade name Диокор, Диокор Соло
- Marks JW (2007-02-15). “Valsartan, Diovan”. MedicineNet. Retrieved 2010-03-04.
- “Diovan prescribing information”. Novartis.
- J “Novartis Annual Report”. Novartis. 2010. Retrieved June 15, 2011.
- Philip Moeller (April 29, 2011). “Blockbuster Drugs That Will Go Generic Soon”. U.S.News & World Report.
- Eva Von Schaper (August 5, 2011). “Novartis’s Jimenez Has Blockbuster Plans For Diovan After Patent Expires”. Bloomberg.
- Li NC, Lee A, Whitmer RA, et al. (January 2010). “Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis”. BMJ 340: b5465. doi:10.1136/bmj.b5465.PMC 2806632. PMID 20068258.
- Wang J, Ho L, Chen L, et al. (November 2007). “Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease” (PDF). J. Clin. Invest. 117 (11): 3393–402. doi:10.1172/JCI31547.PMC 2040315. PMID 17965777. Retrieved 2009-11-11.
- Verma S, Strauss M (November 2004). “Angiotensin receptor blockers and myocardial infarction: These drugs may increase myocardial infarction—and patients may need to be told”. BMJ329 (7477): 1248–9. doi:10.1136/bmj.329.7477.1248.PMC 534428. PMID 15564232.
- Strauss MH, Hall AS (August 2006). “Angiotensin receptor blockers may increase risk of myocardial infarction: unraveling the ARB-MI paradox”. Circulation 114 (8): 838–54.doi:10.1161/CIRCULATIONAHA.105.594986.PMID 16923768.
- Tsuyuki RT, McDonald MA (August 2006). “Angiotensin receptor blockers do not increase risk of myocardial infarction”. Circulation 114 (8): 855–60.doi:10.1161/CIRCULATIONAHA.105.594978.PMID 16923769.
- Julius S, Kjeldsen SE, Weber M, et al. (June 2004). “Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial”. The Lancet 363 (9426): 2022–31.doi:10.1016/S0140-6736(04)16451-9. PMID 15207952.
- Granger CB, McMurray JJ, Yusuf S, et al. (September 2003). “Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial”. The Lancet 362 (9386): 772–6.doi:10.1016/S0140-6736(03)14284-5. PMID 13678870.
- Levy BI (September 2005). “How to explain the differences between renin angiotensin system modulators”. Am. J. Hypertens. 18 (9 Pt 2): 134S–141S.doi:10.1016/j.amjhyper.2005.05.005. PMID 16125050.
- Levy BI (January 2004). “Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system”. Circulation 109 (1): 8–13.doi:10.1161/01.CIR.0000096609.73772.C5.PMID 14707017.
- Reudelhuber TL (December 2005). “The continuing saga of the AT2 receptor: a case of the good, the bad, and the innocuous”. Hypertension 46 (6): 1261–2.doi:10.1161/01.HYP.0000193498.07087.83.PMID 16286568.
- McMurray JJ, Holman RR, Haffner SM, et al. (April 2010).“Effect of valsartan on the incidence of diabetes and cardiovascular events” (PDF). The New England Journal of Medicine 362 (16): 1477–90. doi:10.1056/NEJMoa1001121.PMID 20228403.
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag.ISBN 3-85200-196-X.
- Briggs GG, Nageotte MP (2001). “Fatal fetal outcome with the combined use of valsartan and atenolol”. The Annals of Pharmacotherapy 35 (7–8): 859–61. doi:10.1345/aph.1A013.PMID 11485133.
.................................................................................................
4 CANDESARTAN

Candesartan cilexetil Candesartan cilexetil, Candesartan hexetil, H212/91, TCV-116, Kenzen, Blopress 16 mg Plus, Parapres, Ratacand, Blopress, Amias, Atacand
ATACAND
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT1 subtype angiotensin II receptor antagonist. Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1-[p-(o-1H-tetrazol-5ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester). Its empirical formula is C33H34N6O6, and its structural formula is:
 |
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral. ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant. 
Drug Patent Expiration and Exclusivity
| ACTIVE INGREDIENT | FORM | DOSAGE | DRUG TYPE | APPLICATION | PRODUCT | |
|---|---|---|---|---|---|---|
| CANDESARTAN CILEXETIL | TABLET; ORAL | 4MG | RX | 020838 | 001 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 8MG | RX | 020838 | 002 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 16MG | RX | 020838 | 003 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 32MG | RX | 020838 | 004 |
Patents
There are 6 patent(s) protecting ASTRAZENECA’s ATACAND. The last patent 5534534*PED expires on 2014-01-09.View patent at USPTO
| PATENT US | US | EXPIRATION |
|---|---|---|
| 5534534*PED | 2014-1-9 | |
| 5534534 | Pharmaceutical compositions for oral use and method of preparing them
A pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) having antagonistic action to angiotensin II ##STR1## (wherein the ring W is an optionally substituted N-containing heterocyclic residue; R.sup.3 is a group capable of forming an anion or a group convertible thereinto; X is a direct bond or a spacer having an atomic length of two or less between the phenylene group and the phenyl group; and n is an integer of 1 or 2) and an oily substance having a lower melting point, and a method for preparing a pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) and an oily substance having a lower melting point, which comprises admixing the compound of the formula (I) with an oily substance having a lower melting point and then subjecting the mixture to molding.
| 2013-7-9(expired) |
| 5196444*PED | 2012-12-4(expired) | |
| 5196444 | 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-c arboxylate and compositions and methods of pharmaceutical use thereof
1-(Cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-car boxylate or a pharmaceutically acceptable salt thereof has potent angiotensin II antihypertensive activity, thus being useful as therapeutic agents for treating circulatory system diseases such as hypertensive diseases, heart diseases (e.g. hypercardia, heart failure, cardiac infarction, etc.), strokes, cerebral apoplexy, nephritis, etc.
| 2012-6-4(expired) |
| 7538133*PED | 2011-10-18(expired) | |
| 5705517*PED | 2011-10-18(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ASTRAZENECA.
| DATE | SUPPLEMENT NO. | ACTION | DOCUMENTS |
|---|---|---|---|
| 2013-04-26 | 038 | Labeling Revision | |
| 2012-04-27 | 035 | Labeling Revision | |
| 2012-04-13 | 032 | Labeling Revision | |
| 1998-06-04 | 000 | Approval | |
| 2011-06-24 | 033 | Labeling Revision | |
| 2009-10-22 | 031 | Patient Population Altered | |
| 2006-08-17 | 026 | Labeling Revision | |
| 2005-05-18 | 022 | New or Modified Indication | |
| 2005-02-22 | 024 | New or Modified Indication | |
| 2004-12-16 | 023 | Labeling Revision | |
| 2000-06-14 | 008 | Labeling Revision | |
| 2002-09-13 | 015 | Comparative Efficacy Claim | |
| 2003-01-22 | 017 | Labeling Revision | |
| 2003-04-23 | 019 | Labeling Revision | |
| 2013-02-21 | 037 | Manufacturing Change or Addition | |
| 1999-08-11 | 005 | Package Change | |
| 2000-12-27 | 009 | Manufacturing Change or Addition | |
| 2001-05-24 | 012 | Manufacturing Change or Addition | |
| 2001-11-28 | 016 | Labeling Revision | |
| 1999-07-28 | 004 | Control Supplement | |
| 2001-04-02 | 011 | Manufacturing Change or Addition | |
| 2001-10-04 | 014 | Control Supplement | |
| 1998-11-16 | 002 | Manufacturing Change or Addition | |
| 1999-12-08 | 006 | Package Change | |
| 2001-06-07 | 010 | Manufacturing Change or Addition | |
| 2001-03-29 | 013 | Package Change | |
| 1998-12-07 | 001 | Manufacturing Change or Addition |
 Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.Synthesis
Candesartan is synthesised as follows:  kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
 kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
The method has technical problems as follows: a) the starting material is obtained by a minor reaction, b) its yield is relatively low and its industrial applicability is poor (due to N2 gas formation) because the Curtius rearrangement reaction is involved, and c) materials industrially hard to handle such as SOCI2 or NaH are used. In addition, methods for preparing an intermediate of candesartan cilexetil are disclosed in Organic Process Research & Development 11:490-493(2007), as represented by the following Reaction Scheme 2: Reaction Scheme 2

3:1 s

However, the preparation process has also shortcomings of a) undesired byproducts formed by nitrogenation at ortho- or para-position, b) safety problems from strong acids (sulfuric acid and nitric acid) used twice when introducing and rearranging nitrogen groups, and c) utilization of high-flammable Raney Ni.
cut paste
Novel and Practical Synthesis of Candesartan Cilexetil
Yongjun Mao, Ruisheng Xiong, Zheng Liu, Haihong Li, Jingkang Shen, and Jingshan Shen* *Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Rd., Zhangjiang Hi-Tech Park, Shanghai, 201203, China
Abstract
A novel and convergent synthetic route of candesartan cilexetil (API of Atacand), an effective angiotensin II receptor blocker, is described. Cleavage of the N-Boc and N-trityl protective group are implemented simultaneously and formation of the benzimidazole ring is conducted at the last step of this route, which gives candesartan cilexetil in 55% yield over six steps with 99.1% purity (HPLC).  Full Text HTMLPDF (567KB)PDF with Links (932KB)
Full Text HTMLPDF (567KB)PDF with Links (932KB)  This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.| Benzimidazole derivs., their production and use | |
| Naka, T.; Nishikawa, K.; Kato, T. (Takeda Chemical Industries, Ltd.) | |
| EP 0459136; EP 0720982; JP 1992364171; JP 1996099960; US 5196444; US 5328919; US 5401764; US 5703110; US 5705517; US 5962491; US 6004989 |
more info Candesartan cilexetil of Formula I, disclosed in U.S. Patent No. 5,196,444 as crystalline form, i.e., Form-I (C-type crystals), is chemically described as 1- cyclohexyloxycarbonyloxyethyl 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate.

H Formula I It is useful in the treatment of cardiovascular complaints such as hypertension and heart failure. Candesartan cilexetil is poorly soluble in water, which is attributed to its hydrophobic nature. Solubility plays an important role in achieving the desired concentration of a drug in systemic circulation for accomplishing the pharmacological response. Various techniques are known in literature to increase the solubility of poorly-soluble drugs, including decreasing the particle size, complexation, changing the surface characteristics of the particles, and incorporation of drug particles into colloidal systems like nanoparticles and liposomes. Among these, the most commonly used technique to increase the solubility is particle size reduction. Sometimes the rate of dissolution of a poorly-soluble drug is the rate limiting factor in its rate of absorption by the body. These drugs may be more readily bioavailable if administered in a finely divided state. Particle size reduction increases the surface area causing an increase in the dissolution rate of the compound, and hence, its bioavailability. There are certain techniques reported in literature to reduce the particle size of such poorly-soluble drugs. PCT Publication No. WO 2006/122254 discloses stable candesartan cilexetil of fine particle size, wherein the stable micronized candesartan cilexetil is prepared by slurrying a sample of candesartan cilexetil of fine particle size in a suitable solvent for a suitable amount of time. In this application, candesartan cilexetil of fine particle size is obtained directly from the synthesis of candesartan cilexetil or by comminuting candesartan cilexetil using milling. PCT Publication No. WO 2005/123720 describes fine particles of candesartan cilexetil having improved pharmacokinetic profile and a process for their production, wherein fine particle size is obtained by a) dissolving candesartan cilexetil in an organic solvent; b) cooling the solution obtained in step a) under stirring to crystallize candesartan cilexetil from the solution; and c) isolating candesartan cilexetil having a particle size of with d90 not more than about 25 μ. U.S. Patent Application No. 2006/0165806 describes compositions comprising a candesartan, such as candesartan cilexitil. The candesartan particles of the composition have an effective average particle size of less than about 2000 nm. U.S. Patent Application No. 2008/0038359 describes a nanoparticle pharmaceutical formulation comprising a poorly soluble drug substance having an average particle size of less than about 1000 nm, a solid or semisolid dispersion vehicle, and optionally a non-surface modifying excipient. U.S. Patent No. 7,828,996 discloses the methods for forming nanoparticles of a material of narrow polydispersity with ultrasonic waves using a partially submersed sonicator that does not touch any part of the apparatus and the point of addition of organic solvent is in the wave funnel produced by sonication and within the selected distance from the wave-source depending on the desired particle size. U.S. Patent No. 7,780,989 discloses the preparation of a dispersion of nanocrystalline particles in an aqueous medium using ultrasound. U.S. Patent No. 5,314,506 describes a crystallization process in which a jet of a solution containing a substance is impinged with a second jet containing an anti-solvent for the substance. The rapid mixing produced by the impinging jets results in a reduction of the crystals so formed compared to conventional slow crystallization processes. The smallest crystals disclosed are about 3 μ and the majority are in the range of from about 3 μ to about 20 μ. PCT Publication No. WO 00/44468 describes a modification to the apparatus described in U.S. Patent No. 5,314,506, wherein ultrasound energy is applied at the point of impingement of the two jets to further enhance localized mixing and is stated to give direct formation of small crystals with a diameter of less than 1 μ. Generally, the crystalline particles described have an average size of 0.5 μ. Conventional particle size reduction methods such as high energy milling may result in loss of yield, noise and dusting, as well as unwanted exposure to highly potent pharmaceutical compounds. Also, in the case of crystalline compounds, stress generated on crystal surfaces during milling can adversely affect labile compounds. Therefore, there is a need for a process for particle size reduction of candesartan cilexetil, which is industrially advantageous, easy to handle and is cost effective.
- Candesartan is a potent, selective AT1 subtype angiotensin II receptor antagonist and used for treatment of hypertension. Due to poor absorption of Candesartan in body, the prodrug candesartan cilexetilwas developed. The candesartan cilexetil is rapidly and completely hydrolyzed to candesartan in gastrointestinal tract.
- [0004]U.S. Pat. No. 5,196,444 discloses Candesartan cilexetil and a process for its preparation by the reaction of 2-ethoxy-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid with trityl chloride in presence of triethyl amine in methylene chloride and purification by column chromatography gives 2-ethoxy-1-[[2'-(N-triphenylmethyltetrazol-5-yl)-biphenyl-4-yl]methyl] benzimidazole -7-carboxylic acid, which upon condensation with cyclohexyl 1-iodoethyl carbonate in presence of potassium carbonate in DMF followed by purification with column chromatography gives a colorless powder which is recrystallized in ethanol yields ‘C’ type crystals of Candesartancilexitil.
- [0005]U.S. Pat. Application No. 2005/131027 discloses a process for preparation of candesartan cilexetil by reaction of trityl candesartanwith cilexetil halide and at least one base in a low boiling solvent in presence of phase transfer catalyst to give Trityl candesartan cilexetil, which upon deprotection with at least one organic acid in at least one organic solvent. U.S. Pat. Application 2005/131027 further discloses the deprotection of Trityl candesartan cilexetil in methanol without an acid.
- [0006]The PCT publication WO 2005/021535 discloses the deprotection of Trityl candesartan cilexetil with neutral or slightly basic medium in alcohol.
- [0007]Chem.Pharm.Bull. 47(2), 182-186 (1999) discloses two novel crystalline forms of Candesartan cilexetil, form-I and form-II.
- [0008]PCT publication WO 04/085426 discloses Candesartan cilexetil 1,4-Dioxane solvate and two more crystalline forms, designated as form-III and form-IV. The disclosed process for preparation of form-III involves crystallization of Candesartan cilexetil in toluene and for form-IV involves crystallization in a mixture of methyl tert-butyl ether and methanol.
- [0009]PCT publication WO 2005/077941 discloses several crystalline forms, solvates of Candesartan cilexetil along with a process for preparation of form-I (type-C).
- [0010]The prior art disclosed methods for preparation of Candesartan cilexetilinvolves purification of Trityl candesartan and Candesartan cilexetil by column chromatography or involves the use of strong acids like IN HCl or the use of organic acids or without an acid in methanol for detrytilation of Trityl candesartan cilexetil.
- [0011]There is a requirement of a process for preparation of Candesartancilexetil which yields a pure Candesartan cilexetil without involving the purification by column chromatography and the usage of strong acids for deprotection.
Candesartan cilexetil of formula (I) shown beiow is chemicaily described as (+/-)-1- [[(cyclohexyloxy)carbonyl]oxy]ethyl-2-ethoxy-1 -[[2'-(1 H-tetrazol-5-yl)-1 , 1 '-biphenyl- 4-yl]methyl]-1 H-benzimidazoie-7-carboxylate. An alternative designation is (+-)-1- hydroxyethyf 2~Ethoxy-1 -{p-(o-1 H-tetrazo!-5-yIphenyi)benzyJ)-7-benzϊmidazoie~ carboxyiic acid cyclohexyl carbonate (ester), with candesartan being the underlying carboxylic acid, i.e. 2-Ethoxy-1 -(p-(o-1 H-tetrazol-5-ylphenyl)benzy!)-7-benz- imidazolecarboxylic acid.

Because of its ability to inhibit the angiotensin-converting enzyme it is widely used for the treatment of hypertension and related diseases and conditions. As an angiotensin Ii receptor antagonist, candesartan ciiexetil avoids the side-effects of calcium antagonists, and shows high stability and obvious curative effects. Currently candesartan ciiexetil is soid as racemic mixture, it is produced according to published patents, e.g. EP 0 720 982 B1 and EP 0 459 136. in Chem. Pharm. Bull. 47(2), 182-186 (1999) two crystalline forms (Form I and II), together with an amorphous form, are disclosed and characterized by their DSC thermograms, X-ray diffraction patterns and IR spectra. US 5,196,444 disclosed the C-type crystal (Form I) of candesartan cilexetif, and processes for producing it under acidic conditions. WO 04/085426 discloses the dioxane solvate of candesartan ciiexetil, together with two additional crystalline forms. WO 2005/077941 discloses hydrates and solvates of candesartan ciiexetil, together with processes for their preparation. WO 2006/048237 also describes the preparation of new polymorphic forms ofcandesartan ciiexetil, together with processes for their preparation, including the preparation of amorphous candesartan ciiexetil by precipitating it with a liquid cyclic hydrocarbon from a solution of candesartan ciiexetil in a chlorinated solvent. in WO 2005/123721 processes for the preparation of amorphous candesartanciiexetil are provided, comprised of spray-drying and precipitation. HPLC CUT PASTE , READER TO PICK ONLY REQUIRED INFO Candesartan cilexetil (60 g) is dissolved in isopropanol (900 m!_) at 60-65 0C. Solution is hot filtered into reactor and quickly cooled to 35 0C. At this temperature nucleation is provoked with 300 mg of candesartan cilexetil form I and stirring is enforced. Suspension is cooled to 3O0C in 1 hour and rigorous stirring is continued at this temperature for additional 5 hours. Then stirring power is reduced and the suspension is cooled to 2O0C in 8 hours. The product is filtered, washed with isopropanoi and dried for 2 hours at 38°C. Yield: 48.7 g of candesartan cilexetil form I. Area % HPLC: candesartan cilexetil: 99.73%, alky ester of candesartan cilexetil 0.08%, candesartan cilexetii pyran below 0.05%, tritylcandesartan cϋexetil 0.09% Average particle size: 19 /vm, no agglomerates present (see Figure 2) B) Detection of impurities in candesartan cilexetil Example 6 Detection of candesartan cϊlexetil pyran in candesartan cilexetii by HPLC HPLC (external standard method) was performed using the following specifications : Column: Zorbax Eclipse XDB-C18, 50 mm x 4.6 mm i.d.τ 1.8 μm particles Eluent A: 0.01 M NaH2PO4, pH 2.5 Eluent B: acetonitriie Gradient of Eluent:

Flow rate: about 1.2 ml/min Diluent: acetonitriie : water = 70 : 30 (V/V). Detection: UV, wavelength 225 nm injection volume: 5 μl Column temperature : 500C Autosampler temperature: 7°C Example 7 Detection of cilexetil pyran in 1 -chloroethyl cyclohexylcarbonate by GC GC/FID (area percent method) was performed using the following specifications: Column: capillary (fused-silica) AT-WAX or adequate Length: 30 m ID: 0.32 mm Film thickness: 0.25 μm Carrier gas: helium Carrier gas flow rate: 2.0 ml/mi n Split ratio: 10 : 1 Air flow rate: 400 ml/min Hydrogen flow rate: 40 ml/min Make up gas flow ISb rate: 25 ml/min Column temperature 100°C (0 min) → 10°C/min → 2000C (10 min or prolonged if necessary) Injector temperature: 21 O0C Detector temperature: 250OC Injection volume : 1 μl Diluent: Acetonithle: chromatography grade. Chromatographic system suitability Signal/noise of 1 -chloroethyl cyclohexyl carbonate: not less than 10
………………
Seki M * Mitsubishi Tanabe Pharma Corporation, Osaka, Japan An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012; 44: 3231-3237 
Significance
Candesartan cilexetil (Atacand®) is an angiotensin II receptor antagonist that is prescribed for the treatment of hypertension. It is a prodrug that is hydrolyzed to candesartan in the gut. The synthesis depicted, features an efficient protocol for ruthenium-catalyzed C–H arylation of the tetrazole A. Comment A significant challenge in this small-scale synthesis was the final removal of the benzyl protecting group from the tetrazole unit using transfer hydrogenation. Best results were obtained using a ‘thickshell’ Pd/C catalyst from Evonik
;.......................................................................
5 EPROSARTAN
EPROSARTAN MESYLATE

TEVETEN® (eprosartan mesylate) is a non-biphenyl non-tetrazole angiotensin II receptor (AT1) antagonist. A selective non-peptide molecule, TEVETEN® is chemically described as the monomethanesulfonate of (E)-2-butyl-1 -(p-carboxybenzyl)-α-2-thienylmethylimid-azole-5 -acrylic acid.
Its empirical formula is C23H24N2O4S•CH4O3S and molecular weight is 520.625. Its structural formula is:
 |
EPROSARTAN MESYLATE
tevetenEprosartan mesilate, SK&F-108566-J(?, SK&F-108566, Teveten SB, Navixen, Regulaten, Tevetenz, Teveten
| US 5656650 | exp Aug 12, 2014 |
CAS EPROSARTAN
144143-96-4
133040-01-4
| Chemical Name: | Eprosartan mesylate |
| Synonyms: | EPROSARTAN MESYLATE;Eprosartan Methanesulfonate;4-[[2-butyl-5-(2-carboxy-3-thiophen-2-yl-prop-1-enyl)-imidazol-1-yl]methyl]benzoic acid mesylate;4-({2-butyl-5-[(1E)-2-carboxy-2-(thiophen-2-ylMethyl)eth-1-en-1-yl]-1H-iMidazol-1-yl}Methyl)benzoic acid;(E)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid Methanesulfonate;(αE)-α-[[2-Butyl-1-[(4-carboxyphenyl)Methyl]-1H-iMidazol-5-yl]Methylene]-2-thiophenepropanoic Acid MonoMethanesulfonate |
| CBNumber: | CB4842192 |
| Molecular Formula: | C24H28N2O7S2 |
| Formula Weight: | 520.61832 |
Eprosartan is an angiotensin II receptor antagonist used for the treatment of high blood pressure. It is marketed as Teveten byAbbott Laboratories in the United States.It is marketed as Eprozar by INTAS Pharmaceuticals in India and by Abbott Laboratorieselsewhere. It is sometimes paired with hydrochlorothiazide, marketed in the US as Teveten HCT and elsewhere as TevetenPlus.
The drug acts on the renin-angiotensin system in two ways to decrease total peripheral resistance. First, it blocks the binding ofangiotensin II to AT1 receptors in vascular smooth muscle, causing vascular dilatation. Second, it inhibits sympatheticnorepinephrine production, further reducing blood pressure.
As with other angiotensin II receptor antagonists, eprosartan is generally better tolerated than enalapril (an ACE inhibitor), especially among the elderly.[1]
- Ruilope L, Jäger B, Prichard B (2001). “Eprosartan versus enalapril in elderly patients with hypertension: a double-blind, randomized trial”. Blood Press. 10 (4): 223–9. doi:10.1080/08037050152669747. PMID 11800061.
PAT APR EXP
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| United States | 5185351 | 1993-02-09 | 2010-02-09 |
| Canada | 2115170 | 2004-05-25 | 2012-08-12 |
| United States | 5656650 | 1994-08-12 | 2014-08-12 |
| Canada | 2250395 | 2005-09-06 | 2017-03-26 |
J Med Chem1991,34,(4):1514-7
J Med Chem1993,36,(13):1880-92
Synth Commun1993,23,(22):3231-48
AU 9056901, EP 403159, JP 91115278, US 5185351.
Drugs Fut1997,22,(10):1079

Eprosartan mesylate was developed successfully by SmithKline Beecham Corporation in 1997, and marketed in Germany in 1998 under the trade-name Teveten and in the United States later in 1999. Eprosartan mesylate, as an angiotensin II receptor blocker, is an antihypertensive drug of the latest generation. Eprosartan mesylate is potent to lower systolic and diastolic pressures in mild, moderate and severe hypertensive patients, and is safe and tolerable. Eprosartan mesylate is rapidly absorbed when administrated orally, with a bioavailability of 13% and a protein binding rate of 98%. The blood peak concentration and AUC (Area Under Curve) can be elevated by about 50% in patients with liver and kidney dysfunction, or fullness after administration, and can be elevated by 2 to 3 folds in elderly patients. Eprosartan mesylate has a structure shown as follows:

U.S. Pat. No. 5,185,351 discloses a method for preparing eprosartan mesylate using Eprosartan and methanesulfonic acid in isopropanol (U.S. Pat. No. 5,185,351, Example 41 (ii)). However, it is found when following this method for preparing eprosartan mesylate in industry, an esterification reaction can occur between eprosartan and isopropanol and the following two impurities can be generated:

In addition to the above two esterification impurities, the salifying method provided by the above patent is prone to produce isopropyl mesylate. Considering currently known potential risk of gene toxicity of methylsulfonic acid ester on human as well as the stringent requirements of methylsulfonic acid ester from the Europe and the America authorities, it is important to produce eprosartan mesylate in a non-alcohol solvent during the process of producing eprosartan mesylate, since it avoids the formation of methylsulfonic acid ester and the residue thereof in the final product. Since the dosage of eprosartan mesylate is high, it is particularly important to strictly control methylsulfonic acid ester in eprosartan mesylate.
In addition, for the above salifying method, solid eprosartan is suspended in propanol at a low temperature, then methanesulfonic acid is added, about ten seconds later a great deal of eprosartan mesylate precipitate is obtained. Therefore, solid eprosartan may be embedded by the precipitated eprosartan mesylate. Since isopropyl alcohol has a high viscosity at low temperature, a heavy filtering operation burden is needed to obtain solid from isopropanol, and the obtained solid contains quite an amount of isopropanol.

Eprosartan has been obtained by several different ways: 1) The iodination of 2-butylimidazole (I) with I2 and Na2CO3 in dioxane/water gives 2-butyl-4,5-diiodoimidazole (II), which is treated with benzyl chloromethyl ether (III) and K2CO3 in DMF yielding the imidazole derivative (IV). The condensation of (IV) with N-methyl-N-(2-pyridyl)formamide (V) by means of butyllithium in THF affords 1-(benzyloxymethyl)-2-butyl-4-iodoimidazole-5-carbaldehyde (VI), which is deprotected with concentrated HCl ethanol to give 2-butyl-4-iodoimidazole-5-carbaldehyde (VII). The acylation of (VII) with methyl 4-(bromomethyl)benzoate (VIII) by means of K2CO3 in hot DMF yields 4-(2-butyl-5-formyl-4-iodoimidazol-1 ylmethyl)benzoic acid methyl ester (IX), which is deiodinated by hydrogenation with H2 over Pd/C in methanol affording compound (X). The condensation of (X) with methyl 3-(2-thienyl)propionate (XI) by means of lithium diisopropylamide (LDA) in THF gives (XII), which is acylated with acetic anhydride and dimethylaminopyridine (DMAP) in dichloromethane yielding the corresponding acetate (XIII). Elimination of acetic acid from (XIII) with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in hot toluene affords the expected propenoic ester (XIV), which is finally saponified with NaOH or KOH in ethanol/water.
…………………………………………………………………………………………………….
WO 1998035962 A1

…………………………………………………………………………………………..



...................................................
6 OLMESARTAN
6 OLMESARTAN

OLMESARTAN
Formula C29H30N6O6
Mol. mass 558.585 g/mol
Mol. mass 558.585 g/mol
(5-methyl-2-oxo-2H-1,3-dioxol-4-yl)methyl 4-(2-hydroxypropan-2-yl)-2-propyl-1-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1H-imidazole-5-carboxylate
Olmesartan medoxomil is a prodrug that hydrolysed to olmesartan during absorption. It is an angiotensin II receptor antagonist used for hypertension and is chemically designated as 5-methyl-2-oxo-1,3-dioxolen-4-y1)methoxy-4-(1-hydroxy-1-methylethyl)-2-propyl-l-{4-[2-(tetrazol-5-y1)-phenyl]phenyl}methylimidazol-5-carboxylate. Olmesartan is also beneficial in animáis and is a strong agent to show activity against atherosclerosis, liver disorders and diabetic nephropathy
Olmesartan medoxomil (trade names: Benicar in the US, Olmetec in EU, Canada and Japan, WinBP, Golme in India, Erastapex in Egypt) is an angiotensin II receptor antagonist used to treat high blood pressure.
Olmesartan is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.[1] The U.S. Food and Drug Administration (FDA) has determined that the benefits of Benicar continue to outweigh its potential risks when used for the treatment of patients with high blood pressure according to the drug label.[2]
Angiotensin-II receptor antagonists should be used with caution in renal artery stenosis. Monitoring of plasma-potassium concentration is advised, particularly in the elderly and in patients with renal impairment; lower initial doses may be appropriate in these patients. Angiotensin-II receptor antagonists should be used with caution in aortic or mitral valve stenosis and in hypertrophic cardiomyopathy. Those with primary aldosteronism, and Afro-Caribbean patients (particularly those with left ventricular hypertrophy), may not benefit from an angiotensin-II receptor antagonist.
The usual recommended starting dose of olmesartan is 20 mg once daily. The dose may be increased to 40 mg after two weeks of therapy, if further reduction in blood pressure is desirable. Doses above 40 mg do not appear to have greater effect, and twice-daily dosing offers no advantage over the same total dose given once daily.[1] No adjustment of dosage is typically necessary for advanced age, renal impairment, or hepatic dysfunction. For patients with possible depletion of intravascular volume (e.g., patients treated with diuretics), olmesartan should be initiated with caution; consideration should be given to use of a lower starting dose in such cases.[1] If blood pressure is not controlled by Benicar alone, a diuretic may be added. Benicar may be administered with other antihypertensive agents. Benicar may be administered with or without food.[1]
Olmesartan and Sevikar HCT is marketed worldwide by Daiichi Sankyo, in India by Abbott Healthcare Pvt. Ltd. under the trade name WinBP, by Zydus Cadila under the trade name Olmy, by Ranbaxy Laboratories Ltd. under the trade name Olvance, and in Canada by Schering-Plough as Olmetec. Benicar HCT is the brand name of a medication containing olmesartan medoxomil in combination with hydrochlorothiazide, a thiazide diuretic. Three dosage combinations are available: 20 mg or 40 mg of olmesartan medoxomil combined with 12.5 mg of hydrochlorothiazide, or 40 mg of olmesartan medoxomil combined with 25 mg of hydrochlorothiazide. Benitec H, another medication containing olmesartan medoxomil and hydrochlorothiazide, is marketed by GlaxoSmithKline in India. In Poland as Olesartan Medoxomil by TEVA, Olimestra and Co-Olimestra (with HCTZ) by Miklich Lab., Elestar (with amlodipine) and Elestar HCT (with amlodipine, HCTZ) by Menarini, Sevikar HCT (with amoldipine, HCTZ) by Aiichi Sankyo.

Olmesartan is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.[1] The U.S. Food and Drug Administration (FDA) has determined that the benefits of Benicar continue to outweigh its potential risks when used for the treatment of patients with high blood pressure according to the drug label.[2]
Angiotensin-II receptor antagonists should be used with caution in renal artery stenosis. Monitoring of plasma-potassium concentration is advised, particularly in the elderly and in patients with renal impairment; lower initial doses may be appropriate in these patients. Angiotensin-II receptor antagonists should be used with caution in aortic or mitral valve stenosis and in hypertrophic cardiomyopathy. Those with primary aldosteronism, and Afro-Caribbean patients (particularly those with left ventricular hypertrophy), may not benefit from an angiotensin-II receptor antagonist.
Structure
The olmesartan molecule includes one tetrazole group (a 5-member heterocyclic ring of four nitrogen and one carbon atom) and one imidazole group (a 5-membered planar heterocyclic aromatic ring of two nitrogen and three carbon atoms, classified as an alkaloid).Olmesartan as the starting material can be easily produced according to the method described in Japanese Examined Patent Application (Kokoku) No. Hei 7-121918 (Japanese Patent No. 2082519 ; US Patent No. 5616599 ) or the like
Olmesartan is a prodrug that works by blocking the binding of angiotensin II to the AT1 receptors in vascular muscle; it is therefore independent of angiotensin II synthesis pathways, unlike ACE inhibitors. By blocking the binding rather than the synthesis of angiotensin II, olmesartan inhibits the negative regulatory feedback on renin secretion. As a result of this blockage, olmesartan reduces vasoconstriction and the secretion of aldosterone. This lowers blood pressure by producing vasodilation, and decreasing peripheral resistance.The usual recommended starting dose of olmesartan is 20 mg once daily. The dose may be increased to 40 mg after two weeks of therapy, if further reduction in blood pressure is desirable. Doses above 40 mg do not appear to have greater effect, and twice-daily dosing offers no advantage over the same total dose given once daily.[1] No adjustment of dosage is typically necessary for advanced age, renal impairment, or hepatic dysfunction. For patients with possible depletion of intravascular volume (e.g., patients treated with diuretics), olmesartan should be initiated with caution; consideration should be given to use of a lower starting dose in such cases.[1] If blood pressure is not controlled by Benicar alone, a diuretic may be added. Benicar may be administered with other antihypertensive agents. Benicar may be administered with or without food.[1]

Research
Two clinical studies (MORE [6] and OLIVUS [7])[8] report that Benicar reduced arterial plaque during therapy for high-blood pressure (hypertension).- RxList Inc. (5 July 2007). "Benicar (olmesartan medoxomil)". RxList Inc. Retrieved 22 July 2010.
- "FDA Alert: Benicar (olmesartan): Ongoing Safety Review". Drugs.com. Retrieved 2013-06-27.
- Angiotensin II receptor blocker induced fetopathy: 7 cases. Hünseler C, Paneitz A, Friedrich D, Lindner U, Oberthuer A, Körber F, Schmitt K, Welzing L, Müller A, Herkenrath P, Hoppe B, Gortner L, Roth B, Kattner E, Schaible T. Klin Padiatr. 2011 Jan;223(1):10-4. Epub 2011 Jan 26.
- "BENICAR Prescribing Information". Retrieved 2011-01-20.
- Rubio-Tapia, Alberto; Herman, Margot L.; Ludvigsson, Jonas F.; Kelly, Darlene G.; Mangan, Thomas F.; Wu, Tsung-Teh; Murray, Joseph A. (NaN undefined NaN). "Severe Spruelike Enteropathy Associated With Olmesartan". Mayo Clinic Proceedings 87 (8): 732–738. doi:10.1016/j.mayocp.2012.06.003.
- as referenced in http://www.medicalnewstoday.com/releases/91285.php "Olmetec(R) Is First Angiotensin Receptor Blocker (ARB) To Suggest Atherosclerosis Regression (In Hypertensives With Cardiovascular Risk), UK"
- Cardiovascular Research Foundation (2008, October 16). Drug May Reduce Coronary Artery Plaque. ScienceDaily. Retrieved January 5, 2013, from http://www.sciencedaily.com /releases/2008/10/081012121318.htm
- (Review) R Preston Mason, Cardiovascular Division, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, and Elucida Research, Beverly, MA, USA. Vascular Health and Risk Management, Dovepress, Published Date June 2011 Volume 2011:7 Pages 405 - 416. Optimal therapeutic strategy for treating patients with hypertension and atherosclerosis: focus on olmesartan medoxomil. Retrieved January 5, 2013, from http://www.dovepress.com/optimal-therapeutic-strategy-for-treating-patients-with-hypertension-a-peer-reviewed-article-VHRM
4-Isopropenyl-2-propyl-1-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-5-carboxylic acid [olmesartan dehydrate, compound 34b described in J. Med. Chem., 39, 323-338 (1996)]
- Daiichi-Sankyo Benicar page
- Benicar HCT from RXlist.com
- Mayo Clinic Proceedings vol.87 Issue 8, pages 732-738
- Olmesartan medoxomil is known by two names,
- (a)(5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-[4-(2(tetrazole-5yl)phenyl]phenyl]methylimidazole-5-carboxylate
- (b)4-(1-Hydroxy-1-methylethyl)-2-propyl-1-[[2'-(1H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl]methyl]-1H-imidazol-5-carboxylic acid (5-methyl-2-oxo-1,-3 dioxol-4-yl)methyl ester, and has a CAS No. [144689-63-4].
- The structural formula is represented below:
 Olmesartan medoxomil, which is an angiotensin II receptor antagonist, is useful as an active ingredient in medicaments for treatment and prophylaxis of hypertension (for example, Patent documents 1 to 5 or Non-patent document 1 and 2). Techniques for producing high-purity olmesartan medoxomil are necessary for use of olmesartan medoxomil as a medicament.
Olmesartan medoxomil, which is an angiotensin II receptor antagonist, is useful as an active ingredient in medicaments for treatment and prophylaxis of hypertension (for example, Patent documents 1 to 5 or Non-patent document 1 and 2). Techniques for producing high-purity olmesartan medoxomil are necessary for use of olmesartan medoxomil as a medicament. - Olmesartan medoxomil is produced from olmesartan by the steps described below, but there is the problem that olmesartan which is a starting material, olmesartan medoxomil dehydrate which is a by-product, or the like, is present as an impurity.


- Patent document 1: Japanese Examined Patent Application (Kokoku) No. Hei 7-121918 (Japanese Patent No. 2082519 )
- Patent document 2: US Patent No. 5616599
- Patent document 3: International Patent Publication No. WO2006/029056
- Patent document 4: International Patent Publication No. WO2006/029057
- Patent document 5: International Patent Publication No. WO2006/073519
- Non-patent document 1: J. Med. Chem., 39, 323-338 (1996)
- Non-patent document 2: Annu. Rep. Sankyo Res. Lab. (Sankyo Kenkyusho Nempo) 55, 1-91 (2003)
- The prior art synthetic methods involve coupling reaction between the substituted imidazole and substituted biphenyl methyl bromide. J. Med. Chem. 1996 vol. 39 No.1, page 323-38 describes the synthesis of Olmesartan medoxomil as follow. [Scheme-1 ]

- US 5616599 describes a process for the preparation of olmesartan medoxomil as follows.
- [0006]4-(1-hydroxyl-1-methylethyl)-2-propyl imidazole-5carboxylic acid is reacted with 5-methyl-2-oxo-1, 3-dioxolene-4-yl)methyl chloride using N,N-diisopropylethyl amine as base in N,N-dimethyl acetamide at 60°C to give (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl4-(1-hydroxy-1-methylethyl)-2-propyl imidazole-5-carboxylate. The resulting product is coupled with N-(triphenylmethyl)-5-[4'-(bromomethyl)biphenyl-2-yl]tetrazole [herein referred to as TTBB] at 60°C in N, N-dimethyl acetamide using potassium carbonate as base to give protected olmesartan medoxomil. The protected olmesartan medoxomil is deprotected using 75 % acetic acid to give olmesartan medoxomil.
- This process involves column chromatographic purification of intermediates which is not desirable on commercial scale operation.
- US 5616599 describes another process for the preparation of olmesartan Medoxomil which involves addition of methyl Magnesium chloride on diethyl 2-propylimidazole-4, 5-dicarboxylate in tetrahydrofuran at -30 to -20°C to give ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate, which is coupled with TTBB using sodium hydride as base in N, N-dimethylformamide at 60°C to give ethyl-4-(1-hydroxy-1-methylethyl)2-propyl-1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-. 5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate. The product thus formed is hydrolyzed using lithium hydroxide monohydrate as base in 1,4-dioxane at 5-10°C to give lithium salt of 4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylic acid, which is then coupled with 5-methyl-2-oxo-(1,3-dioxolene-4-yl)methyl chloride using K2CO3 as base in N,N-dimethylacetamide at 50°C to give trityl protected olmesartan medoxomil which on deprotection using 75% acetic acid gives Olmesartan Medoxomil.
- During the condensation of ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate, with TTBB using sodium hydride as base in N, N-dimethylformamide, various impurities are formed, and isolation of the product involves extractive workup.
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
Marcus Baumann, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin
Innovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW Cambridge, UK
Corresponding author email
Editor-in-Chief: J. Clayden
Beilstein J. Org. Chem. 2011, 7, 442–495.
The structurally related imidazole core of olmesartan is formed in a different fashion (Scheme 36). Condensation between diaminomaleonitrile and trimethyl orthobutyrate furnishes the trisubstituted imidazole 181 in high yield [53,54]. Acid-mediated nitrile hydrolysis followed by esterification results in the corresponding diester unit 182. Treatment of 182 with four equivalents of methylmagnesium chloride in a mixture of diethyl ether and dichloromethane selectively provides tertiary alcohol 183. In subsequent steps this imidazole is alkylated with the tetrazole containing biphenyl appendage, followed by ester hydrolysis and alkylation of the resulting carboxylate with 4-(chloromethyl)-5-methyl-2-oxo-1,3-dioxole to yield olmesartan (Scheme 36).
![[1860-5397-7-57-i36]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i36.png?max-width=550&background=FFFFFF)
Scheme 36: Preparation of the imidazole ring in olmesartan.
- 53.......Yanagisawa, H.; Fujimoto, K.; Amemiya, Y.; Shimoji, Y.; Kanazaki, T.; Koike, H.; Sada, T. Angiotensin II Antagonist 1-Biphenylmethylimidazole Compounds and their Therapeutic Use. U.S. Patent 5,616,599, April 1, 1997.
Return to citation in text: [1] - 54..........Yanagisawa, H.; Amemiya, Y.; Kanazaki, T.; Shimoji, Y.; Fujimoto, K.; Kitahara, Y.; Sada, T.; Mizuno, M.; Ikeda, M.; Miyamoto, S.; Furukawa, Y.; Koike, H. J. Med. Chem. 1996, 39, 323–338. doi:10.1021/jm950450f
Return to citation in text: [1]
....................
Olmesartan medoxomil of high purity (99.3-99.7% by HPLC ) is prepared using an improved process of its intermediate, namely- ethyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2'-(2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate, comprising:
Reacting ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole-5-carboxylate with N-(Triphenylmethyl)-5-[4'-(bromomethyl)biphenyl-2- yl]tetrazole in an organic solvent in presence of a base and a phase transfer catalyst in non-aqueous system to give after workup, ethyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate, which is further processed, by following improved reaction conditions in three steps to provide substantially pure [HPLC purity 99.3 to 99.7 %] olmesartan medoxomil.
Reacting ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole-5-carboxylate with N-(Triphenylmethyl)-5-[4'-(bromomethyl)biphenyl-2- yl]tetrazole in an organic solvent in presence of a base and a phase transfer catalyst in non-aqueous system to give after workup, ethyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate, which is further processed, by following improved reaction conditions in three steps to provide substantially pure [HPLC purity 99.3 to 99.7 %] olmesartan medoxomil.
Example-1Preparation of Ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole-5-carboxylate
- To a 3M solution of MeMgCl(55.86 g, 0.74 mol) in tetrahydrofuran was added a solution of diethyl 2-propyl imidazole- 4,5-dicarboxylate (50 g,0.19 mol) in tetrahydrofuran (200 ml) at -10 to 0°C under N2 atmosphere. The mixture was stirred at -5 to 0°C for 10 minutes. Reaction mass was quenched into 400 ml 25 % ammonium chloride solution followed by extraction with ethyl acetate (300 ml). The organic phase was separated, washed with brine, dried over Na2SO4, and concentrated in vacuo to give a syrup, which was crystallized using diisopropyl ether.
Yield: 85-90 %,
Purity by HPLC: 88-93 %.
1H-NMR (CDCl3) δ: 7.8-8.1 (s, 1H), 5.8(s, 1H)., 4.35(q, 2H), 2.68(t, 2H), 1.78(m, 2H), 1.61(s, 6H), 1.36(t, 3H), 0.96(t, 3H).
- -5-
- Mixture of Ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate (41 g, 0.17 mol), potassium carbonate (47g, 0.34 mol) and tetrabutylammonium bromide (4.9 g, 0.01 mol) in acetone was stirred at room temperature for 1hr. Then TTBB (93% Purity, 92.89g, 0.15 mol) was charged, refluxed for 14hrs. Potassium salts were filtered off from the reaction mass and the filtrate was charcoalised for 1hr. It was filtered over celite bed and the filtrate was distilled off completely to get a semi solid mass. 250 ml of Methanol was added to the residue and stirred for 2-3 hrs to give a solid product, which was filtered and washed with chilled methanol and dried.
Yield: 80-85%,
Purity by HPLC: 85-90%.
1H-NMR (CDCl3) δ: 7.8-8.1 (m, 1H), 6.7-7.61 (m, 22H), 5.78 (bs, 1H), 5.38(s, 2H), 4.12 (q, 2H), 2.52 (t, 2H), 1.64(s, 6H), 1.5-1.8(m, 2H), 1.08(t, 3H), 0.88(t, 3H).
- To a solution of Ethyl-4-(1-hydroxy-1-methylethyl) 2-propyl- 1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate (105 g, 0.14 mol) in tetrahydrofuran , was added LiOH.H2O (7.8 g, 0.18 mol) solution below 10°C. The reaction mixture was stirred at room temperature for 15 hours. Reaction mass was concentrated under vacuum at 35°C to 1/4 th of its volume. 300 ml of ethyl acetate and NaCl (130 g) were added to the residue under stirring. The organic phase was separated, dried over sodium sulphate and concentrated under vacuum to get the product. The crude product was taken as such to the next stage.
- To the solution of lithium salt of 4-(1-hydroxy-1-methylethyl) 2-propyl- 1-[[2'-[2-(triphenylmethyl)-2H-tetrazol-5yl] biphenyl-4-yl] methyl] imidazole -5-carboxylic acid , (97 g, 0.13 mol) in N, N-dimethyl acetamide(200 ml) was added triethylamine(12.7 g, 0.12 mol), stirred at room temperature for 0.5 hours. 5-methyl-2-oxo-(1,3-dioxolene-4-yl)methyl chloride (85% purity, 37.3 g, 0.25 mol) was added below 10°C. The mixture was stirred at 50-55°C for 4 hours, checked TLC. Dichloromethane (400 ml) and chilled water (500 ml) were added under stirring. The organic phase was separated, given brine wash (50 ml), dried over sodium sulphate and concentrated under vacuum to get a residue. To the residue methanol was added, stirred for 1hr, cooled to 5-10°, filtered and washed with chilled methanol and dried.
Yield: 75-80%,
Purity by HPLC: 96-98%.
1H-NMR (CDCl3) δ: 7.87(d, 1H), 6.90-7.52(m, 20H), 6.68(d, 2H), 5.61(s, 1H), 5.3(s, 2H), 4.7(s, 2H), 2.54(t, 2H), 1.97(s, 3H), 1.6-1.75(m, 2H), 1.62(s, 6H), 0.87(t, 3H).
- To the suspension of trityl protected olmesartan medoxomil (50g, 0.06 mol) in 250 ml 75% acetic acid was stirred at 50-55°C for 1.5hrs and cooled to 5-10°C. The by-product trityl alcohol was filtered off and washed with 75% acetic acid. The filtrate was concentrated under vacuum to get syrup, which was crystallized using isopropyl alcohol.
Yield: 85-88%,
Purity by HPLC: 95-98%.
The material was further purified with ethyl methyl ketone. It was filtered and washed with ethyl methyl ketone and dried to give substantially pure olmesartan medoxomil.
Yield: 70-75%,
Purity by HPLC: 99.3-99.7%.
1H-NMR (CDCl3) δ: 7.81(dd, 1H), 7.43-7.6(m, 3H), 7.09d, 2H), 6.79(d, 2H), 5.41(s, 1H),4.95(s, 1H), 2.56(t, 3H), 2.17(s, 3H), 1.58-1.69(m, 2H), 1.58(s, 6H), 0.92(t,3H).
....................
NMR
OLMESARTAN MEDOXIMIL
1H-NMR (CDCl3) δ: 7.81(dd, 1H), 7.43-7.6(m, 3H), 7.09d, 2H), 6.79(d, 2H), 5.41(s, 1H),4.95(s, 1H), 2.56(t, 3H), 2.17(s, 3H), 1.58-1.69(m, 2H), 1.58(s, 6H), 0.92(t,3H).
......................................................................................
7
AMG 925
AMG 925
1401033-86-0
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone
2-Hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone
2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (AMG 925)
FLT3/CDK4 inhibitor,potent and selective
AMG 925 is a dual kinase inhibitor of FLT3 and CDK4 with IC50 value of 1 nM and 3 nM, respectively
C26H29N7O2., 471.55
BY
SECTION 1
STEP A

STEP B

STEP C

STEP D

9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine
COUPLER 1

tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)-carboxylate
STEP E

tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridine-6(5H)-carboxylate
STEP F
 COMPD 1
COMPD 1
9-((l r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro- l,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1)
SECTION B
COUPLER2

2,5-dioxopyrrolidin-l-yl 2-acetoxyacetate
STEP G

2-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)-2-oxoethyl acetate
STEP H
 AMG 925
AMG 925
STEP I
AMG 925 is a potent, selective, and orally available FLT3/CDK4 dual inhibitor. It also inhibits CDK6 potently in kinase assay. In acute myeloid leukemia (AML) cell lines MOLM13 and Mv4-11, AMG 925 inhibits cell growth (IC50 values of 19nM and 18nM, respectively) through inhibiting P-FLT3 and P-STAT5 and inducing apoptosis. FLT3 mutants cause resistance to the current FLT3 inhibitors. AMG 925 is reported to inhibit cell growth in AML cells with FLT3 mutants FLT3-D835Y and FLT3-D835V. In AML tumor –bearing mice, administration of AMG 925 shows inhibition of P-STAT5 and P-RB as well as cell growth both in subcutaneous MOLM13 xenograft tumor model and systemic MOLM13-Luc xenograft tumor model. AMG 925 is also reported to have antitumor activity in a dose-dependent manner in theRB-positive Colo205 colon adenocarcinoma xenograft model
AMGEN
cute myeloid leukemia (AML) represents a significant unmet medical need. It is a hematological malignancy characterized by a block in differentiation and aberrant proliferation of the myeloid lineage of hematopoietic progenitor cells. There are approximately 13,000 new cases and 9,000 deaths per year in the United States. The survival rate is 25-70% in patients younger than 60 years and 5-15% in older patients, with worse outcomes in patients with poor risk cytogenetics. Current standard of care treatment is daunorubicin and cytarabine chemotherapy with induction and consolidation phases. Bone marrow stem cell transplant is also used for treating AML in younger patients.
Cyclin-dependent kinases (CDKs) are a family of serine/ threonine protein kinases playing important cellular functions. The cyclins are the regulatory subunits that activate the catalytic CDKs. CDKl/Cyclin B 1 , CDK2/Cyclin A, CDK2/Cyclin E, CDK4/Cyclin D, CDK6/Cyclin D are critical regulators of cell cycle progression. CDKs also regulate transcription, DNA repair, differentiation, senescence and apoptosis (Morgan, D. O., Annu. Rev. Cell. Dev. Biol., 13:261-291 (1997)).
Small molecule inhibitors of CDKs have been developed to treat cancer
(de Career, G. et al., Curr. Med. Chem., 14:969-85 (2007)). A large amount of genetic evidence supports that CDKs, their substrates or regulators have been shown to be associated with many human cancers (Malumbres, M. et al, Nature Rev. Cancer, 1 :222- 231 (2001)). Endogenous protein inhibitors of CDKs including p 16, p21 and p27 inhibit CDK activity and their overexpression results in cell cycle arrest and inhibition of tumor growth in preclinical models (Kamb, A., Curr. Top. Microbiolo. Immunol., 227: 139- 148 (1998)).
Small molecule inhibitors of CDKs may also be used to treat variety of other diseases that result from aberrant cell proliferation, including cardiovascular disorders, renal diseases, certain infectious diseases and autoimmune diseases. Cell proliferation pathways including genes involved in the cell cycle Gl and S phase checkpoint (p53, pRb, pi 5, pi 6, and Cyclins A, D, E, CDK 2 and CDK4) have been associated with plaque progression, stenosis and restenosis after angioplasty. Over- expression of the CDK inhibitor protein p21 has been shown to inhibit vascular smooth muscle proliferation and intimal hyperplasia following angioplasty (Chang, M. W. et al., J. Clin. Invest, 96:2260 (1995); Yang, Z-Y. et al., Proc. Natl. Acad. Sci. (USA) 93:9905 (1996)). A small molecule CDK2 inhibitor CVT-313 (Ki = 95 nM) was shown to cause significant inhibition of neointima formation in animal models (Brooks, E. E. et al., J. Biol. Chem., 272:29207-2921 1 (1997)). Disregulation of cell cycle has been associated with polycystic kidney diseases, which are characterized by the growth of fluid-filled cysts in renal tubules. Treatment with small molecule inhibitors of CDKs yielded effective arrest of cystic disease in mouse models (Bukanov, N. O., et al., Nature, 4444:949-952 (2006)).
Infection by a variety of infectious agents, including fungi, protozoan parasites such as Plasmodium falciparum, and DNA and RNA viruses may be treated with CDK inhibitors. CDKs have been shown to be required for replication of herpes simplex virus (HSV) (Schang, L. M. et al., J. Virol., 72:5626 (1998)). Synovial tissue hyperplasia plays important roles in the development of rheumatoid arthritis; inhibition of synovial tissue proliferation may suppress inflammation and prevent joint destruction. It has been shown that over-expression of CDK inhibitor protein pl6 inhibited synovial fibroblast growth (Taniguchi, K. et al., Nat. Med., 5:760-767 (1999)) and joint swelling was substantially inhibited in animal arthritis models.
Selective inhibitors of some CDKs may also be used to protect normal untransformed cells by inhibiting specific phases of cell cycle progression (Chen, et al., J. Natl. Cancer Institute, 92: 1999-2008 (2000)). Pre-treatment with a selective CDK inhibitor prior to the use of a cytotoxic agent that inhibits a different phase of the cell cycle may reduce the side effects associated with the cytotoxic chemotherapy and possibly increase the therapeutic widow. It has been shown that induction of cellular protein inhibitors of CDKs (pi 6, p27 and p21) conferred strong resistance to paclitaxel- or cisplatin-mediated cytotoxicity on the inhibitor-responsive cells but not on the inhibitor-unresponsive cells (Schmidt, M, Oncogene, 2001 20:6164-71).
CDK4 and CDK6 are two functionally indistinguishable cyclin D dependent kinases. They are widely expressed with high levels of expression observed in cells of hematopoeitic lineage (CDK4/6 will be used throughout this document to reference both CDK4 and CDK6). CDK4/6 promotes Gl-S transition of the cell cycle by phosphorylating the retinoblastoma protein (Rb). CDK4 and CDK6 single knockout mice are viable and double knockout mice die around birth with defective
hematopoiesis (Satyanarayana, A. et al., Oncogene, 28:2925-39 (2009); Malumbres, M. et al., Cell, 1 18:493-504 (2004)). Strong evidence supports a significant involvement of the cyclin D-CDK4-pl6INK4A-Rb pathway in cancer development (Malumbres, M. et al., Nature Rev. Cancer, 1 :222-31 (2001)). Rb negatively regulates the cell cycle at Gl by sequestering E2F proteins that are required for initiation of S phase, p 1 is a key member of the ΓΝΚ4 family of CDK4/6 cellular inhibitors. The genes for Rb and pl6INK4A are tumor suppressors that are often deleted or silenced in cancer cells.
Additionally CDK4, CDK6 and cyclin D are reported to be amplified in hematologic malignancies and solid tumors. The importance of this pathway in oncogenesis is further supported by the finding that depletion or inactivation of CDK4 inhibits tumor growth in mouse tumor models (Yu, Q. et al., Cancer Cell, 9:23-32 (2006); Puyol, M. Cancer Cell, 18:63-73 (2010)). Rb and p 16^^ are rarely deleted in AML. However, the plS1^^ gene, another member of the ΓΝΚ4 family, has been reported to be down regulated by hypermethylation in up to 60% of AML (Naofumi, M. et al., Leukemia Res., 29:557-64 (2005); Drexler, H. G. Leukemia, 12:845-59 (1998); Herman, J. G. et al., Cancer Res., 57:837-41 (1997)), suggesting a possible critical role for CDK4/6 in AML cells.
FLT3 (Fms-like tyrosine kinase 3, FLK2) is a class III receptor tyrosine kinase. It is activated by the FLT3 ligand (FL) and signals through the PI3K, RAS, and JAK/STAT pathways (Scholl C. et al., Semin. Oncol., 35:336-45 (2008); Meshinchi S. et al., Clin. Cancer Res., 15:4263-9 (2009)). FLT3 plays a role in early hematopoiesis and FLT3 deficient mice have reduced numbers of progenitors of multiple lymphoid lineages (Mackarehtschian K, et al., Immunity, 3: 147-61 (1995). Activating mutations in FLT3 are found in approximately 30% of AML patients, representing the most frequent genetic alteration in the disease. About 75% of the activating mutations are internal tandem duplications (ITD) and 25% are point mutations in the activation loop of the kinase domain.
The most frequently identified activating point mutation is D835Y (Yamamoto et al., Blood, 97(8): 2434-2439 (2001)). However, mutations have also been found at N841I (Jiang, J. et al., Blood, 104(6): 1855-1858 (2004)) and Y842C (Kindler et al., Blood, 105(1): 335-340 (2005)). Additional point mutations have been identified in the juxtamembrane domain and kinase domain, although these have been shown to result in lower transforming potential (Reindel et al., Blood 107(9): 3700- 3707 (2006)).
Murine bone marrow transplanted with a retrovirus expressing the
FLT3-ITD has been shown to result in the production of a lethal myeloproliferative disease in mice (Kelly et al., Blood 99: 310-318 (2002)) characterized by leukocytosis consisting of mature neutrophils. This disease did not show a block in differentiation as seen in human AML suggesting that FLT3 mutations confer a proliferative or survival advantage to the cells. Additional oncogene mutation producing a block in
differentiation such as AML1/ETO is hypothesized to be required to produce disease that is more similar to human AML.
A number of FLT3 inhibitors have been tested in clinical trials.
Although they have shown initial clinical responses in AML, the responses observed were transient and resistance can develop rapidly (Weisberg, E. et al., Oncogene, 29:5120-34 (2010)). The major resistance mechanism appears to be through the acquisition of secondary mutations in FLT3, which may interfere with the binding of FLT3 inhibitors to the FLT3 receptor (Weisberg, E. et al., Oncogene, 29:5120-34 (2010); Chu, S. H. et al., Drug Resist. Update, 12:8-16 (2009)). One such resistance mutation (N676K) was identified in a patient at the time of clinical relapse while on multi-kinase FLT3 inhibitor midostaurin (PKC412) monotherapy (Heidel, F. et al., Blood, 107:293-300 (2006)). Combinations of FLT3 inhibitors with chemotherapy are being tested in clinical trials despite the recognition that chemotherapy is poorly tolerated. Additional possible mechanisms for lack of durable responses include inadequate target coverage (Pratz, K. W., et al., Blood, 139:3938-46 (2009)) and protection of AML cells in the bone marrow where stromal growth factors may provide proliferative signals in addition to FLT3 activation (Tarn, W. F. et al., Best Pract. Res. Clin. Haematol., 21 : 13-20 (2008)). Inhibitors with combined FLT3 and CDK4/6 inhibitory activities are novel and may prove beneficial in treating various cancers including, but not limited to, AML.
Fused tricyclic pyridine, pyrimidine, and triazine compounds useful for treating diseases mediated by CDK4 are disclosed in WO 2009/085185, published on July 9, 2009, which is hereby incorporated by reference in its entirety and for all purposes as if fully set forth herein. Various gem-disubstituted and spirocyclic compounds useful for treating diseases mediated by CDK4 are disclosed in WO 2009/0126584, published on October 15, 2009, which is hereby incorporated by reference in its entirety and for all purposes as if fully set forth herein.
A continued need exists for new compounds that can be used to modulate CDK4, CDK6, and/or FLT3 and can be used to treat various disease conditions associated with these kinases. The compounds of the present invention provide significant improvements in inhibition in one or more of these kinases and have properties making them excellent therapeutic candidates.
………………………
[01 16] In some embo f Formula I, the compound is

SCHEME 3
R1′-CI




3G …………………………………………………………..3H
Example 1. 9-((lr,4r)-4-Methylcyclohexyl)-N-(5,6,7,8-tetrahydro-1 ,6-naphthyridin-2-yl)-9H-py 3-d] pyrimidin-2-amine
 KEY INTERMEDIATE 1
KEY INTERMEDIATE 1
……………………………………..SECTION 1 BELOW
STEP A
4-Chloropyrimidine-2-amine (commercially available from Sigma-Aldrich, St. Louis, MO) (1000 g, 7.72 mol, 1.0 eq), trans- 4-methylcyclohexylamine hydrochloride (commercially available from TCI America, M1780) (1500 g, 10.03 mol, 1.3 eq) and TEA (3.23 L, 23.2 mol, 3.0 eq) were mixed together in n-butanol (8 L). The reaction mixture was heated at reflux for 36 hours and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, diluted with water (8 L) and extracted with EtOAc (2 x 10 L). The organic layers were combined, dried over Na2S04, and concentrated under reduced pressure to give the title compound (1770 g) which was us
STEP B
Synthesis of 5-iodo-A^-((lr,4r)-4-methylcyclohexyl)pyridine-2,4- diamine. N4-((lr,4r)-4-Methylcyclohexyl)pyridine-2,4-diamine (1770 g, 8.58 mol, 1.0 eq) was dissolved in anhydrous DMF (8 L). To this solution under N2 atmosphere at 10 °C was added NIS (1.93 kg, 8.58 mol, 1.0 eq) in portions over 10 minutes. Upon completion of the addition, the reaction mixture was stirred at room temperature for 2 hours. The reaction was monitored using LCMS. Upon completion, the reaction mixture was cooled using an ice bath, quenched with saturated aqueous sodium carbonate (5 L) and extracted with EtOAc (2 x 15 L). The combined organic extracts were washed with saturated aqueous sodium carbonate (2 x 5 L), water (3 x 2 L), dried over Na2S04, and concentrated under reduced pressure. The residue was purified using column chromatography eluting with 25% to 40% EtOAc in hexanes to provide the title compound (1.47 kg, 57% over two steps). ^-NMR (300 MHz, DMSO-d6) δ ppm 0.85 (3H, d, J= 7.2 Hz), 0.98 (1H, dd, J= 12.9, 2.7 Hz), 1.41 – 1.27 (3H, m), 1.66 (2H, d, J = 12.3 Hz), 1.78 (2H, d, J= 12.3 Hz), 3.85 (1H, m), 5.48 (1H, d, J= 8.1 Hz), 6.16 (2H, br s), 7.86
STEPC
Synthesis of 5-(3-fluoropyridin-4-yl)-N -((lr,4r)-4- methylcyclohexyl)pyrimidine-2,4-diamine. To a solution of 2,2,6,6- tetramethylpiperidine (commercially available from Sigma-Aldrich, St. Louis, MO) (997 mL, 5.87 mol, 3 eq) in anhydrous THF (6 L) under N2 atmosphere at 0 °C, was added n-BuLi (2.5 M in hexanes, 2.35 L, 5.87 mol, 3 eq) via an addition funnel over 30 minutes. Upon completion of the addition, the reaction mixture was stirred at 0 °C for 1 hour. The reaction mixture was cooled to -74 °C (acetone/ dry ice bath) and a solution of 3-fluoropyridine (commercially available from Sigma-Aldrich, St. Louis, MO) (561 g, 5.773 mol, 2.95 eq) in anhydrous THF (500 mL) was added over 15 minutes keeping the temperature below -63 °C. Upon completion of the addition, the reaction mixture was stirred at -74 °C for an additional 2 hours. A solution of ZnBr2 (1422 g, 6.32 mol, 3.22 eq) in anhydrous THF (3 L) was then added dropwise over 35 minutes keeping the temperature below -60 °C. Upon completion of the addition, the cold bath was removed and the reaction mixture was allowed to warm to room temperature. Then 5- iodo-N4-((lr,4r)-4-methylcyclohexyl)pyridine-2,4-diamine (650 g, 1.95 mol, 1.0 eq) was added in one portion followed by Pd(PPh3)4 (113 g, 97.8 mmol, 0.05 eq). The reaction mixture was heated at reflux overnight and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, quenched with saturated aqueous NaHC03 (6 L) and extracted with EtOAc (10 L x 2). The organic extracts were washed with saturated NaHC03 (2.5 L x 2) and brine (2.5 L), and were then concentrated under vacuum. The residue was dissolved in 2N HC1 (2.5 L) and washed with DCM (1.25 L x 3). The aqueous phase was adjusted to pH 10-12 by addition of aqueous 4N NaOH and extracted with DCM (1.5 L x 3). The organic extracts were washed with water (1.25 L x 2), dried and concentrated to give the title compound (540 g, 92%). ^-NMR (300 MHz, DMSO-d6) δ ppm 0.85 (3H, d, J= 7.2 Hz), 0.98 (1H, dd, J= 12.9, 2.7 Hz), 1.30 – 1.18 (3H, m), 1.64 (2H, d, J= 12.3 Hz), 1.74 (2H, d, J= 1 1.7 Hz), 3.96 (1H, m), 5.00 (1H, d, J= 8.4 Hz), 6.24 (2H, br s), 7.35 (1H, dd, J= 6.6, 4.4 Hz), 7.58 (1H, s), 8.37 (1H, d, J= 4.8 Hz), 8.50 (1H, d, J= 6.6 Hz) ppm.
STEPD
Synthesis of 9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine. To a solution of 5-(3- fluoropyridin-4-yl)-N4-((lr,4r)-4-methylcyclohexyl)pyrimidine-2,4-diamine (854 g, 2.84 mol, 1.0 eq) in anhydrous 1 -methyl-2-pyrrolidinone (8 L) under N2atmosphere at room temperature, was added LiHMDS (1.0 M in toluene, 8.5 L, 8.5 mol, 3.0 eq) over 30 minutes. Upon completion of the addition, the reaction mixture was heated at 90 °C overnight and monitored using LCMS. Upon completion, the reaction mixture was cooled to room temperature, quenched with ice cold water (10 L) and extracted with EtOAc (12 L). The organic phase was washed with saturated aqueous NaHC03 (4 L x 2), and water (2 L x 3). The aqueous layers were combined and back extracted with EtOAc (15 L x 2). The organic layers were combined, dried over Na2SO/t, and concentrated under reduced pressure. The solid thus obtained was suspended in DCM (2.5 L) and agitated using a rotary evaporator for 30minutes. The solid was collected by filtration, washed with DCM and dried to afford the title compound (400 g). The mother liquor was purified by column chromatography (eluting with DCM/MeOH = 50: 1) to afford, after triturating with DCM (750 mL), additional title compound (277 g, total: 677 g, yield: 84%). ¾ NMR (300 MHz, CD3OD) δ ppm 1.02 (d, J= 6.3 Hz, 3H), 1.33-1.20 (m, 2H), 1.67-1.60 (m, 2H), 1.95-1.84 (m, 4H), 1.58-1.45 (m, 2H), 4.87-4.77 (m, 1H), 7.94 (d, J= 5.1 Hz, 1H), 8.31 (d, J= 5.1 Hz, 1H), 8.87 (s, 1H), 8.96 (s, 1H) ppm; MS m/z: 28
……………………………………
COUPLER 1
Synthesis of tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)-carboxylate. To a slurry of 2-chloro-5,6,7,8-tetrahydro-l,6-naphthyridine hydrochloride (106.1 g, 517 mmol, commercially available from D-L Chiral Chemicals, ST-0143) and N,N-diisopropylethylamine (80 g, 108 mL, 621 mmol, 1.2 eq) in DCM (1 L) was added a solution of di-tert-butyl dicarbonate (119 g, 543 mmol, 1.05 eq) in
DCM (100 mL) via an addition funnel within 1 hr. The reaction mixture became a clean solution and the solution thus obtained was stirred at room temperature for an additional hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was dissolved in EtOAc (1 L) and washed with water (3 x 300 mL), washed with brine (300 mL) and dried over MgSOzt. The solvent was evaporated under vacuum to give the title compound as an off- white solid (139 g, yield: 100%). lH NMR (400MHz ,CDC13) δ ppm 1.49 (9H, s), 2.97 (2H, t, J= 5.9 Hz), 3.73 (2H, t, J= 6.0 Hz), 4.57 (2H, s), 7.17 (1H, d, J= 8.0 Hz), 7.38 (1H, d, J= 8.0 Hz) ppm;
……………………………
STEP E
Synthesis of tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridine-6(5H)-carboxylate. To a solution of 9-((lr,4r)-4-methylcyclohexyl)- 9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (2.81 g, lO mmol) in 1,4-dioxane (45 mL) were added tert-butyl 2-chloro-7,8-dihydro-l,6-naphthyridine-6(5H)- carboxylate (2.57 g, 9.55 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanene (231 mg, 0.40 mmol), and sodium t-butoxide (1.44 g, 15 mmol). Argon was bubbled through the mixture for 10 minutes. Tris(dibenzylideneacetone)dipalladium (0)(183 mg, 0.20 mmol) was added, and argon was again bubbled through the mixture for 5 minutes. The reaction mixture thus obtained was stirred at 100 °C for 3 hours whereupon HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was cooled to 40 °C and diluted with DCM (90 mL) and treated with Si- triamine (functionalized silica gel, from Silicycle, FR31017TR130B) (2.8 g) overnight at room temperature. Celite® brand filter aid 545 (6 g) was added, and the mixture was filtered with a sintered glass funnel and the solid phase was rinsed with DCM (100 mL). The filtrate was concentrated to 25 mL on a rotary evaporator and diluted with a mixture of EtOAc and hexane (20 mL, 4: 1). The resulting slurry was stirred at room temperature for 5 hours. The solid was collected by filtration, washed with a mixture of EtOAc and hexane (20 mL, 1 : 1) and air dried for a few hours to provide the title compound as an off-white solid (4.90 g, 100% yield). lH NMR (500 MHz, CD2C12) δ ppm 1.06 (3H, d, J= 6.4 Hz), 1.34 – 1.22 (2H, m), 1.48 (9H, s), 1.67 (1H, br. s), 2.02 – 1.93 (4H, m), 2.63 (2H, dq, J= 3.1, 12.8 Hz), 2.88 (2H, t, J= 5.7 Hz), 3.74 (2H, t, J= 6.0 Hz), 4.57 (2H, s), 7.51 (1H, d, J= 8.6 Hz), 7.85 (1H, d, J= 5.1 Hz), 8.10 (1H, br. s), 8.42 (1H, d, J= 8.3 Hz), 8.46 (1H, d, J= 4.9 Hz), 8.97 (1H, s), 9.10 (1H, s) ppm;
…………………………
STEP F
1
Synthesis of 9-((l r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro- l,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1).
To a suspension of tert-butyl 2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amm^
6(5H)-carboxylate: 9-((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3- d]pyrimidin-2-amine (4.65 g, 9.05 mmol) in MeOH (30 mL) were added concentrated HC1 (6.74 mL) and water (14 mL). The mixture thus obtained was stirred at room temperature overnight. 50% NaOH in water (4.8 mL) was added at 0 °C to the reaction mixture to adjust the pH value to 9. The precipitated yellow solid was collected by filtration, rinsed with water (25 mL) and air dried for 3 days to give the title compound (3.75 g, 100%). lH NMR (400 MHz, CDC13) δ ppm 1.07 (3H, d, J= 6.5 Hz), 1.29 – 1.25 (3H, m), 2.00 – 1.95 (3H, m), 2.02 (2H, s), 2.69 – 2.53 (2H, m), 2.89 (2H, t, J= 6.0 Hz), 3.26 (2H, t, J= 6.0 Hz), 4.04 (2H, s), 4.71 (1H, m, J= 12.8, 12.8 Hz), 7.41 (1H, d, J= 8.4 Hz), 7.84 (1H, d, J= 6.1 Hz), 7.84 (1H, d, J= 6.1 Hz), 8.03 (1H, s), 8.34 (1H, d, J= 8.4 Hz), 8.50 (1H, d, J= 5.3 Hz), 8.96 (1H, s), 9.08 (1H, s) ppm; LCMS m/z: 414 (M+l).
SECTION 2 BELOW
SYNTHESIS OF LABEL 5 FROM 1
Example 5. 2-Hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone
 LABEL 5
LABEL 5
…………………………
COUPLER 2
Synthesis of 2,5-dioxopyrrolidin-l-yl 2-acetoxyacetate.
A 3-neck round-bottom flask equipped with a mechanical stirrer, thermocouple and addition funnel with nitrogen inlet was charged with N-hydroxysuccinimide (commercially available from Sigma- Aldrich, St. Louis, MO) (21 1 g, 1.83 mol) and DCM (2.25 L) at room temperature, resulting in a suspension. Pyridine (178 mL, 2.2 mol) was added in one portion with no change in the internal temperature. A solution of acetoxyacetyl chloride (commercially available from Sigma-Aldrich, St. Louis, MO) (197 mL, 1.83 mol) in DCM (225 mL) was added dropwise over 60 minutes and the temperature rose to 35 °C. Stirring was continued at room temperature for 2.5 hours. The reaction mixture was washed with water (IxlL), IN HCl (2xlL) and brine (IxlL). The organic layer was concentrated under vacuum and azeotroped with toluene (IxlL) to obtain the product as a white solid (367 g, 93%). lH NMR (400MHz, CDC13) δ 4.96 (2H, s), 2.86 (4H s), 2.19 (3H, s) ppm; LCMS m/z: 238 (M+Na).
…………………………………
STEP G
Synthesis of 2-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)-2-oxoethyl acetate.
To a suspension of 9-((lr,4r)-4- methylcyclohexyl)-N-(5,6,7,8-tetrahydro-l,6-naphthyridin-2-yl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1) (827 mg, 2.0 mmol) in chloroform (10 mL) were added diisopropylethylamine (258 mg, 348 uL, 2.0 mmol) and 2,5- dioxopyrrolidin- l-yl 2-acetoxyacetate (560 mg, 2.6 mmol). The reaction mixture thus obtained was stirred at room temperature for 30 minutes whereupon the mixture became a yellow solution. HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was concentrated. MeOH (5 mL) and water (6 mL) were added to form a slurry which was stirred at room temperature for 1 hour. The solid was collected by filtration to give the title compound as a light yellow solid (1.04 g, 98% yield). lH NMR (400 MHz, CDC13, rotamers) δ ppm 1.08 (3H, d, J= 6.5 Hz), 1.37 – 1.20 (2H, m), 2.03 – 1.97 (4H, m), 2.22 (3H, s), 2.69 – 2.52 (2H, m, J= 2.9, 12.8, 12.8, 12.8 Hz), 3.08 – 2.93 (2H, m), 3.75 (1H, t, J= 5.9 Hz), 3.97 (1H, t, J= 5.6 Hz), 4.59 (1H, s), ), 4.80 – 4.65 (2H, m), ), 4.90 – 4.82 (2H, m), 7.57 – 7.45 (1H, m), 7.86 (1H, d, J= 5.7 Hz), 8.21 – 8.10 (1H, m), 8.49 – 8.40 (1H, m), 8.52 (1H, d, J= 5.3 Hz), 8.
…………………………………..
STEPH
LABEL 5
Synthesis of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone (5).
To a solution of 2-(2-((9-((lr,4r)-4- methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8- dihydro-l,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate (514 mg, 1.0 mmol) in DCM (7.5 mL) and MeOH (2.5 mL) was added 0.5 M sodium methoxide solution in MeOH (0.30 mL, 0.15 mmol), and the reaction mixture was stirred at room temperature for 1 hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was treated with EtOH (5 mL) and water (10 mL) to provide a solid which was collected by filtration, washed with water, and dried in a vacuum oven at 55 °C overnight to give the title compound (5) as a white solid (468 mg, 99% yield).
lH NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 1.09 (3H, d, J= 6.5 Hz), 1.31-1.43 (2H, m), 1.70-1.80 (1H, m), 1.99-2.03 (2H, m), 2.06-2.13 (2H, m), 2.68 (2H, dq, J= 3.3, 12.7 Hz), 3.10 (2H, t, J= 5.4 Hz), 3.88 (2H, br. s.), 4.46 (2H, br. s.), 4.77 (2H, br. s), 4.90 (1H, tt, J= 3.9, 12.4 Hz), 7.76 (1H, d, J= 8.5 Hz), 8.33 (1H, d, J= 8.5 Hz), 8.40 (1H, d, J= 6.0 Hz), 8.63 (1H, d, J= 6.0 Hz), 9.35 (1H, s), 9.43 (1H, s) ppm; L
………………………………………………
STEP I

5 LABEL HCI Dihydrate
[0222] Synthesis of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido [4′,3 ‘ :4,5] pyrrolo [2,3-d] pyrimidin-2-yl)amino)-7,8-dihydro-l ,6- naphthyridin-6(5H)-yl)ethanone monohydrochloride dihydrate. To a suspension of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3- d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone (472 mg, 1.0 mmol) in water (2 mL) was added 2 N HCI (2 mL). The mixture became a clear solution. The pH value of the solution was adjusted to 4 by addition of 2 N NaOH at 0 °C and the precipitated light yellow solid was collected by filtration. The collected solid was washed with cold water three times. The solid was dried under vacuum to give the title compound as a light yellow solid (469 mg, 92% yield).
¾ NMR (500 MHz, DMSO-d6) δ 1.02 (3H, d, J= 5.0 Hz), 1.20- 1.30 (2H, m), 1.64 (1H, m), 1.88-1.90 (4H, m), 2.59-2.66 (2H, m), 2.85-2.95 (2H, m), 3.71(1H, m), 3.83 (1H, m), 4.19-4.22 (2H, m), 4.60-4.67 (2H, m), 4.85 (1H, m), 7.75 (1H, d, J= 8.5 Hz), 8.19 (1H, d, J= 8.5 Hz), 8.55 (1H, d, J= 5.0 Hz), 8.63 (1H, d, J= 5.0 Hz), 9.47 (1H, s), 9.58 (1H, s), 10.59 (1H, br.s) ppm; LCMS m/z: 472 (M+l). Anal.
Calc: C = 57.40, H = 6.30, N = 18.02; Found: C = 57.06, H = 6.31, N = 17.92. [0223] Alternative Synthesis of Hydrochloride Salt of 2-Hydroxy-l-(2-((9-
((lr,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2- yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone. To a suspension of 2- hydroxy- 1 -(2-((9-(( 1 r,4r)-4-methylcyclohexyl)-9H-pyrido [4′,3 ‘ :4,5]pyrrolo [2,3 – d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)ethanone (2.385 g, 5.0 mmol) in water (10 mL) was added 2N HC1 (10 mL) at 20°C. The mixture became a clear light yellow solution. The pH value of the solution was adjusted to 4 by addition of 2N NaOH through addition funnel at 0° C, and the precipitated yellow solid was collected by filtration. The resulting solid was washed with cold water three times. The solid was dried under vacuum at 50° C for two days to provide 2.49 g of the hydrochloride salt of 2-hydroxy-l-(2-((9-((lr,4r)-4-methylcyclohexyl)-9H- pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-l,6-naphthyridin- 6(5H)-yl)ethanone as a solid. This salt was also obtained as a hydrate.
………………
Example 5
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone


Synthesis of 2,5-dioxopyrrolidin-1-yl 2-acetoxyacetate
A 3-neck round-bottom flask equipped with a mechanical stirrer, thermocouple and addition funnel with nitrogen inlet was charged with N-hydroxysuccinimide (commercially available from Sigma-Aldrich, St. Louis, Mo.) (211 g, 1.83 mol) and DCM (2.25 L) at room temperature, resulting in a suspension. Pyridine (178 mL, 2.2 mol) was added in one portion with no change in the internal temperature. A solution of acetoxyacetyl chloride (commercially available from Sigma-Aldrich, St. Louis, Mo.) (197 mL, 1.83 mol) in DCM (225 mL) was added dropwise over 60 minutes and the temperature rose to 35° C. Stirring was continued at room temperature for 2.5 hours. The reaction mixture was washed with water (1×1 L), 1N HCl (2×1 L) and brine (1×1 L). The organic layer was concentrated under vacuum and azeotroped with toluene (1×1 L) to obtain the product as a white solid (367 g, 93%). 1H NMR (400 MHz, CDCl3) δ 4.96 (2H, s), 2.86 (4H, s), 2.19 (3H, s) ppm; LCMS m/z: 238 (M+Na).

Synthesis of 2-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate
To a suspension of 9-((1r,4r)-4-methylcyclohexyl)-N-(5,6,7,8-tetrahydro-1,6-naphthyridin-2-yl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-amine (1) (827 mg, 2.0 mmol) in chloroform (10 mL) were added diisopropylethylamine (258 mg, 348 uL, 2.0 mmol) and 2,5-dioxopyrrolidin-1-yl 2-acetoxyacetate (560 mg, 2.6 mmol). The reaction mixture thus obtained was stirred at room temperature for 30 minutes whereupon the mixture became a yellow solution. HPLC-MS analysis indicated that the reaction was complete. The reaction mixture was concentrated. MeOH (5 mL) and water (6 mL) were added to form a slurry which was stirred at room temperature for 1 hour. The solid was collected by filtration to give the title compound as a light yellow solid (1.04 g, 98% yield). 1H NMR (400 MHz, CDCl3, rotamers) δ ppm 1.08 (3H, d, J=6.5 Hz), 1.37-1.20 (2H, m), 2.03-1.97 (4H, m), 2.22 (3H, s), 2.69-2.52 (2H, m, J=2.9, 12.8, 12.8, 12.8 Hz), 3.08-2.93 (2H, m), 3.75 (1H, t, J=5.9 Hz), 3.97 (1H, t, J=5.6 Hz), 4.59 (1H, s),), 4.80-4.65 (2H, m),), 4.90-4.82 (2H, m), 7.57-7.45 (1H, m), 7.86 (1H, d, J=5.7 Hz), 8.21-8.10 (1H, m), 8.49-8.40 (1H, m), 8.52 (1H, d, J=5.3 Hz), 8.98 (1H, s), 9.11 (1H, s) ppm; LCMS m/z: 514 (M+1).

Synthesis of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (5)
To a solution of 2-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)-2-oxoethyl acetate (514 mg, 1.0 mmol) in DCM (7.5 mL) and MeOH (2.5 mL) was added 0.5 M sodium methoxide solution in MeOH (0.30 mL, 0.15 mmol), and the reaction mixture was stirred at room temperature for 1 hour and monitored using LCMS. Upon completion, the reaction mixture was concentrated. The residue was treated with EtOH (5 mL) and water (10 mL) to provide a solid which was collected by filtration, washed with water, and dried in a vacuum oven at 55° C. overnight to give the title compound (5) as a white solid (468 mg, 99% yield). 1H NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 1.09 (3H, d, J=6.5 Hz), 1.31-1.43 (2H, m), 1.70-1.80 (1H, m), 1.99-2.03 (2H, m), 2.06-2.13 (2H, m), 2.68 (2H, dq, J=3.3, 12.7 Hz), 3.10 (2H, t, J=5.4 Hz), 3.88 (2H, br. s.), 4.46 (2H, br. s.), 4.77 (2H, br. s), 4.90 (1H, tt, J=3.9, 12.4 Hz), 7.76 (1H, d, J=8.5 Hz), 8.33 (1H, d, J=8.5 Hz), 8.40 (1H, d, J=6.0 Hz), 8.63 (1H, d, J=6.0 Hz), 9.35 (1H, s), 9.43 (1H, s) ppm; LCMS m/z: 472 (M+1).

Synthesis of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone monohydrochloride dihydrate
To a suspension of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (472 mg, 1.0 mmol) in water (2 mL) was added 2 N HCl (2 mL). The mixture became a clear solution. The pH value of the solution was adjusted to 4 by addition of 2 N NaOH at 0° C. and the precipitated light yellow solid was collected by filtration. The collected solid was washed with cold water three times. The solid was dried under vacuum to give the title compound as a light yellow solid (469 mg, 92% yield). 1H NMR (500 MHz, DMSO-d6) δ 1.02 (3H, d, J=5.0 Hz), 1.20-1.30 (2H, m), 1.64 (1H, m), 1.88-1.90 (4H, m), 2.59-2.66 (2H, m), 2.85-2.95 (2H, m), 3.71 (1H, m), 3.83 (1H, m), 4.19-4.22 (2H, m), 4.60-4.67 (2H, m), 4.85 (1H, m), 7.75 (1H, d, J=8.5 Hz), 8.19 (1H, d, J=8.5 Hz), 8.55 (1H, d, J=5.0 Hz), 8.63 (1H, d, J=5.0 Hz), 9.47 (1H, s), 9.58 (1H, s), 10.59 (1H, br.s) ppm; LCMS m/z: 472 (M+1). Anal. (C26H29N7O2—HCl.2H2O) Calc: C=57.40, H=6.30, N=18.02. Found: C=57.06, H=6.31, N=17.92.
Alternative Synthesis of Hydrochloride Salt of 2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl) amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl) ethanone
To a suspension of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (2.385 g, 5.0 mmol) in water (10 mL) was added 2N HCl (10 mL) at 20° C. The mixture became a clear light yellow solution. The pH value of the solution was adjusted to 4 by addition of 2N NaOH through addition funnel at 0° C., and the precipitated yellow solid was collected by filtration. The resulting solid was washed with cold water three times. The solid was dried under vacuum at 50° C. for two days to provide 2.49 g of the hydrochloride salt of 2-hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone as a solid. This salt was also obtained as a hydrate.
………………………
J. Med. Chem., 2014, 57 (8), pp 3430–3449
DOI: 10.1021/jm500118j
2-Hydroxy-1-(2-((9-((1r,4r)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (28)
compound 28 as a white solid (468 mg, 99% yield).
1H NMR (500 MHz, acetic acid-d4, 373 K) δ ppm 9.43 (1 H, s), 9.35 (1 H, s), 8.63 (1H, d, J = 6.0 Hz), 8.40 (1 H, d, J = 6.0 Hz), 8.33 (1 H, d, J = 8.5 Hz), 7.76 (1 H, d, J = 8.5 Hz), 4.90 (1 H, m), 4.77 (2 H, br s), 4.46 (2 H, br s), 3.88 (2 H, br s), 3.10 (2 H, t, J = 5.4 Hz), 2.68 (2 H, dq, J = 12.7, 3.3 Hz), 2.06–2.13 (2 H, m), 1.99–2.03 (2 H, m), 1.70–1.80 (1 H, m), 1.31–1.43 (2 H, m), 1.09 (3H, d, J = 6.5 Hz).
HRMS (ESI) m/z: calculated for [M + H]+ 472.2455, found 472.2461.
…………………………
PAPER
OPRD
† Chemical Process R&D, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California91320
‡ Norchim S.A.S., 33 Quai d’Amont, Saint Leu d’Esserent, France 60340
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op500367p

The development of a synthetic route to manufacture the drug candidate AMG 925 on kilogram scale is reported herein. The hydrochloride salt of AMG 925 was prepared in 23% overall yield over eight steps from commercially available raw materials, and more than 8 kg of the target molecule were delivered. The synthetic route features a Buchwald–Hartwig amination using BrettPhos as ligand and conducted to afford 12 kg of product in a single batch. In addition, this work highlights the challenges associated with the use of poorly soluble process intermediates in the manufacture of active pharmaceutical ingredients. Creative solutions had to be devised to conduct seemingly routine activities such as salt removal, pH adjustment, and heavy metal scavenging due to the low solubility of the process intermediates. Finally, a slurry-to-slurry amidation protocol was optimized to allow for successful scale-up.
Manufacture of 2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone (AMG 925)
AMG 925 was isolated in 91.5% yield (8.31 kg), 95.5% overall mass balance, 99.9 wt %, and 99.7 LCAP.
Mp 213–215 °C;
1H NMR (400 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 9.47–9.59 (m, 2H), 8.76 (d, 1H, J = 6 Hz), 8.55 (d, 1H, J = 6 Hz), 8.48 (d, 1H,J = 9 Hz), 7.79–7.92 (m, 1H), 4.95 (t, 1H,J = 12 Hz), 4.87 and 4.68 (2 singlets, 2H), 4.47–4.59 (m, 2H), 4.04 and 3.80 (2 triplets, 2H, J= 6 Hz), 3.03–3.17 (m, 2H), 2.65–2.82 (m, 2H), 1.96–2.15 (m, 4H), 1.77 (br s, 1H), 1.39 (q, 2H, J = 12 Hz), 1.09 (d, 3H, J = 7 Hz);
13C NMR (100 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 171.9, 171.8, 158.4, 157.8, 154.7, 149.0, 148.9, 141.6, 135.2, 132.9, 126.3, 124.1, 123.6, 117.7, 113.7, 113.6, 107.5, 107.4, 60.1, 59.9, 56.3, 43.7, 42.6, 40.5, 38.7, 34.0, 31.5, 29.8, 28.8, 28.1, 21.5.
Manufacture of 2-Hydroxy-1-(2-((9-((1R,4R)-4-methylcyclohexyl)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-2-yl)amino)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)ethanone Hydrochloride (AMG 925 HCl)
AMG 925 HCl was isolated in 92.5% yield (7.96 kg), 99.1% overall mass balance, 83.8 wt % AMG 925, 99.75 LCAP AMG 925, 6.2 wt % Cl, 9.6 wt % water, 3800 ppm AcOH, d10 4.0 μm,d50 15.2 μm, d90 38.8 μ, Vm 18.7 μm, BET surface area 1.5 m2/g.
1H NMR (400 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 9.63 (s, 1H), 9.56 (s, 1H), 8.71–8.76 (m, 1H), 8.60–8.66 (m, 1H), 8.20–8.29 (m, 1H), 7.90–7.98 (m, 1H), 4.90–5.01 (m, 1H), 4.86 and 4.70 (2 singlets, 2H), 4.53 and 4.51 (2 singlets, 2H), 4.05 and 3.82 (2 triplets, 2H, J = 6 Hz), 3.11–3.26 (m, 2H), 2.68 (q, 2H, J = 12 Hz), 1.95–2.13 (m, 4H), 1.74 (br s, 1H), 1.36 (q, 2H, J = 12 Hz), 1.06 (d, 3H, J = 8 Hz);
13C NMR (100 MHz, acetic acid-d4, mixture of two rotamers at 20 °C) 174.9, 174.8, 161.3, 161.2, 160.5, 157.5, 151.6, 151.5, 149.3, 148.9, 145.5, 138.1, 136.0, 129.3, 129.2, 127.1, 126.6, 120.9, 116.7, 116.6, 110.8, 110.7, 63.0, 62.9, 59.3, 46.4, 45.3, 43.2, 41.3, 36.9, 34.3, 32.6, 31.3, 30.6, 24.4; exact mass [C26H29N7O2 + H]+: calculated = 472.2461, measured = 472.2451.
References:
1. K. Keegan et al, Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol Cancer Ther. 2014 Apr;13(4):880-9.
2. ZH Li, et al, Discovery of AMG 925, a FLT3 and CDK4 Dual Kinase Inhibitor with Preferential Affinity for the Activated State of FLT3, J. Med Chem, March 18, 2014
8 ALLISARTAN

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
 Shanghai Allist Pharmaceutical, Inc.
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension


CN200710094021.4 and CN201110289695.6 disclose the preparation of
Alicante medoxomil, the inventor repeated, the proceeds of crystal and
Chinese patent CN200710094131.0 consistent disclosed.
 Allisartan isoproxil
Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.
 As regards to the solid physical properties of the compound of
formula (I), the patent document of CN101024643A discloses that it is a
white solid, and its melting point is 134.5-136° C. However,
CN101024643A dose not disclose the crystalline structure of the compound
of formula (I).
As regards to the solid physical properties of the compound of
formula (I), the patent document of CN101024643A discloses that it is a
white solid, and its melting point is 134.5-136° C. However,
CN101024643A dose not disclose the crystalline structure of the compound
of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.
 CN200710094021.4 discloses another method
for preparing the compounds of formula I, the following route B, the
starting material by nucleophilic substitution, oxidation, an ester, a
tetrazole ring to obtain a compound of formula I, the first step of the
method nucleophilic substitution easy to generate an imidazole ring -3
para isomer impurities difficult to remove; the last step into the ring
to use sodium azide, operating dangerous.
CN200710094021.4 discloses another method
for preparing the compounds of formula I, the following route B, the
starting material by nucleophilic substitution, oxidation, an ester, a
tetrazole ring to obtain a compound of formula I, the first step of the
method nucleophilic substitution easy to generate an imidazole ring -3
para isomer impurities difficult to remove; the last step into the ring
to use sodium azide, operating dangerous.
 CN201210020174.5 disclosed a series of
anti-hypertensive compound and preparation method, the following line C,
the temperature control in the first step of its preparation O ~ 5 ° C,
a mixed solution of acetone and water, with a 5% aqueous solution of
sodium hypochlorite oxidation, yield 70%, the second step use of
potassium permanganate, manganese dioxide will produce the same, and a
yield of only 40%, the first two steps total yield of 28%, is very low,
and the post-treatment methods are by column separation, the first two
steps are used are organic and inorganic mixed solvent is not conducive
to recovery, not suitable for scale-up.
CN201210020174.5 disclosed a series of
anti-hypertensive compound and preparation method, the following line C,
the temperature control in the first step of its preparation O ~ 5 ° C,
a mixed solution of acetone and water, with a 5% aqueous solution of
sodium hypochlorite oxidation, yield 70%, the second step use of
potassium permanganate, manganese dioxide will produce the same, and a
yield of only 40%, the first two steps total yield of 28%, is very low,
and the post-treatment methods are by column separation, the first two
steps are used are organic and inorganic mixed solvent is not conducive
to recovery, not suitable for scale-up.






Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
Allist Shanghai Pharmaceutical Co., Ltd.
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension
The prior art discloses Arleigh medoxomil
illiquid, low bulk density, electrostatic phenomena evident. Chinese
patent discloses a CN200710094131.0 Alicante medoxomil polymorph and
method of preparation. Allie medoxomil based crystal prepared by the
method has high stability characteristics, but relatively small bulk
density of the crystal clear after the electrostatic phenomenon and poor
liquidity dried, crushed and used for easy dispensing generate dust,
operating the site clean and labor protection inconvenience, on the
other hand also for accurate weighing and packaging products
inconvenience.
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.
CHINANEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
2-butyl-4-chloro -1- [2 ‘- (1H- tetrazol-5-yl)
-1,1’-biphenyl- – methyl] – imidazole-5-carboxylic acid, 1 –
[(isopropoxy) – oxy] -, methyl ester, is a novel angiotensin Ⅱ receptor
antagonist. China Patent CN200680000397.8 disclosed structural formula
Alicante medoxomil compound. Allie medoxomil toxicity, blood pressure
better than the same type of products (such as losartan), which by
generating active metabolite (EXP3174) in vivo metabolism, and thus play
its antihypertensive effect.
The prior art discloses Arleigh medoxomil
illiquid, low bulk density, electrostatic phenomena evident. Chinese
patent discloses a CN200710094131.0 Alicante medoxomil polymorph and
method of preparation. Allie medoxomil based crystal prepared by the
method has high stability characteristics, but relatively small bulk
density of the crystal clear after the electrostatic phenomenon and poor
liquidity dried, crushed and used for easy dispensing generate dust,
operating the site clean and labor protection inconvenience, on the
other hand also for accurate weighing and packaging products
inconvenience.
CN200710094021.4 and CN201110289695.6 disclose the
preparation of Alicante medoxomil, the inventor repeated, the proceeds
of crystal and Chinese patent CN200710094131.0 consistent disclosed.
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.
Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)
 To a 20L reactor 9800ml of methanol,
stirring was started, the rotational speed is added at 200r / min
1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The
reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20
° C with stirring 3h, the reaction mixture was filtered to give a pale
yellow clear filtrate. The filtrate was
concentrated under reduced pressure to move 20L flask, vacuum degree of
0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
To a 20L reactor 9800ml of methanol,
stirring was started, the rotational speed is added at 200r / min
1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The
reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20
° C with stirring 3h, the reaction mixture was filtered to give a pale
yellow clear filtrate. The filtrate was
concentrated under reduced pressure to move 20L flask, vacuum degree of
0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)
 850ml of absolute ethanol was added to the
3L reaction vessel was charged with crude Alicante medoxomil (800.lg,
1.45mol), heated to reflux. After
completely dissolved clear, slow addition of n-heptane 1300ml, reflux
15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered
under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake
was washed / 3, the back pressure dry vacuum filtration, the purified
Alicante medoxomil (780.9g, 97.6% yield).
850ml of absolute ethanol was added to the
3L reaction vessel was charged with crude Alicante medoxomil (800.lg,
1.45mol), heated to reflux. After
completely dissolved clear, slow addition of n-heptane 1300ml, reflux
15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered
under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake
was washed / 3, the back pressure dry vacuum filtration, the purified
Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)
 950ml of absolute ethanol was added to the
5L reaction vessel was charged with crude Alicante medoxomil (549.9g,
1.72mol), heated to reflux. After
completely dissolved clear, slow addition of n-heptane 1200ml, reflux
15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered
under reduced pressure, ethanol / n-heptane = cake was washed with a
mixture of 1/3, and dried under reduced pressure after filtration to
obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
950ml of absolute ethanol was added to the
5L reaction vessel was charged with crude Alicante medoxomil (549.9g,
1.72mol), heated to reflux. After
completely dissolved clear, slow addition of n-heptane 1200ml, reflux
15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered
under reduced pressure, ethanol / n-heptane = cake was washed with a
mixture of 1/3, and dried under reduced pressure after filtration to
obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)
……………….
PATENT
Example
122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic
acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)
 To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of
potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn.
The solution was stirred at room temperature for 20 minutes. Then 0.562 g
of 1-chloromethyl isopropyl carbonate was added and the mixture was
reacted at 45-50° C. for 16 hours. After the reaction was completed, the
mixture solution was filtered, and 30 ml of water was added into the
filtrate. The resulting mixture was extracted with 30 ml of ethyl
acetate twice. The organic phase was dried and concentrated to give
1.724 g of oil, which was directly used in the next reaction without
purification.
To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of
potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn.
The solution was stirred at room temperature for 20 minutes. Then 0.562 g
of 1-chloromethyl isopropyl carbonate was added and the mixture was
reacted at 45-50° C. for 16 hours. After the reaction was completed, the
mixture solution was filtered, and 30 ml of water was added into the
filtrate. The resulting mixture was extracted with 30 ml of ethyl
acetate twice. The organic phase was dried and concentrated to give
1.724 g of oil, which was directly used in the next reaction without
purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.
USE CTRLand + Key, or click on picture
| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
.......................................................


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
You may search for our products through the search bar on our website. If you would like to receive a copy of our product catalog, please contact us at info@alfa-chemistry.com. Imidazoles
ReplyDelete