PART 2 AThttp://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
1 TEGOBUVIR
2 DANOPREVIR
3 CILUPREVIR
4 SOVAPREVIR
5 VEDROPREVIR
6 VANIPREVIR
7 NARLAPREVIR
8 DELDEPREVIR, NECEPREVIR
9 FALDAPREVIR
10 LEDIPASVIR
11DACLATASVIR
12 DELEOBUVIR
13 FILIBUVIR
14 FAVIPIRAVIR
15 ASUNAPREVIR
SEE MORE AT
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
1 TEGOBUVIR

 tegobuvir
tegobuvir
1 TEGOBUVIR
2 DANOPREVIR
3 CILUPREVIR
4 SOVAPREVIR
5 VEDROPREVIR
6 VANIPREVIR
7 NARLAPREVIR
8 DELDEPREVIR, NECEPREVIR
9 FALDAPREVIR
10 LEDIPASVIR
11DACLATASVIR
12 DELEOBUVIR
13 FILIBUVIR
14 FAVIPIRAVIR
15 ASUNAPREVIR
SEE MORE AT
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
VIR SERIES ..HEP C VIRUS 2/2
1 TEGOBUVIR
TEGOBUVIR
A non-structural protein 5B polymerase inhibitor
for Treatment of chronic hepatitis C
for Treatment of chronic hepatitis C
5-[6-[2,4-Bis(trifluoromethyl)phenyl]pyridazin-3-ylmethyl]-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine
CHEMICAL NAMES
1. 5H-Imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis(trifluoromethyl)phenyl]-3-pyridazinyl]methyl]-
2-(2-fluorophenyl)-
2-(2-fluorophenyl)-
2. 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-
imidazo[4,5-c]pyridine
imidazo[4,5-c]pyridine
MOLECULAR FORMULA C25H14F7N5
MOLECULAR WEIGHT 517.4
MANUFACTURER Gilead Sciences, Inc.
CODE DESIGNATION
- GS 333126
- GS 9190
- GS-333126
- GS-9190
- Tegobuvir
- UNII-5NOK5X389M
CAS REGISTRY NUMBER 1000787-75-6
GS-9190, an RNA-directed RNA polymerase (NS5B) inhibitor, is in phase II clinical evaluation at Gilead for the treatment of hepatitis C virus (HCV) infection. A clinical trial with GS-9190 in combination with peginterferon alfa-2a and ribavirin and with GS-9451 or with GS-9256 in treatment-naive subjects with chronic genotype 1 HCV infection was discontinued due to serious adverse events.
Gilead (Originator)
Katholieke Universiteit Leuven (Originator)
Katholieke Universiteit Leuven (Originator)
……………………………………….
PATENTS
WO 2005063744
WO 2008005519
WO 2009009001
WO 2010151488
WO 2010151487
WO 2010151472
WO 2011072370
WO 2011156757
WO 2012087596
WO 2013101550
Hebner CM, Han B, Brendza KM, Nash M, Sulfab M, Tian Y, Hung M, Fung W, Vivian RW, Trenkle J, Taylor J, Bjornson K, Bondy S, Liu X, Link J, Neyts J, Sakowicz R, Zhong W, Tang H, Schmitz U.
PLoS One. 2012;7(6):e39163. doi: 10.1371/journal.pone.0039163. Epub 2012 Jun 13.
Wong KA, Xu S, Martin R, Miller MD, Mo H.
Virology. 2012 Jul 20;429(1):57-62. doi: 10.1016/j.virol.2012.03.025. Epub 2012 Apr 28.
Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski JP, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR.
Hepatology. 2012 Mar;55(3):749-58. doi: 10.1002/hep.24744.
Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W.
Antimicrob Agents Chemother. 2011 Sep;55(9):4196-203. doi: 10.1128/AAC.00307-11. Epub 2011 Jul 11.
- ……………………..
- http://www.google.com/patents/WO2013040492A2
- ompound 1 can be prepared using synthetic methods and intermediates like those described in US 7,754,720. Compound 1 can also be prepared as described in the following Example.
- Compound 1 is:
 Compound 1 may also be referred to as 5-((6-(2,4-bis(trifluoromethyl)phenyl)pyridazin-3-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis (trifluoromethyl)phenyl]pyridazin=3-yl]methyl]-2-(2-fluorophenyl).
Compound 1 may also be referred to as 5-((6-(2,4-bis(trifluoromethyl)phenyl)pyridazin-3-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis (trifluoromethyl)phenyl]pyridazin=3-yl]methyl]-2-(2-fluorophenyl). - Example 1 : 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H- imidazo[4,5-c]pyridi

 Compound 103 was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid 105 and a 2N aq. Na2C03 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80°C for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6g Si02 using MeOH/CH2CI2 to elute compound. The compound thus obtained was contaminated with PPh3(0). The product was repurified on a 1 mm Chromatotron plate with 0 to 5%MeOH/CH2CI2 in 1 % steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound 1 was obtained with no PPh3 contamination. 1H NMR (300MHz,CD3OD) δ 6.20 (s, 2), 7.32 (m, 3), 7.52 (m, 1 ), 7.78 (d, 1), 7.89 (d, 1), 7.95 (s, 2), 8.15 (m, 3), 8.35 (d, 1), 9.12 (s, 1); LC/MS M+H = 518.The intermediate compound 104 was prepared as follows, a. Preparation of Compound 10
Compound 103 was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid 105 and a 2N aq. Na2C03 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80°C for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6g Si02 using MeOH/CH2CI2 to elute compound. The compound thus obtained was contaminated with PPh3(0). The product was repurified on a 1 mm Chromatotron plate with 0 to 5%MeOH/CH2CI2 in 1 % steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound 1 was obtained with no PPh3 contamination. 1H NMR (300MHz,CD3OD) δ 6.20 (s, 2), 7.32 (m, 3), 7.52 (m, 1 ), 7.78 (d, 1), 7.89 (d, 1), 7.95 (s, 2), 8.15 (m, 3), 8.35 (d, 1), 9.12 (s, 1); LC/MS M+H = 518.The intermediate compound 104 was prepared as follows, a. Preparation of Compound 10 101 102
101 102 To a solution of the commercially available starting material 101 in CHCI3, trichloroisocyanuric acid (TCCA) was added at 60°C. Then the solution was stirred for 1.5 hrs, cooled, and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g of compound 102. b. Preparation of Compound 104.
To a solution of the commercially available starting material 101 in CHCI3, trichloroisocyanuric acid (TCCA) was added at 60°C. Then the solution was stirred for 1.5 hrs, cooled, and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g of compound 102. b. Preparation of Compound 104. 102 104
102 104 To a solution of compound 103 in DMF (dimethylformamide), NaOH was added.Compound 102 was dissolved in DMF (20 mL) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2S0 . The solvent was removed and the product recrystallized withdichloromethane. The yield was 5.7 g of compound 103.
To a solution of compound 103 in DMF (dimethylformamide), NaOH was added.Compound 102 was dissolved in DMF (20 mL) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2S0 . The solvent was removed and the product recrystallized withdichloromethane. The yield was 5.7 g of compound 103. - ……………………………
- US7754720
- Example 1a Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineIn this method, dimethoxyethane or its related solvents, all having the general formula R1OR2O(R4O)aR3 wherein each of R1, R2, R3 and R4 are independently selected from C1-C6 alkyl and a is 0 or 1, have been found to be particularly advantageous over the conventional solvent DMF. Typically, each of R1, R2, R3 and R4 are independently C1-C2 alkyl and usually a is 0. C1-C6 alkyl includes fully saturated primary, secondary or tertiary hydrocarbon groups with 1 to 6 carbon atoms and thereby includes, but is not limited to methyl, ethyl, propyl, butyl, etc.Step 1



Compound MW Amount mmoles Equivalents SM 128.56 5 g 38.9 1 TCCA 232.41 3.62 g 15.6 0.4 CHCl3 130 ml To a solution of the commercially available starting material (SM) in CHCl3, trichloroisocyanuric acid (TCCA) was added at 60° C. Then the solution was stirred for 1.5 hrs., cooled down and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g.Step 2


Compound MW Amount mmoles Equivalents S.M. 163 5.073 g 31.12 1 Core 213.2 6.635 g 31.12 1 NaOH (10%) 40 1.245 g 31.12 1 DMF 320 ml To a solution of core (obtained as described in literature in DMF (dimethylformamide), NaOH was added. Then SM for this step (obtained from step 1) was dissolved in DMF (20 ml) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2SO4. The solvent was removed and the product recrystallized with DCM (dichloromethane). The yield was 5.7 g.Step 3


Compound MW Amount Moles Equivalents A 453.79 95 mg 0.209 1 DME 500 ul 2 N aq. Na2CO3 313ul 0.626 3 2,4-bisCF3- 257.93 80.9 mg 0.313 1.5 phenylboronic acid Pd(PPh3)4 1155 12 mg 0.0104 0.05 Compound A was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid and a 2N aq. Na2CO3 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80° C. for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6 g SiO2 using MeOH/CH2Cl2 to elute compound. The compound thus obtained was contaminated with PPh3(O). The product was repurified on a 1 mm Chromatotron plate with 0 to 5% MeOH/CH2Cl2 in 1% steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound (1) was obtained with no PPh3 contamination.1H NMR (300 MHz, CD3OD)6.20 (s, 2)7.32 (m, 3)7.52 (m, 1)7.78 (d, 1)7.89 (d, 1)7.95 (s, 2)8.15 (m, 3)8.35 (d, 1)9.12 (s, 1)LC/MS M+H=518Example 1b Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineThis example is directed to an additional method for making compound (1), employing the following schemes. Methanesulfonic acid was added to 2-fluorobenzoic acid in a reactor with active cooling keeping T≦50° C. 3,4-Diaminopyridine was then added portionwise to this cooled slurry, keeping T≦35° C. The contents of the reactor were then heated to 50° C. Phosphorus pentoxide was added in a single charge. The reaction was then heated at 90-110° C. for at least 3 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature and water was added portionwise slowly to quench the reaction. The reaction was then diluted with water. In solubles were removed by filtration. The pH of the filtrate was adjusted to 5.5-5.8 with ammonium hydroxide. The reaction was allowed to self-seed and granulate for ˜4 hours at ambient temperature. The pH was then adjusted to 8.0-9.3 with ammonium hydroxide. The slurry was held at ambient temperature for at least 2 hours. The solids were isolated by filtration and washed with water, followed by IPE. The wet cake was dried in vacuo at not more than 60° C. until ≦1% water remains. The dry product is core (2).
Methanesulfonic acid was added to 2-fluorobenzoic acid in a reactor with active cooling keeping T≦50° C. 3,4-Diaminopyridine was then added portionwise to this cooled slurry, keeping T≦35° C. The contents of the reactor were then heated to 50° C. Phosphorus pentoxide was added in a single charge. The reaction was then heated at 90-110° C. for at least 3 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature and water was added portionwise slowly to quench the reaction. The reaction was then diluted with water. In solubles were removed by filtration. The pH of the filtrate was adjusted to 5.5-5.8 with ammonium hydroxide. The reaction was allowed to self-seed and granulate for ˜4 hours at ambient temperature. The pH was then adjusted to 8.0-9.3 with ammonium hydroxide. The slurry was held at ambient temperature for at least 2 hours. The solids were isolated by filtration and washed with water, followed by IPE. The wet cake was dried in vacuo at not more than 60° C. until ≦1% water remains. The dry product is core (2).Summary of Materials M.W. Wt. Ratio Mole ratio 3,4-Diaminopyridine 109.13 1.0 1.0 2-Fluorobenzoic acid 140.11 1.4 1.1 Methanesulfonic acid 96.1 7.0 8.0 Phosphorus pentoxide 141.94 1.3 1.0 Water 18.02 40 — Isopropyl ether 102.17 5.0 — Ammonium hydroxide 35.09 ~10 —  A solution of compound (2a) in 1,2-dichloroethane was heated to 40-45° C. Trichloroisocyanuric acid was added and the mixture was heated at 60-70° C. for at least 2 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature. Celite was added to absorb insolubles, then solids were removed by filtration. The filtrate was washed with 0.5 N sodium hydroxide solution. The organic layer was concentrated to lowest stirrable volume and displaced with DMF. Core (2) and 10% aqueous sodium hydroxide solution were added. The reaction was stirred at ambient temperature for at least 8 hours. The reaction was sampled for completion by HPLC analysis. An additional 10% charge of 10% sodium hydroxide solution was added to the reaction. The reaction was then charged into water to isolate the crude product. After granulating for at least 1 hour, the solids were isolated and washed with water and isopropyl ether. Ethyl acetate was added and refluxed (internal T=70-77° C.) for 1-5 hours to dissolve product, then cooled to 18-23° C. slowly over 4-8 hours. The reactor contents were agitated at 18-23° C. for 8-20 hours and solids collected by filtration and rinsed with ethyl acetate. Low melt (i.e., DSC about 220 degrees C.) amorphous compound (1) was discharged. Amorphous compound (1) was dissolved in ethyl acetate by heating at reflux (internal T=70-77° C.) for 1-5 hours. Water content is controlled to about 0.2% by azeotropically removing water (with ethyl acetate the upper limit on water content is about 0.6% by weight; at about 0.9% by weight water the amorphous material will reprecipitate and crystals will not be obtained). The reactor contents are cooled slowly to 18-23° C. over 4-8 hours, then agitated at 18-23° C. for 8-20 hours and solids collected by filtration. The solids were rinsed with ethyl acetate and dried in vacuo at not more than 60° C. to obtain the dry crystalline compound (1).
A solution of compound (2a) in 1,2-dichloroethane was heated to 40-45° C. Trichloroisocyanuric acid was added and the mixture was heated at 60-70° C. for at least 2 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature. Celite was added to absorb insolubles, then solids were removed by filtration. The filtrate was washed with 0.5 N sodium hydroxide solution. The organic layer was concentrated to lowest stirrable volume and displaced with DMF. Core (2) and 10% aqueous sodium hydroxide solution were added. The reaction was stirred at ambient temperature for at least 8 hours. The reaction was sampled for completion by HPLC analysis. An additional 10% charge of 10% sodium hydroxide solution was added to the reaction. The reaction was then charged into water to isolate the crude product. After granulating for at least 1 hour, the solids were isolated and washed with water and isopropyl ether. Ethyl acetate was added and refluxed (internal T=70-77° C.) for 1-5 hours to dissolve product, then cooled to 18-23° C. slowly over 4-8 hours. The reactor contents were agitated at 18-23° C. for 8-20 hours and solids collected by filtration and rinsed with ethyl acetate. Low melt (i.e., DSC about 220 degrees C.) amorphous compound (1) was discharged. Amorphous compound (1) was dissolved in ethyl acetate by heating at reflux (internal T=70-77° C.) for 1-5 hours. Water content is controlled to about 0.2% by azeotropically removing water (with ethyl acetate the upper limit on water content is about 0.6% by weight; at about 0.9% by weight water the amorphous material will reprecipitate and crystals will not be obtained). The reactor contents are cooled slowly to 18-23° C. over 4-8 hours, then agitated at 18-23° C. for 8-20 hours and solids collected by filtration. The solids were rinsed with ethyl acetate and dried in vacuo at not more than 60° C. to obtain the dry crystalline compound (1).Summary of Materials M.W. Wt. Ratio Mole ratio 3-chloro-6-methylpyridazine 128.56 1.0 1.0 2,4bis(trifluromethyl)phenylboronic 257.93 4.0 2.0 acid X-Phos 476.72 0.18 0.05 Palladium acetate 224.49 0.04 0.025 1,2-Dimethoxyethane 90.12 16.7 — Potassium carbonate 138.21 2.15 2.0 Water 18.02 7.8 — Copper iodide 190.45 0.037 0.025 Celite — 0.25 — Heptane 100.2 22.4 — Nuclear Magnetic Resonance (1H-, 13C-, and 19F-NMR) SpectraNuclear magnetic resonance (NMR) spectra of compound (1) is consistent with the proposed structure. The 13C, 19F, and 1H-NMR spectra of compound (1) in DMSO-d6 were measured using a Varian UnityInova-400 FT-NMR spectrometer. Spectra are shown in the table below. The NMR chemical shift assignments were established using 2D correlation experiments (COSY, HSQC, HMBC and HSQCTOCSY).1H- and 13C-NMR Chemical Shift Assignments for Compound (1) Reference StandardAtom δC/ppm (DMSO-d6) δF/ppm (DMSO-d6) δH/ppm (DMSO-d6) 1A 140.16 2A 128.32 (qa, JCF = 32 Hz) 3A 123.61, m 8.24 (m, 1 H) 4A 130.27 (q, JCF = 34 Hz) 5A 129.54 (q, JCF = 3 Hz) 8.22 (m, 1 H) 6A 133.36 7.88 (m, 1 H) 7A 123.20 (q, JCF = 273 Hz) −56.4b 8A 123.02 (q, JCF = 275 Hz) −62.0b 1B 158.76 2B 128.16 8.01 (d, 1 H, J = 8.4 Hz) 3B 126.20 7.95 (d, 1 H, J = 8.8 Hz) 4B 157.70 5B 60.49 6.17 (s, 2 H) 2C 131.86 8.31 (m, 1 H) 3C 112.63 7.86 (m, 1 H) 4C 155.44 6C 168.11 (d, JCF = 6 Hz) 8C 145.08 9C 133.06 9.25 (s, 1 H) 1D 123.11 (d, JCF = 10 Hz) 2D 160.46 (d, JCF = 254 Hz) −111.7 3D 116.59 (d, JCF = 22 Hz) 7.29 (m, 1 H) 4D 130.84 (d, JCF = 8 Hz) 7.46 (m, 1 H) 5D 124.13 (d, JCF = 4 Hz) 7.31 (m, 1 H) 6D 131.72 (d, JCF = 2 Hz) 8.35 (m, 1 H) amultiplicity, s: singlet, d: doublet, q: quartet, m: multiplet binterchangeable signals
...............................................
2 DANOPREVIR
 DanoprevirDanoprevir(ITMN-191) is a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.Array BioPharma (Originator)
DanoprevirDanoprevir(ITMN-191) is a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.Array BioPharma (Originator)RG7227 ITMN-191 RO5190591 2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-
dioxocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester2. (2R,6S,12Z,13aS,14aR,16aS)-14a-[(cyclopropylsulfonyl)carbamoyl]-6-{[(1,1-
dimethylethoxy)carbonyl]amino}-5,16-dioxo-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate
Treatment of hepatitis CMOLECULAR FORMULA C35H46FN5O9SMOLECULAR WEIGHT 731.8MANUFACTURER GenentechCODE DESIGNATION R05190591CAS REGISTRY NUMBER 850876-88-9, 916881-67-9Danoprevir(ITMN-191) is a peptidomimeticITMN-191 (R-7227), a macrocyclic protease inhibitor, is in phase II clinical evaluation for the treatment of chronic hepatitis C virus (HCV) infection as monotherapy and in combination with Pegasys(R) (pegylated interferon alpha-2a) and Copegus(R) (ribavirin). The product candidate is also being evaluated in combination with R-7128 in treatment-naive patients infected with HCV genotype 1.Danoprevir (ITMN-191; RG-7227), under development by InterMune Inc and Roche Holding AG, is a promising, potent NS3/4A protease inhibitor for the oral treatment of HCV infection. Preclinical data demonstrated that danoprevir binds with high affinity and dissociates slowly from the HCV NS3 protease, allowing high liver drug exposure with only modest plasma drug exposure.In 2006, originator InterMune and licensee Roche entered into an exclusive worldwide collaboration agreement to develop and commercialize products from InterMune’s hepatitis C (HCV) protease inhibitor program, including ITMN-191. In 2010, the licensing agreement was terminated. Also in 2010, Roche acquired worldwide development and commercialization rights to R-7227 from InterMune. Preclinical pharmacokinetic results support the exploration of twice-daily oral dosing in HCV.A phase Ib, ‘IFN-free’ clinical trial demonstrated that danoprevir, combined with the HCV polymerase inhibitor RG-7128 (Pharmasset Inc/Roche Holding AG), was effective in reducing HCV-RNA levels in a large proportion of treatment-naïve patients with HCV infection and in approximately half of previously non-responsive patients with HCV-1 infection, without resistance or safety concerns. In a phase IIb trial in treatment-naïve patients with HCV-1 infection, danoprevir plus pegylated IFNalpha2a and ribavirin resulted in undetectable levels of HCV-RNA in the majority of patients, without any evidence of viral resistance; however, the high-dose danoprevir arm was prematurely terminated because of grade 4 ALT elevations. Phase I trials have also demonstrated that ritonavir boosting improved the pharmacokinetic profile of danoprevir; therefore, at the time of publication, a phase IIb trial to evaluate ritonavir-boosted, low-dose danoprevir in combination with RG-7128 was planned. (source:inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.SODIUM SALTHERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES1. 2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-dioxocyclopropa
[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester, sodium salt (1:1)2. sodium (cyclopropylsulfonyl){[(2R,6S,12Z,13aS,14aR,16aS)-6-{[(1,1-dimethylethoxy)
carbonyl]amino}-2-{[(4-fluoro-1,3-dihydro-2H-isoindol-2-yl)carbonyl]oxy}-5,16-dioxo-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecine-14a(5H)-yl]formyl}azanideMOLECULAR FORMULA C35H45FN5NaO9SMOLECULAR WEIGHT 753.8SPONSOR GenentechCODE DESIGNATION- Danoprevir sodium
- ITMN-191
- R 7227 sodium
- R7227
- RO 5190591-001
- RO5190591-001
- UNII-217RJI972K
CAS REGISTRY NUMBER 916826-48-7DANOPREVIR SODIUM
The HCV protease mediates the cleavage of the HCV polyprotein to release the functional proteins that are essential for viral propagation. The inhibition of the HCV protease activity is expected to block HCV replication in infected host cells. Numberous HCV protease inhibitors have been identified. Non- limiting examples of HCV protease inhibitors are described in U.S. Patent Application Pub. Nos. 20040106559, 20040180815, 20040266668, 2004038872, 20050090432, 20050267018, 20070054842, 20070281885, 2007299078, 20080032936, 20080125444, 20080279821, 20090111757, 20090148407, 20090202480, 20090269305, 20090285773, 20090285774, 20100081700, 20100144608, 2010018355, 20100183551, 20100221217, 20100260710, 20100286185 and 20110135604, and U.S. Patent Nos. 6608027, 6767991, 7091184, 7119072, 7544798, 7642235 and 7829665, as well as WO2007014919, WO2007014926, WO2008046860, WO2008095058,………………………………danoprevirpatents and journal ref1. WO 2005037214..2. WO 20050954033. WO 2007015824..4. WO 20081289215. WO 20090805426. WO 20091428427. WO 20100155458. WO 20130794249. WO 201206268510.WO 201310663111. Concise asymmetric synthesis of a (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid-derived sulfonamide and ethyl ester
Org Biomol Chem 2013, 11(39): 6796http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob41394b/unauth#!divAbstract12.J. Med. Chem., Article ASAP,DOI: 10.1021/jm400164cKazmierski WM, Hamatake R, Duan M, Wright LL, Smith GK, Jarvest RL, Ji JJ, Cooper JP, Tallant MD, Crosby RM, Creech K, Wang A, Li X, Zhang S, Zhang YK, Liu Y, Ding CZ, Zhou Y, Plattner JJ, Baker SJ, Bu W, Liu L.J Med Chem. 2012 Apr 12;55(7):3021-6. doi: 10.1021/jm201278q. Epub 2012 Apr 3.Duan M, Kazmierski W, Crosby R, Gartland M, Ji J, Tallant M, Wang A, Hamatake R, Wright L, Wu M, Zhang YK, Ding CZ, Li X, Liu Y, Zhang S, Zhou Y, Plattner JJ, Baker SJ.Bioorg Med Chem Lett. 2012 Apr 15;22(8):2993-6. doi: 10.1016/j.bmcl.2012.02.039. Epub 2012 Feb 22.……………….J. Med. Chem., Article ASAPDOI: 10.1021/jm400164c (2R,6S,13aS,14aR,16aS,Z)-6-(tert-Butoxycarbonylamino)-14a-(cyclopropylsulfonylcarbamoyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoroisoindoline-2-carboxylate (49)
(2R,6S,13aS,14aR,16aS,Z)-6-(tert-Butoxycarbonylamino)-14a-(cyclopropylsulfonylcarbamoyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoroisoindoline-2-carboxylate (49)
1H NMR (500 MHz, acetone-d6) δ 10.70 (br s, 1 H), 8.34 (d, 1 H), 7.39–7.33 (m, 1 H), 7.20 (d, 1 H), 7.10–7.02 (m, 2 H), 6.13 (d, 1 H), 5.70 (q, 1 H), 5.44 (br s, 1 H), 4.99 (t, 1H), 4.78–4.59 (m, 5 H), 4.18–4.08 (m, 1 H), 3.88–3.81 (m, 1 H), 2.86–2.78 (m, 3 H), 2.71–2.60 (m, 1 H), 2.52–2.35 (m, 3 H), 1.92–1.81 (m, 2 H), 1.75 (t, 1 H), 1.61–1.14 (m, 17 H), 1.04–0.95 (m, 2 H). 13C NMR (DMSO-d6, 100 MHz) δ174.0, 172.3, 170.8, 157.5 (d, J = 244 Hz), 155.8, 154.0, 140.9 (d, J = 64.8 Hz), 130.6, 130.2, 129.6, 123.8 (d, J = 55.7 Hz), 119.6, 114.4 (d, J = 18.3 Hz), 78.4, 74.7, 59.7, 53.4, 52.7, 52.4, 49.4 (d, J = 37.1 Hz), 44.6, 34.9, 31.8, 30.4, 28.8, 28.6, 27.0, 25.7, 22.6, 4.8. APCI MS m/z730.4 (M – 1).………………………………………………………..Bioorg Med Chem Lett. 2012 Apr 15;22(8):2993-6. doi: 10.1016/j.bmcl.2012.02.039. Epub 2012 Feb 22. …………………………..PICK UP 12 REF COMPD FROM LIST= DANOPREVIR
…………………………..PICK UP 12 REF COMPD FROM LIST= DANOPREVIR
 NMR AND SYNExample 58 Synthesis of Compound 12-RefCompound 11-Ref (0.18 mmol) was dissolved in 10 mL anhydrous dichloromethane, EDCI (69.8 mg, 0.36 mmol, 2 eq.) was added and stirred at room temperature overnight until completed. The reaction mixture was worked out and concentrated.The obtained solid was dissolved in 10 mL of anhydrous dichloromethane, DBU (61.0 mg, 0.40 mmol) and RSO2NH2 (0.363 mmol, R=cyclopropyl) were added and stirred at room temperature overnight until completed. The reaction mixture was worked out and purified by flash column to obtain the product 12-Ref (62 mg; Yield: 53%).1H-NMR for the product 12-Ref (CDCl3, 500 MHz): δ 10.28-10.29 (d, 1H), 6.87-7.07 (m, 3H), 5.72-5.74 (m, 1H), 5.48 (br , 1H), 4.99-5.03 (m, 2H), 4.58-4.79 (m, 5H), 4.42 (m, 1H), 4.21 (m, 1H), 3.83-3.85 (m, 1H), 2.90-2.93 (m, 1H), 2.48-2.57 (m, 3H), 2.27-2.30 (m, 1H), 1.88-1.97 (m, 2H), 1.67-1.79 (m, 2H), 1.45-1.58 (m, 6H), 1.34 -1.40 (m, 2H), 1.27 (s, 4H), 1.24 (s, 5H), 1.08-1.15 (m, 2H), 0.91-0.94 (m, 1H). ESI-MS (M+H+): m/z calculated 732.3, founded 732.5.………………………..SYNTHESISFor certain NS3 inhibitors shown in this section, additional chemical transformations are utilized to obtain the final products. The preparations of two such examples are described for compounds 153 and 154 below:
NMR AND SYNExample 58 Synthesis of Compound 12-RefCompound 11-Ref (0.18 mmol) was dissolved in 10 mL anhydrous dichloromethane, EDCI (69.8 mg, 0.36 mmol, 2 eq.) was added and stirred at room temperature overnight until completed. The reaction mixture was worked out and concentrated.The obtained solid was dissolved in 10 mL of anhydrous dichloromethane, DBU (61.0 mg, 0.40 mmol) and RSO2NH2 (0.363 mmol, R=cyclopropyl) were added and stirred at room temperature overnight until completed. The reaction mixture was worked out and purified by flash column to obtain the product 12-Ref (62 mg; Yield: 53%).1H-NMR for the product 12-Ref (CDCl3, 500 MHz): δ 10.28-10.29 (d, 1H), 6.87-7.07 (m, 3H), 5.72-5.74 (m, 1H), 5.48 (br , 1H), 4.99-5.03 (m, 2H), 4.58-4.79 (m, 5H), 4.42 (m, 1H), 4.21 (m, 1H), 3.83-3.85 (m, 1H), 2.90-2.93 (m, 1H), 2.48-2.57 (m, 3H), 2.27-2.30 (m, 1H), 1.88-1.97 (m, 2H), 1.67-1.79 (m, 2H), 1.45-1.58 (m, 6H), 1.34 -1.40 (m, 2H), 1.27 (s, 4H), 1.24 (s, 5H), 1.08-1.15 (m, 2H), 0.91-0.94 (m, 1H). ESI-MS (M+H+): m/z calculated 732.3, founded 732.5.………………………..SYNTHESISFor certain NS3 inhibitors shown in this section, additional chemical transformations are utilized to obtain the final products. The preparations of two such examples are described for compounds 153 and 154 below: (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-2-(4-fluoroisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecine-14a-carboxylic acid (0.10 g, 0.16 mmol) and TEA (0.024 mL, 0.18 mmol) in THF (5 mL) was added ethyl carbonochlridate (0.016 mL, 0.17 mmol) at 0° C. The reaction was stirred at 0° C. for 2 hrs. Sodium boronhydride (0.012 g, 0.32 mmol) was added and the reaction was stirred at rt for 3 days. Water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was purified by column chromatography (ethyl acetate) to give the product (0.060 g, 61.4%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.47 (b, 1H), 7.35 (m, 1H), 7.10-7.20 (m, 2H), 7.03 (m, 1H), 5.47 (m, 1H), 5.28 (b, 1H), 4.98 (m, 1H), 4.67 (b, 4H), 4.56 (m, 1H), 4.46 (m, 1H), 4.26 (m, 1H), 3.92 (m, 1H), 3.66 (m, 2H), 3.16 (m, 1H), 2.67 (m, 1H), 2.21 (m, 2H), 1.80 (m, 1H), 1.68 (m, 1H), 1.30 (m, 8H), 1.11-1.20 (m, 9H), 0.85 (m, 1H), 0.77 (m, 1H).
(2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-2-(4-fluoroisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecine-14a-carboxylic acid (0.10 g, 0.16 mmol) and TEA (0.024 mL, 0.18 mmol) in THF (5 mL) was added ethyl carbonochlridate (0.016 mL, 0.17 mmol) at 0° C. The reaction was stirred at 0° C. for 2 hrs. Sodium boronhydride (0.012 g, 0.32 mmol) was added and the reaction was stirred at rt for 3 days. Water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was purified by column chromatography (ethyl acetate) to give the product (0.060 g, 61.4%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.47 (b, 1H), 7.35 (m, 1H), 7.10-7.20 (m, 2H), 7.03 (m, 1H), 5.47 (m, 1H), 5.28 (b, 1H), 4.98 (m, 1H), 4.67 (b, 4H), 4.56 (m, 1H), 4.46 (m, 1H), 4.26 (m, 1H), 3.92 (m, 1H), 3.66 (m, 2H), 3.16 (m, 1H), 2.67 (m, 1H), 2.21 (m, 2H), 1.80 (m, 1H), 1.68 (m, 1H), 1.30 (m, 8H), 1.11-1.20 (m, 9H), 0.85 (m, 1H), 0.77 (m, 1H). A solution of oxalyl chloride 90.045 mL, 0.089 mmol) in DCM (5 mL) at −78° C. was added a solution of DMSO (0.015 g, 0.020 mmol) in DCM (2 mL) dropwise over 2 ninytes. The reaction was stirred at −78° C. for 10 minutes and the a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-14a-(hydroxymethyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecin-2-yl-4-fluoroisoindoline-2-carboxylate (0.050 g, 0.081 mmol) in DCM (2 mL) was added. After stirred at −78° C. for 40 min, TEA (0.051 mL, 0.37 mmol) was added. The reaction was warmed to rt, water (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in MeOH (5 mL) and ammonium hydroxide (0.085 g, 2.45 mmol) and acetic acid (0.014 mL, 0.25 mmol) were added. The reaction stirred at rt for 3 minutes. NaCNBH3 90.015 g, 0.245 mmol) was added and stirred at rt for 30 minutes. The MeOH was removed. DCM (20 mL) and saturated sodium bicarbonate (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in DCM (5 mL). TEA (0.017 mL, 0.122 mmol) was added and followed by the cyclopropanesulfonyl chloride (0.015 g, 0.098 mmol). The reaction was stirred at rt for 5 hrs. The solvent was removed. The residue was purified by column chromatography (ethyl acetate) to give the product (0.017 g, 28.2%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.52 (m, 1H), 7.35 (m, 1H), 7.02-7.20 (m, 4H), 5.56 (m, 1H), 4.99 (m, 1H), 4.97 (m, 1H), 4.67 (m, 2H), 4.66 (s, 2H), 4.46 (m, 1H), 4.24 (m, 1H), 3.92 (m, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 2.74 (m, 1 h), 2.67 (m, 1H), 2.22 (m, 2H), 1.84 (m, 1H), 1.68 (m, 1H), 1.08-1.36 (m, 20H), 0.89 (m, 2H), 0.81 (m, 2H).………………………………………..
A solution of oxalyl chloride 90.045 mL, 0.089 mmol) in DCM (5 mL) at −78° C. was added a solution of DMSO (0.015 g, 0.020 mmol) in DCM (2 mL) dropwise over 2 ninytes. The reaction was stirred at −78° C. for 10 minutes and the a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-14a-(hydroxymethyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecin-2-yl-4-fluoroisoindoline-2-carboxylate (0.050 g, 0.081 mmol) in DCM (2 mL) was added. After stirred at −78° C. for 40 min, TEA (0.051 mL, 0.37 mmol) was added. The reaction was warmed to rt, water (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in MeOH (5 mL) and ammonium hydroxide (0.085 g, 2.45 mmol) and acetic acid (0.014 mL, 0.25 mmol) were added. The reaction stirred at rt for 3 minutes. NaCNBH3 90.015 g, 0.245 mmol) was added and stirred at rt for 30 minutes. The MeOH was removed. DCM (20 mL) and saturated sodium bicarbonate (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in DCM (5 mL). TEA (0.017 mL, 0.122 mmol) was added and followed by the cyclopropanesulfonyl chloride (0.015 g, 0.098 mmol). The reaction was stirred at rt for 5 hrs. The solvent was removed. The residue was purified by column chromatography (ethyl acetate) to give the product (0.017 g, 28.2%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.52 (m, 1H), 7.35 (m, 1H), 7.02-7.20 (m, 4H), 5.56 (m, 1H), 4.99 (m, 1H), 4.97 (m, 1H), 4.67 (m, 2H), 4.66 (s, 2H), 4.46 (m, 1H), 4.24 (m, 1H), 3.92 (m, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 2.74 (m, 1 h), 2.67 (m, 1H), 2.22 (m, 2H), 1.84 (m, 1H), 1.68 (m, 1H), 1.08-1.36 (m, 20H), 0.89 (m, 2H), 0.81 (m, 2H).……………………………………….. Hoffmann-La Roche and Genentech’s danoprevir/r (RG7227) is a twice-daily, ritonavir-boosted HCV protease inhibitor with activity against HCV genotypes 1, 4 and 6. DAUPHINE, an ongoing phase II trial in 421 treatment-naive people with HCV genotypes 1 and 4, is comparing doses (200, 100, and 50 mg danoprevir, boosted with 100 mg ritonavir, twice-daily) and response-guided therapy with danoprevir/r plus PEG-IFN/RBV. At 12 weeks after treatment completion, HCV RNA was undetectable in 86% of the highest-dosing arm, 77% of the 100 mg arm, and 65% of the 50 mg arm.Response to treatment in the 200 mg dosing arm did not differ according to HCV subtype or IL28B genotype; at 12 weeks after treatment completion, 88% of people with HCV subtype 1a and an IL28B non-CC genotype had undetectable HCV RNA. Across all dosing arms, HCV RNA remained undetectable 12 weeks after treatment completion in 100% of people with HCV genotype 4.In the response-guided therapy arm, 76% of early responders (who were treated for 12 weeks) and 67% of late responders (treated for 24 weeks) maintained undetectable HCV RNA 12 weeks after treatment completion, bringing the overall total to 72%.One death occurred during the trial—from sudden heart attack, in a participant with preexisting diabetes and hypertension—it was considered unrelated to study drugs. Adverse events were reported in virtually all study participants. Side effects from ritonavir, which is used to boost danoprevir levels, increased the likelihood of more than one serious adverse event among people in the danoprevir/r arms (range 4–9% vs. 1% for placebo). The rate of danoprevir/r-related treatment discontinuations was similar to the rate of PEG-IFN/RBV-associated discontinuations (3–7%, and 3–8%, respectively).Common side effects (experienced by more than 15% of study participants) included fatigue, fever, chills, weakness, nausea, diarrhea, itching, rash, hair loss, headache, aching muscles and joints, insomnia, cough, and appetite loss. Diarrhea was the only side effect associated with danoprevir/r. Adding danoprevir/r did not increase rates of rash or anemia (known side effects of other HCV protease inhibitors). Most grade 3 and grade 4 lab abnormalities were neutropenia, reported in 22% to 38% of study participants.
Hoffmann-La Roche and Genentech’s danoprevir/r (RG7227) is a twice-daily, ritonavir-boosted HCV protease inhibitor with activity against HCV genotypes 1, 4 and 6. DAUPHINE, an ongoing phase II trial in 421 treatment-naive people with HCV genotypes 1 and 4, is comparing doses (200, 100, and 50 mg danoprevir, boosted with 100 mg ritonavir, twice-daily) and response-guided therapy with danoprevir/r plus PEG-IFN/RBV. At 12 weeks after treatment completion, HCV RNA was undetectable in 86% of the highest-dosing arm, 77% of the 100 mg arm, and 65% of the 50 mg arm.Response to treatment in the 200 mg dosing arm did not differ according to HCV subtype or IL28B genotype; at 12 weeks after treatment completion, 88% of people with HCV subtype 1a and an IL28B non-CC genotype had undetectable HCV RNA. Across all dosing arms, HCV RNA remained undetectable 12 weeks after treatment completion in 100% of people with HCV genotype 4.In the response-guided therapy arm, 76% of early responders (who were treated for 12 weeks) and 67% of late responders (treated for 24 weeks) maintained undetectable HCV RNA 12 weeks after treatment completion, bringing the overall total to 72%.One death occurred during the trial—from sudden heart attack, in a participant with preexisting diabetes and hypertension—it was considered unrelated to study drugs. Adverse events were reported in virtually all study participants. Side effects from ritonavir, which is used to boost danoprevir levels, increased the likelihood of more than one serious adverse event among people in the danoprevir/r arms (range 4–9% vs. 1% for placebo). The rate of danoprevir/r-related treatment discontinuations was similar to the rate of PEG-IFN/RBV-associated discontinuations (3–7%, and 3–8%, respectively).Common side effects (experienced by more than 15% of study participants) included fatigue, fever, chills, weakness, nausea, diarrhea, itching, rash, hair loss, headache, aching muscles and joints, insomnia, cough, and appetite loss. Diarrhea was the only side effect associated with danoprevir/r. Adding danoprevir/r did not increase rates of rash or anemia (known side effects of other HCV protease inhibitors). Most grade 3 and grade 4 lab abnormalities were neutropenia, reported in 22% to 38% of study participants.Interferon-free DAA Combinations
Danoprevir/r and Mericitabine, plus Ribavirin (HCV Genotypes 1 and 4)
Roche’s phase IIb study, INFORM-SVR, is combining response-guided therapy with danoprevir/r, a twice-daily ritonavir-boosted HCV protease inhibitor, and mericitabine, a twice-daily nucleoside polymerase inhibitor, with or without ribavirin for 12 to 24 weeks in non-cirrhotic people with HCV genotype 1. The original study design was modified after high relapse rates were observed in the 12-week treatment and ribavirin-free arms. Treatment was extended to 24 weeks, and ribavirin was given to all participants.The majority of INFORM-SVR participants were male, had HCV genotype 1a, and non-CC genotypes. Of the 64 people treated for 24 weeks with all three drugs, 41% experienced SVR-12. People with HCV genotype 1b were more likely to achieve SVR-12 (71% versus 26% in HCV genotype 1a). In contrast, SVR-12 was more likely among people with non-CC genotypes (32% for CC versus 44% for non-CC), although only 4 people had HCV genotype 1b and CC genotype. Breakthrough rates were higher in people who did not receive ribavirin, and in HCV genotype 1a versus 1b. Resistance to danoprevir/r was observed in all patients who experienced viral breakthrough; mericitabine resistance was found in one person.Almost all participants had more than one adverse event; a total of 567 mild-to-moderate events were reported among 83 people. The most common side effects, occurring in >10% of people were headache, fatigue, nausea, diarrhea, colds, insomnia, itching, weakness, dizziness, irritability, shortness of breath, cough, upset stomach, painful joints, and vomiting. As for laboratory abnormalities, one person experienced grade 3 anemia, four people had grade 3 lipid elevations, and one case each of grade 3 elevations in phosphate and lipase were observed.A single serious adverse event, multiple myeloma, occurred 53 days after treatment completion and one person discontinued due to pain in the back of the throat (it was not specified whether or not this was a treatment-related adverse event).
- Everson G, Cooper C, Shiffman ML, et al. Rapid and sustained achievement of undetectable HCV RNA during treatment with ritonavir-boosted danoprevir/PEG-IFNa-2A/RBV in HCV genotype 1 or 4 patients: Dauphine week 36 interim analysis (Abstract 1177). Paper presented at: 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from: http://mobile.ilcapp.eu/EASL_161/poster_24544/program.aspx. (Accessed 2012 June 25)
- Gane EJ, Pockros P, Zeuzem S, et al. Interferon-free treatment with combination of mericitabine and danoprevir/r with or without ribavirin in treatment-naïve HCV genotype-1 infected patients (Abstract 1412). 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from:http://mobile.ilcapp.eu/EASL_161/poster_24848/program.aspx. (Accessed 2012 June 25)
Non- limiting examples of suitable HCV protease inhibitors include ACH-1095(Achillion), ACH-1625 (Achillion), ACH-2684 (Achillion), AVL-181 (Avila), AVL-192 (Avila), BI-201335 (Boehringer Ingelheim), BMS-650032 (BMS), boceprevir, danoprevir, GS- 9132 (Gilead), GS-9256 (Gilead), GS-9451 (Gilead), IDX-136 (Idenix), IDX-316 (Idenix), IDX- 320 (Idenix), MK-5172 (Merck), narlaprevir, PHX-1766 (Phenomix), telaprevir, TMC-435 (Tibotec), vaniprevir, VBY708 (Virobay), VX-500 (Vertex), VX-813 (Vertex), VX-985 (Vertex), or a combination thereof. Non-limiting examples of suitable HCV polymerase inhibitors include ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), GSK625433 (Glaxo SmithKline), BCX-4678 (BioCryst), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination thereof. A polymerase inhibitor may be a nucleotide polymerase inhibitor, such as GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination therefore. A polymerase inhibitor may also be a non- nucleoside polymerase inhibitor, such as ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), or a combination thereof. Non-limiting examples of suitable NS5A inhibitors include GSK62336805 (Glaxo SmithKline), ACH-2928 (Achillion), AZD2836 (Astra-Zeneca), AZD7295 (Astra-Zeneca), BMS-790052 (BMS), BMS- 824393 (BMS), GS-5885 (Gilead), PPI-1301 (Presidio), PPI-461 (Presidio), or a combination thereof. Non-limiting examples of suitable cyclophilin inhibitors include alisporovir (Novartis & Debiopharm), NM-811 (Novartis), SCY-635 (Scynexis), or a combination thereof. Non-limiting examples of suitable HCV entry inhibitors include ITX-4520 (iTherx), ITX-5061 (iTherx), or a combination thereof.WO 2007015824WO 2003053349WO 2005095403WO 2005037214WO 2005095403WO 2005037214WO 2003053349WO 2007015824WO 2008128921US8048862 14 Apr 2009 1 Nov 2011 Intermune, Inc. Macrocyclic inhibitors of hepatitis C virus replication US8119592 10 Oct 2006 21 Feb 2012 Intermune, Inc. Compounds and methods for inhibiting hepatitis C viral replication US8232246 30 Jun 2009 31 Jul 2012 Abbott Laboratories Anti-viral compounds US8299021 19 Apr 2012 30 Oct 2012 Intermune, Inc. Macrocyclic inhibitors of hepatitis C virus replication US8420596 10 Sep 2009 16 Apr 2013 Abbott Laboratories Macrocyclic hepatitis C serine protease inhibitors WO2013106631A1 11 Jan 2013 18 Jul 2013 Abbvie Inc. Processes for making hcv protease inhibitors Danoprevir Clinical Trial Information( data fromhttp://clinicaltrials.gov)
NCT NUMBER RECRUITMENT CONDITIONS SPONSOR
/COLLABORATORSSTART DATE PHASES NCT01331850 Completed Hepatitis C, Chronic Hoffmann-La Roche 2011-05 Phase 2 NCT01531647 Completed Healthy Volunteer Hoffmann-La Roche 2012-01 Phase 1 NCT01588002 Completed Healthy Volunteer Hoffmann-La Roche 2012-04 Phase 1 NCT01592318 Active, not recruiting Healthy Volunteer Hoffmann-La Roche 2012-05 Phase 1 NCT01749150 Recruiting Hepatitis C, Chronic Hoffmann-La Roche 2013-04 Phase 2 - .............................................................................
- 3 CILUPREVIR
 CILUPREVIR(1S,4R,6S,7Z,14S,18R)-14- {[(cyclopentyloxy)carbonyl]amino}-18-[(7-methoxy-2- {2-[(propan-2-yl)amino]-1,3-thiazol-4-yl}quinolin-4- yl)oxy]-2,15-dioxo-3,16- diazatricyclo[14.3.0.0{4,6}]nonadec-7-ene-4- carboxylic acidCiluprevir, BILN-2061, BILN 2061, CHEBI:161337, BILN2061, BILN 2061ZW, BILN-2061-ZW,CAS , 300832-84-2Molecular Formula: C40H50N6O8S Molecular Weight: 774.9254
CILUPREVIR(1S,4R,6S,7Z,14S,18R)-14- {[(cyclopentyloxy)carbonyl]amino}-18-[(7-methoxy-2- {2-[(propan-2-yl)amino]-1,3-thiazol-4-yl}quinolin-4- yl)oxy]-2,15-dioxo-3,16- diazatricyclo[14.3.0.0{4,6}]nonadec-7-ene-4- carboxylic acidCiluprevir, BILN-2061, BILN 2061, CHEBI:161337, BILN2061, BILN 2061ZW, BILN-2061-ZW,CAS , 300832-84-2Molecular Formula: C40H50N6O8S Molecular Weight: 774.9254 Ciluprevir is used in the treatment of hepatitis C. It is manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG under the research code of BILN-2061. It is targeted against NS2-3 protease.[1]Ciluprevir is an HCV NS3 protease inhibitor which had been in phase II clinical trials at Boehringer Ingelheim for the treatment of hepatitis C, however, no recent developments from the company have been reported.1. Challenge and Opportunity in Scaling-Up Metathesis Reaction: Synthesis of Ciluprevir (BILN 2061)Peter J. Dunn, et alhttp://onlinelibrary.wiley.com/doi/10.1002/9781118354520.ch10/summary
Ciluprevir is used in the treatment of hepatitis C. It is manufactured by Boehringer Ingelheim Pharma GmbH & Co. KG under the research code of BILN-2061. It is targeted against NS2-3 protease.[1]Ciluprevir is an HCV NS3 protease inhibitor which had been in phase II clinical trials at Boehringer Ingelheim for the treatment of hepatitis C, however, no recent developments from the company have been reported.1. Challenge and Opportunity in Scaling-Up Metathesis Reaction: Synthesis of Ciluprevir (BILN 2061)Peter J. Dunn, et alhttp://onlinelibrary.wiley.com/doi/10.1002/9781118354520.ch10/summary
DOI: 10.1002/9781118354520.ch10
2. Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
3. Efficient synthesis of (S)-2-(cyclopentyloxycarbonyl)-amino-8-nonenoic acid: Key buiding block for BILN 2061, an HCV NS3 protease inhibitor
Org Process Res Dev 2007, 11(1): 60
4. Chinese Journal of Chemistry, 2011 , vol. 29, 7 pg. 1489 - 1502
DOI: 10.1002/cjoc.201180270
http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201180270/abstract;jsessionid=F5F4331F5A95D00728394A254C2B1AE7.f01t04
................................
US 8222369
WO 2006071619
WO 2000059929
WO 2004092203
WO 2004039833
WO 2004037855
WO 2006036614
WO 2006033878
WO 2005042570
WO 2004093915
...................................................................................
https://www.google.co.in/patents/US8222369


.............................................................................
http://www.google.com/patents/WO2000059929A1
COMPD 822 IS CILUPREVIR IN TABLE 8
EXAMPLE 8 Synthesis of 4-hydroxy-7-methoxy-2[4(2-isopropylaminothiazolyl)] quinoline (8f ) Note: [ A variety of 2-alkylaminothiazolyl substituents were made using the same synthetic scheme where compound 8b was replaced by other alkyl thioureas.]

8b 8c 8d
A. The protocol used for the conversion of -anisidine to 8a was identical to that described in the literature: F.J. Brown et al. J. Med. Chem. 1989, 32 , 807-826. However, the purification procedure was modified to avoid purification by chromatography. The EtOAc phase containing the desired product was treated with a mixture of MgSO4, charcoal and 5% w/w (based on expected mass) silica gel. After filtration on celite, the product was triturated with ether. Compound 8a was obtained as a pale brown solid in >99% purity (as confirmed by HPLC).
B. A suspension of isopropyl thiourea (8b, 3.55 g, 30 mmol) and 3- bromopyruvic acid (8c, 5 g, 1 eq.) in dioxane (300 mL , 0.1 M) was heated to 80 °C.
Upon reaching 80 C the solution became clear and soon after the product precipitated as a white solid. After 2 hours of heating, the solution was cooled to RT and the white precipitate was filtered to obtain compound 8d in high purity (>98% purity as confirmed by NMR) and 94% yield (7.51 g). C. A mixture of the carboxylic acid 8d (4.85 g, 18.2 mmol) and the aniline derivative 8a (3 g, leq.) in pyridine (150 mL, 0.12 M) was cooled to -30 °C (upon cooling, the clear solution became partially a suspension). Phosphorus oxychloride (3.56 ml, 2.1 eq.) was then added slowly over a 5 min period. The reaction was stirred at -30 C for 1 h, the bath was removed and the reaction mixture was allowed to warm-up to RT. After 1.5 h the reaction mixture was poured into ice, the pH was adjusted to 11 with aqueous 3N NaOH, extracted with CH2C12, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The beige solid was then purified by flash chromatography (45% EtOAc in hexane) to give compound 8e as a pale yellow solid in 73% yield (6.07 g). D. A solution of tBuOK (2.42 g, 21.6 mmol) in anhydrous tBuOH (40ml, 0.14 M, distilled from Mg metal) was heated to reflux. Compound 8e (1.8g, 5.4 mmol) was added portion-wise over 5 min and the dark red solution formed was stirred at reflux for an additional 20 min (completion of the reaction was monitored by HPLC). The mixture was cooled to RT and HCl was added (4 N in dioxane, 1.5 eq.). The mixture was then concentrated under vacuum, in order to assure that all of the
HCl and dioxane were removed, the product was re-dissolved twice in CH2C12 and dried under vacuum to finally obtain the HCl salt of compound 8f as a beige solid (1.62 g, 93% pure by HPLC). The product was then poured into a phosphate buffer
(IN NaH2PO4, pH=~4.5) and sonicated. The beige solid was filtered and dried under vacuum to give compound 8f (1.38 g, 81% yield) as a beige solid (91% pure by HPLC).
*H NMR (400 MHz, DMSO) δ 8.27 (s, IH), 8.12 (d, IH, J = 9.2 Hz), 7.97 (br.s, IH), 7.94 (s, IH), 7.43 (s, IH), 7.24 (dd, IH, J = 9.2, 2.2 Hz), 3.97 (m, IH), 3.94 (s, 3H), 1.24 (d, 2H, J = 6.4 Hz)
............
METHYL ESTER
EXAMPLE 34c
Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-iso-propylthiourea gave # 822
Η NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.33-8.23 (bs, IH), 8.21 (d, J = 9.2 Hz, IH), 8.04 (d, J = 8.3 Hz, IH), 7.86 (bs, IH), 7.77 (s, IH), 7.35-7.23 (m, 2H), 5.81 (bs, IH), 5.52 (dd, J = 8.5 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.65 (d, J = 11.8 Hz, IH), 4.51 (dd, J = 7.6 Hz, IH), 4.37 (bs, IH), 4.15 (bs, IH), 4.07-3.98 (m, 2H), 3.97 (s, 3H), 3.88 (d, J = 8.9 Hz, IH), 2.60-2.53 (m, 2H), 2.47-2.37 (m, 2H), 2.19-2.10 (dd, J = 9.2 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.29 (m, 13H), 1.27 and 1.25 (2 x d, J - 6.5 Hz, 6H), 1.23-1.09 (m, 2H). MS; es+: 775.0 (M + H)+, es : 772.9 (M - H)\
CILUPREVIR IS FREE ACID OF ABOVE AND HAS ENTRY 822 TABLE 8
.........
FREE AMINO COMPD
(Table 8)



A. To a solution of the macrocyclic intermediate 23b (13.05 g, 27.2 mmol, 1.0 eq.), Ph3P (14.28 g, 54.4 mmol, 2.0 eq) and 2-carboxymethoxy-4-hydroxy-7- methoxyquinoline (WO 00/09543 & WO 00/09558) (6.67 g, 28.6 mmol, 1.05 eq) in
THF (450 mL) at 0°C, DIAD (10.75 mL, 54.6 mmol, 2.0 eq) was added dropwise over a period of 15 min. The ice bath was then removed and the reaction mixture was stirred at RT for 3 h. After the complete conversion of starting material to products, the solvent was evaporated under vacuum, the remaining mixture diluted with
EtOAc, washed with saturated NaHCO3 (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness. Pure compound 34a was obtained after flash column chromatography; the column was eluted first with hexane/EtOAc (50:50), followed by CHCl3/EtOAc (95:5) to remove Ph3PO and
DIAD byproducts and elution of the impurities was monitored by TLC. Finally, the desired product 34a was eluted from the column with CHC13/ EtOAc (70:30).
Usually, the chromatography step had to be repeated 2-3 times before compound 34a could be isolated in high purity as a white solid with an overall yield of 68% (12.8 g, 99.5% pure by HPLC).
B. To a solution of the Boc-protected intermediate 34a (1.567g) in CH2C12 (15 mL), 4N HCl in dioxane (12 mL) was added and the reaction mixture was stirred at RT for 1 h. [In the event that a thick gel would form half way through the reaction period, an additional 10 mL CH2C12 was added.] Upon completion of the deprotection the solvents were evaporate to dryness to obtain a yellow solid and a paste like material. The mixture was redissolved in approximately 5% MeOH in
CH2C12 and re-evaporated to dryness under vacuum to obtain compound 34b as a yellow solid, which was used in the next step without any purification. C. To a solution of cyclopentanol (614 μL, 6.76 mmoL) in THF (15 mL), a solution of phosgene in toluene (1.93 M, 5.96 mL, 11.502 mmol) was added dropwise and the mixture was stirred at R.T. for 2 h to form the cyclopentyl chloroformate reagent (z). After that period, approximately half of the solvent was removed by evaporation under vacuum, the remaining light yellow solution was diluted by the addition of CH2C12 (5 mL) and concentrated to half of its original volume, in order to assure the removal of all excess phosgene. The above solution of the cyclopentyl chloroformate reagent was further diluted with THF (15 mL) and added to the amine-2HCl salt 34b. The mixture was cooled to 0 C in an ice bath, the pH was adjusted to -8.5-9 with the addition of Et3N (added dropwise) and the reaction mixture was stirred at 0 C for 1 h. After that period, the mixture was diluted with
EtOAc, washed with water (lx), saturated NaHCO3 (2x), H2O (2x) and brine (lx).
The organic layer was dried over anhydrous MgSO4, filtered and evaporated under vacuum to obtain a yellow-amber foam. Compound 34c was obtained as a white foam after purification by flash column chromatography (using a solvent gradient from 30% hexane to 20% hexane in EtOAc as the eluent) in 80% yield (1.27 g) and >93% purity. D. The dimethyl ester 34c (1.17g) was dissolved in a mixture of
THF/MeOH/H2O (20 mL, 2:1:1 ratio), and an aqueous solution of NaOH (1.8 mL,
IN, 1 eq.) was added. The reaction mixture was stirred at RT for 1 h before it was evaporated to dryness to obtain the sodium salt 34d as a white solid (-1.66 mmol). Compound 34d was used in the next step without purification.
E. The crude sodium salt 34d (1.66 mmoL) was dissolved in THF (17 mL), Et3N was added and the mixture was cooled to 0 C in an ice bath. Isobutylchloroformate
(322 μl, 2.5 mmol) was added dropwise and the mixture was stirred at 0 C for 75 min. After that period, diazomethane (15 mL) was added and stirring was continued at 0 C for 30 min and then at RT for an additional 1 h. Most of the solvent was evaporated to dryness under vacuum, the remaining mixture was diluted with EtOAc, washed with saturated NaHCO3 (2x), H2O (2x) and brine (lx), dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain compound 34e as a light yellow foam (1.2g, -1.66 mmol). The diazoketone intermediate 34e was used in the next step without purification.
F. The diazoketone 34e (1.2g, 1.66 mmoL) dissolved in THF (17 mL) was cooled to 0 C in an ice bath. A solution of aqueous HBr (48%, 1.24 mL) was added dropwise and the reaction mixture was stirred at 0 C for 1 h. The mixture was then diluted with EtOAc, wash with saturated NaHCO3 (2x), H2O (2x) and brine (lx), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the β-bromoketone intermediate 34f as a light yellow foam (-1.657 mmol).
G. To a solution of the bromoketone 34f (600 mg,0.779 mmol) in isopropanol (5 mL), thiourea (118 mg, 1.55 mmol) was added and the reaction mixture was placed in a pre-heated oil bath at 75 C where it was allowed to stir for 1 hr. The isopropanol was then removed under vacuum and the product dissolved in EtOAc
(100 mL). The solution was washed with saturated NaHCO3 and brine, the organic layer was dried over anhydrous Na2SO4, filtered and evaporated to afford the crude product 34g (522 mg) as a red-brown solid. This material was used in the final step without any further purification.
H. The crude methyl ester 34g (122 mg, 0.163 mmol) was dissolved in a solution of THF/MeOH/H2O (2:1:1 ratio, 4 mL) and saponified using LiOH»H2O (89 mg, 2.14 mmol). The hydrolysis reaction was carried out over a 12-15 h period at RT. The solvents were then removed under vacuum and the crude product purified by C18 reversed phase HPLC, using a solvent gradient from 10% CH3CN in H2O to 100%
CH3CN, to afford the HCV protease inhibitor #812 as a yellow solid (24 mg, 20% overall yield for the conversion of intermediate 34f to inhibitor #812).
*H NMR (400 MHz, DMSO-d6) δ 8.63 (s, IH), 8.26-8.15 (m, 2H), 7.79 (bs, IH), 7.72
(bs, IH), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, IH), 5.52 (dd, J = 8.3 Hz, IH), 5.27 (dd, J = 9.2 Hz, IH), 4.64 (d, J = 10.8 Hz, IH), 4.50 (dd, J = 8.3 Hz, IH), 4.39-4.31 (m, IH), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J = 9.5 Hz, 2H), 2.65-2.53 (m, 2H), 2.46- 2.36 (m, 2H), 2.20-2.12 (dd, J = 8.6 Hz, IH), 1.80-1.64 (m, 2H), 1.63-1.06 (m, 14H). MS; es+: 733.2 (M + H)+, es": 731.2 (M - H)\
....................
http://www.google.com/patents/WO2006036614A2
(Z)-( 1S,4R, 14S, 18R)- 14-Cyclopentyloxycarbonylamino- 18-[2-(2- isopropylamino-thiazol-4-yl)-7-methoxy-quinolin-4-yloxy]-2,15-dioxo-3,16-diaza- tricyclo[14.3.0.0 ' ]nonadec-7-ene-4-carboxylic acid , whose chemical structure is as follows:
, provided for in Tsantrizos et al., U.S. Patent No. 6,608,027 Bl,
..............................
https://www.google.co.in/patents/WO2005090383A2
ENTRY 218

.......................
http://www.google.com/patents/WO2004039833A1

.................
nmr
Synthesis of BILN 2061, an HCV NS3 protease inhibitor with proven antiviral effect in humans
Org Lett 2004, 6(17): 2901
http://pubs.acs.org/doi/full/10.1021/ol0489907
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032207.pdf procedure
http://pubs.acs.org/doi/suppl/10.1021/ol0489907/suppl_file/ol0489907si20040715_032254.pdf nmr spectra
BILN 2061:
Methyl ester 18 (2.69 g, 3.41 mmol) was dissolved in a mixture of THF
(40 mL), MeOH (20 mL) and water (20 mL) and added LiOH.H2O (1.14 g, 27.3 mmol).The resulting mixture was left to stir at RT for 15 h. The solvents were then removedunder reduced pressure and the crude product was redissolved with EtOAc and dilutedwith brine. The pH of the aqueous layer was adjusted to 6 with aqueous HCl (1N) and theaqueous phase was extracted with EtOAc (3x). The combined organic phase werewashed with water, brine, dried over MgSO4 and concentrated under reduced pressure toafford BILN 2061 as a yellow solid (2.63 g, 99% yield). HPLC(A) 99%, MS m/z (ES+)773 (M+H)+, (ES-) 775 (M-H)-;
1H NMR (DMSO-d6) δ 8.63 (s, 1H), 8.26-8.15 (m, 2H),
7.79 (bs, 1H), 7.72 (bs, 1H), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, 1H), 5.52 (dd, J=8.3 Hz, 1H), 5.27 (dd, J= 9.2 Hz, 1H), 4.64 (d, J= 10.8 Hz, 1H), 4.50 (dd, J= 8.3 Hz, 1H),4.39-4.31 (m, 1H), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J= 9.5 Hz, 2H), 2.65-2.53(m, 2H), 2.46-2.36 (m, 2H), 2.20-2.12 (dd, J= 8.6 Hz, 1H), 1.80-1.64 (m, 2H), 1.63-1.06(m, 14H); HRMS calcd for C40H51N6O8S: 775.3489; found: 775.3476
..............................
WO2007019674A1 Aug 3, 2006 Feb 22, 2007 Boehringer Ingelheim Int Viral polymerase inhibitors WO2010021717A2 * Aug 20, 2009 Feb 25, 2010 Sequoia Pharmaceuticals, Inc. Hcv protease inhibitors WO2010080874A1 Jan 7, 2010 Jul 15, 2010 Scynexis, Inc. Cyclosporine derivative for use in the treatment of hcv and hiv infection EP1455809A2 * Dec 13, 2002 Sep 15, 2004 Bristol-Myers Squibb Co. Inhibitors of hepatitis c virus EP2364984A1 Aug 28, 2006 Sep 14, 2011 Vertex Pharmaceuticals Incorporated Inhibitors of serine proteases EP2366704A1 Aug 28, 2006 Sep 21, 2011 Vertex Pharmaceuticals Incorporated Inhibitors of serine proteases US7368452 Jul 18, 2006 May 6, 2008 Enanta Pharmaceuticals, Inc. Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors US7608590 Jan 28, 2005 Oct 27, 2009 Medivir Ab HCV NS-3 serine protease inhibitors US7671032 Jan 28, 2005 Mar 2, 2010 Medivir Ab HCV NS-3 serine protease inhibitors US7816348 Jan 29, 2007 Oct 19, 2010 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors US7897622 Aug 10, 2007 Mar 1, 2011 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors US8148399 Jul 28, 2006 Apr 3, 2012 Tibotec Pharmaceuticals Ltd. Macrocyclic inhibitors of hepatitis C virus US8153800 Aug 3, 2011 Apr 10, 2012 Tibotec Pharmaceuticals Ltd. Macrocyclic inhibitors of hepatitis C virus US8242140 Jul 31, 2008 Aug 14, 2012 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors US8349869 Mar 6, 2012 Jan 8, 2013 Tibotec Pharmaceuticals Ltd. Macrocylic inhibitors of hepatitis C virus US8476257 Dec 3, 2008 Jul 2, 2013 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors US8541402 May 3, 2012 Sep 24, 2013 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors WO2000059929A1 * Apr 3, 2000 Oct 12, 2000 Boehringer Ingelheim Ca Ltd Macrocyclic peptides active against the hepatitis c virus WO2003053349A2 * Dec 13, 2002 Jul 3, 2003 Squibb Bristol Myers Co Inhibitors of hepatitis c virus WO2003064455A2 * Jan 24, 2003 Aug 7, 2003 Boehringer Ingelheim Ca Ltd Macrocyclic peptides active against the hepatitis c virus WO2003066103A1 * Feb 5, 2003 Aug 14, 2003 Boehringer Ingelheim Pharma Pharmaceutical compositions for hepatitis c viral protease inhibitors - .......................................................................................................

- ......................................................................................................................
- 4 SOVAPREVIR
- SOVAPREVIR(2S, 4R) -1 - [(2S)-2-tert-butyl-4-oxo-4-(piperidin-1-yl) butanoyl]-N-{(1R, 2S) -1 - [(cyclopropanesulfonyl) carbamoyl]-2-ethenylcyclopropyl} -4 - [(7-methoxy-2-phenylquinolin-4-yl) oxy] pyrrolidine-2-carboxamidePATENTUS 2009048297 ENTRY 60WO 2008008502CN 103420991THERAPEUTIC CLAIM ....Treatment of hepatitis CCHEMICAL NAMES1. 2-Pyrrolidinecarboxamide, N-[(1R,2S)-1-[[(cyclopropylsulfonyl)amino]carbonyl]-2-
ethenylcyclopropyl]-1-[(2S)-3,3-dimethyl-1-oxo-2-[2-oxo-2-(1-piperidinyl)ethyl]butyl]-4-
[(7-methoxy-2-phenyl-4-quinolinyl)oxy]-, (2S,4R)-2. (2S,4R)-N-{(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-1-{(2S)-
3,3-dimethyl-2-[2-oxo-2-(piperidin-1-yl)ethyl]butanoyl}-4-[(7-methoxy-2-phenylquinolin-
4-yl)oxy]pyrrolidine-2-carboxamideMOLECULAR FORMULA C43H53N5O8SMOLECULAR WEIGHT 800.0SPONSOR Achillion Pharmaceuticals, Inc.CODE DESIGNATION ACH-0141625CAS REGISTRY NUMBER 1001667-23-7- ACH-0141625
- Sovaprevir
- UNII-2ND9V3MN6O
Sovaprevir (formerly ACH-0141625), an HCV NS3 protease inhibitor, is in phase II clinical trials at Achillion for the oral treatment of naive patients with chronic hepatitis C virus genotype 1.In 2012, fast track designation was assigned by the FDA for the treatment of hepatitis C (HCV). In 2013, a clinical hold was placed for the treatment of hepatitis C (HCV) in combination with atazanavir after elevations in liver enzymes associated with the combination of both compounds.Sovaprevir, previously referred to as ACH-1625, is an investigational, next-generation NS3/4A protease inhibitor discovered by Achillion that is currently on clinical hold. In 2012, Fast Track status was granted by the U.S. Food and Drug Administration (FDA) to sovaprevir for the treatment of chronic hepatitis C viral infection (HCV).Achillion has initiated a Phase 2 clinical trial (007 Study) to evaluate the all-oral, interferon-free combination of sovaprevir and its second-generation NS5A inhibitor, ACH-3102, with ribavirin (RBV), for a 12 week treatment duration, in treatment naïve, genotype 1 (GT1) HCV patients. In July 2013, sovaprevir was placed on clinical hold after elevated liver enzymes were observed in a Phase 1 healthy subject drug-drug interaction study evaluating the effects of concomitant administration of sovaprevir with ritonavir-boosted atazanavir.In accordance with the clinical hold, the FDA provided that no new clinical trials that included dosing with sovaprevir could be initiated, however, the FDA allowed continued enrollment and treatment of patients in the Phase 2 -007 clinical trial evaluating 12-weeks of sovaprevir in combination with ACH-3102 and RBV for patients with treatment-naive genotype 1 HCV. In September 2013, after reviewing Achillion's response, the FDA stated that although all issues identified in the June 2013 letter had been addressed, it had concluded that the removal of the clinical hold was not warranted at this time.The FDA requested, among other things, additional analysis to more fully characterize sovaprevir pharmacokinetics and the intrinsic and extrinsic factors that may lead to higher than anticipated exposures of sovaprevir or other potential toxicities in addition to the observed liver enzyme elevations.The FDA also requested Achillion's proposed plan for future clinical trials in combination with other directly-acting antivirals. At the request of the FDA, Achillion plans to submit a proposed plan for analyzing the additional clinical, non-clinical and pharmacokinetic data requested before the end of 2013, and if that analysis plan is approved by the FDA, submit a complete response during the first half of 2014. Achillion retains worldwide commercial rights to sovaprevir.Sovaprevir has demonstrated activity against all HCV genotypes (GT), including equipotent activity against both GT 1a and 1b (IC50 ~ 1nM) in vitro.With its rapid and extensive partitioning to the liver, as well as high liver/plasma ratios, sovaprevir has been clinically demonstrated to allow for once-daily, non-boosted dosing.The current safety database for sovaprevir includes more than 560 subjects dosed to date and demonstrates that sovaprevir is well tolerated in these subjects.
Sovaprevir has demonstrated high rates of clinical cures in combination with pegylated-interferon and RBV in a challenging, real world, patient population of genotype 1 treatment-naive patients.100% of GT1b subjects achieved a rapid virologic response (RVR) in the 007 Study evaluating the interferon-free combination of sovaprevir + ACH-3102 + RBV for 12 weeks. The Phase 2 study is ongoing.Sovaprevir in vitro retains activity against mutations that confer resistance to 1st-generation protease inhibitors.In clinical studies to date, sovaprevir has demonstrated a high pharmacologic barrier to resistance with no on-treatment viral breakthrough reported to date in GT1b patients.Sovaprevir is believed to be synergistic when combined with other classes of DAAs, including the second-generation NS5A inhibitor, ACH-3102.For more information about the next-generation NS3/4A protease inhibitor, sovaprevir, please see the Related Links on this page or visit Resources.Sovaprevir is an investigational compound. Its safety and efficacy have not been established. (Updated December 2013)SOVAPREVIRAn estimated 3% of the world's population is infected with the hepatitis C virus. Of those exposed to HCV, 80% become chronically infected, at least 30% develop cirrhosis of the liver and 1-4% develop hepatocellular carcinoma. Hepatitis C Virus (HCV) is one of the most prevalent causes of chronic liver disease in the United States, reportedly accounting for about 15 percent of acute viral hepatitis, 60 to 70 percent of chronic hepatitis, and up to 50 percent of cirrhosis, end-stage liver disease, and liver cancer. Chronic HCV infection is the most common cause of liver transplantation in the U.S., Australia, and most of Europe. Hepatitis C causes an estimated 10,000 to 12,000 deaths annually in the United States. While the acute phase of HCV infection is usually associated with mild symptoms, some evidence suggests that only about 15% to 20% of infected people will clear HCV.HCV is an enveloped, single-stranded RNA virus that contains a positive-stranded genome of about 9.6 kb. HCV is classified as a member of the Hepacivirus genus of the family Flaviviridae. At least 4 strains of HCV, GT-1-GT-4, have been characterized.The HCV lifecycle includes entry into host cells; translation of the HCV genome, polyprotein processing, and replicase complex assembly; RNA replication, and virion assembly and release. Translation of the HCV RNA genome yields a more than 3000 amino acid long polyprotein that is processed by at least two cellular and two viral proteases. The HCV polyprotein is:NH2-C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B-COOH.The cellular signal peptidase and signal peptide peptidase have been reported to be responsible for cleavage of the N-terminal third of the polyprotein (C-E1-E2-p7) from the nonstructural proteins (NS2-NS3-NS4A-NS4B-NS5A-NS5B). The NS2-NS3 protease mediates a first cis cleavage at the NS2-NS3 site. The NS3-NS4A protease then mediates a second cis-cleavage at the NS3-NS4A junction. The NS3-NS4A complex then cleaves at three downstream sites to separate the remaining nonstructural proteins. Accurate processing of the polyprotein is asserted to be essential for forming an active HCV replicase complex.Once the polyprotein has been cleaved, the replicase complex comprising at least the NS3-NS5B nonstructural proteins assembles. The replicase complex is cytoplasmic and membrane-associated. Major enzymatic activities in the replicase complex include serine protease activity and NTPase helicase activity in NS3, and RNA-dependent RNA polymerase activity of NS5B. In the RNA replication process, a complementary negative strand copy of the genomic RNA is produced. The negative strand copy is used as a template to synthesize additional positive strand genomic RNAs that may participate in translation, replication, packaging, or any combination thereof to produce progeny virus. Assembly of a functional replicase complex has been described as a component of the HCV replication mechanism. Provisional application 60/669,872 “Pharmaceutical Compositions and Methods of Inhibiting HCV Replication” filed Apr. 11, 2005, is hereby incorporated by reference in its entirety for its disclosure related to assembly of the replicase complex.Current treatment of hepatitis C infection typically includes administration of an interferon, such as pegylated interferon (IFN), in combination with ribavirin. The success of current therapies as measured by sustained virologic response (SVR) depends on the strain of HCV with which the patient is infected and the patient's adherence to the treatment regimen. Only 50% of patients infected with HCV strain GT-1 exhibit a sustained virological response. Direct acting antiviral agents such as ACH-806, VX-950 and NM 283 (prodrug of NM 107) are in clinical development for treatment of chronic HCV. Due to lack of effective therapies for treatment for certain HCV strains and the high mutation rate of HCV, new therapies are needed.................................................(2S,4R)-1-((S)-2-tert-butyl-4-oxo-4-(piperidin-1-yl)butanoyl)-N-((1R,2S)-1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-2-carboxamide SOVAPREVIR IS DESCRIBED AS 60 IN CLAIMSYNTHESIS OF INTERMEDIATE 13 BELOW AND ALSO COMPD 8 IE SOVAPREVIR IN STEP 4Example 1SYNTHESIS OF 1-((2S,4R)-1-((S)-2-TERT-BUTYL-4-OXO-4-(PIPERIDIN-1-YL)BUTANOYL)-4-(7-METHOXY-2-PHENYLQUINOLIN-4-YLOXY)PYRROLIDINE-2-CARBOXAMIDO)-2-VINYLCYCLOPROPANECARBOXYLIC ACIDStep 1. Preparation of N-(cyclopropylsulfonyl)-1-(BOC-amino)-2-vinylcyclopropanecarboxamide
SOVAPREVIR IS DESCRIBED AS 60 IN CLAIMSYNTHESIS OF INTERMEDIATE 13 BELOW AND ALSO COMPD 8 IE SOVAPREVIR IN STEP 4Example 1SYNTHESIS OF 1-((2S,4R)-1-((S)-2-TERT-BUTYL-4-OXO-4-(PIPERIDIN-1-YL)BUTANOYL)-4-(7-METHOXY-2-PHENYLQUINOLIN-4-YLOXY)PYRROLIDINE-2-CARBOXAMIDO)-2-VINYLCYCLOPROPANECARBOXYLIC ACIDStep 1. Preparation of N-(cyclopropylsulfonyl)-1-(BOC-amino)-2-vinylcyclopropanecarboxamide CDI (2.98 g, 18.4 mm, 1.1 eq) is dissolved in ethyl acetate. N-Boc-cyclopropylvinyl acid (3.8 g, 16.7 mm, 1.0 eq), prepared via the procedure given by Beaulieu, P. L. et al. (J. Org. Chem. 70: 5869-79 (2005)) is added to the CDI/ethyl acetate mixture and stirred at RT until the starting material is consumed. Cyclopropyl sulfonamine (2.2 g, 18.4 mm, 1.1 eq) is added to this mixture followed by DBU (2.1 ml, 20.5 mm, 1.23 eq) and the mixture is stirred at RT for 2 h. Workup and purification by silica gel chromatography provides 2g of compound 2.Step 2. Preparation of (2S,4R)-tert-butyl 2-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carboxylate and (2S,4R)—N-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-2-carboxamide
CDI (2.98 g, 18.4 mm, 1.1 eq) is dissolved in ethyl acetate. N-Boc-cyclopropylvinyl acid (3.8 g, 16.7 mm, 1.0 eq), prepared via the procedure given by Beaulieu, P. L. et al. (J. Org. Chem. 70: 5869-79 (2005)) is added to the CDI/ethyl acetate mixture and stirred at RT until the starting material is consumed. Cyclopropyl sulfonamine (2.2 g, 18.4 mm, 1.1 eq) is added to this mixture followed by DBU (2.1 ml, 20.5 mm, 1.23 eq) and the mixture is stirred at RT for 2 h. Workup and purification by silica gel chromatography provides 2g of compound 2.Step 2. Preparation of (2S,4R)-tert-butyl 2-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carboxylate and (2S,4R)—N-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-2-carboxamide Compound 1 (4.3 g, 9.3 mmol, 1.1 eq), prepared according to the method given ins WO 02/060926, in DMF is stirred with O-(Benzotriazol-lyl)-N,N,N′,N′-Tetramethyluronium hexafluorophosphate (4.1 g, 10.5 mmol, 1.3 eq) for 30 minutes, followed by addition of cyclopropylamine 2 (1.92 g, 8.3 mmol, 1.0 eq) and N-methylmorpholine (2.52 g, 25.0 mmol, 3.0 eq). The mixture is stirred over night and the solvent removed under reduced pressure. The resulting residue is diluted with ethyl acetate and washed with saturated aqueous NaHCO3. The organic solvent is dried over MgSO4 and concentrated under reduced pressure to afford crude 3, which is used for next step without further purification.Compound 3 in 10 ml dry CH2Cl2 is treated with 5 mL TFA and stirred over night. The solvent is removed and the residue recrystallized from ethyl acetate to afford 4.12 g Compound 4 (61% yield two steps).Step 3. Preparation of (3S)-3-((2S,4R)-2-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acid
Compound 1 (4.3 g, 9.3 mmol, 1.1 eq), prepared according to the method given ins WO 02/060926, in DMF is stirred with O-(Benzotriazol-lyl)-N,N,N′,N′-Tetramethyluronium hexafluorophosphate (4.1 g, 10.5 mmol, 1.3 eq) for 30 minutes, followed by addition of cyclopropylamine 2 (1.92 g, 8.3 mmol, 1.0 eq) and N-methylmorpholine (2.52 g, 25.0 mmol, 3.0 eq). The mixture is stirred over night and the solvent removed under reduced pressure. The resulting residue is diluted with ethyl acetate and washed with saturated aqueous NaHCO3. The organic solvent is dried over MgSO4 and concentrated under reduced pressure to afford crude 3, which is used for next step without further purification.Compound 3 in 10 ml dry CH2Cl2 is treated with 5 mL TFA and stirred over night. The solvent is removed and the residue recrystallized from ethyl acetate to afford 4.12 g Compound 4 (61% yield two steps).Step 3. Preparation of (3S)-3-((2S,4R)-2-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acid The Acid 5 (58 mg, 0.25 mmol, 1.2 eq), prepared via the procedure given by Evans, D. A., et al. (J. Org. Chem. 64: 6411-6417 (1999)) in 1.2 mL DMF is stirred with 4 (138 mg, 0.21 mmol), HATU (160 mg, 0.42 mmol, 2.0 eq), and DIEA (0.63 mmol, 3.0 eq) overnight. The mixture is subjected to HPLC purification to afford 121 mg 6 (77% yield), which is further treated with 0.5 mL TFA in 1.0 mL DCM overnight. The solvent was removed to provide Compound 7 in 100% yield.Step 4. Preparation of (2S,4R)-1-((S)-2-tert-butyl-4-oxo-4-(piperidin-1-yl)butanoyl)-N-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-2-carboxamide
The Acid 5 (58 mg, 0.25 mmol, 1.2 eq), prepared via the procedure given by Evans, D. A., et al. (J. Org. Chem. 64: 6411-6417 (1999)) in 1.2 mL DMF is stirred with 4 (138 mg, 0.21 mmol), HATU (160 mg, 0.42 mmol, 2.0 eq), and DIEA (0.63 mmol, 3.0 eq) overnight. The mixture is subjected to HPLC purification to afford 121 mg 6 (77% yield), which is further treated with 0.5 mL TFA in 1.0 mL DCM overnight. The solvent was removed to provide Compound 7 in 100% yield.Step 4. Preparation of (2S,4R)-1-((S)-2-tert-butyl-4-oxo-4-(piperidin-1-yl)butanoyl)-N-(1-(cyclopropylsulfonylcarbamoyl)-2-vinylcyclopropyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-2-carboxamide PLEASE NOTE 8 IS SOVAPREVIRThe Acid 7 (0.15 mmol) in 1.0 mL DMF is stirred with pepridine (excess, 0.6 mmol, 4 eq), HATU (115 mg, 0.3 mmol, 2.0 eq), and DIEA (0.45 mmol, 3.0 eq) for 4 hrs. The mixture is subjected to HPLC purification to afford 77.1 mg 8.Step 5. Preparation of (3S)-3-((2S,4R)-2-(1-(ethoxycarbonyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acid
PLEASE NOTE 8 IS SOVAPREVIRThe Acid 7 (0.15 mmol) in 1.0 mL DMF is stirred with pepridine (excess, 0.6 mmol, 4 eq), HATU (115 mg, 0.3 mmol, 2.0 eq), and DIEA (0.45 mmol, 3.0 eq) for 4 hrs. The mixture is subjected to HPLC purification to afford 77.1 mg 8.Step 5. Preparation of (3S)-3-((2S,4R)-2-(1-(ethoxycarbonyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acid Step 5. Preparation of (3S)-3-((2S,4R)-2-(1-(ethoxycarbonyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acidThe Acid 5 (105 mg, 0.46 mmol, 1.2 eq) in 1.2 mL DMF is stirred with 9 (202 mg, 0.38 mmol), HATU (290 mg, 0.76 mmol, 2.0 eq), and DIEA (1.2 mmol, 3.0 eq) overnight. The mixture is subjected to HPLC purification to afford 204.3 mg 10 (75% yield), which is further treated with 0.5 mL TFA in 1.0 mL DCM overnight. The solvent is removed to provide 11 in 100% yield.
Step 5. Preparation of (3S)-3-((2S,4R)-2-(1-(ethoxycarbonyl)-2-vinylcyclopropylcarbamoyl)-4-(7-methoxy-2-phenylquinolin-4-yloxy)pyrrolidine-1-carbonyl)-4,4-dimethylpentanoic acidThe Acid 5 (105 mg, 0.46 mmol, 1.2 eq) in 1.2 mL DMF is stirred with 9 (202 mg, 0.38 mmol), HATU (290 mg, 0.76 mmol, 2.0 eq), and DIEA (1.2 mmol, 3.0 eq) overnight. The mixture is subjected to HPLC purification to afford 204.3 mg 10 (75% yield), which is further treated with 0.5 mL TFA in 1.0 mL DCM overnight. The solvent is removed to provide 11 in 100% yield. Step 6. Preparation of Final ProductThe Acid 11 (30 mg, 0.045 mmol) in 1.0 mL DMF is stirred with pepridine (0.27 mmol, 6 eq), HATU (34 mg, 0.09 mmol, 2.0 eq), and DIEA (0.14 mmol, 3.0 eq) for 2 hrs. The mixture is subjected to HPLC purification to afford 21.2 mg 12 (65% yield), which is hydrolyzed in methanol with 2N NaOH for 6 hrs. The mixture is acidified with 6N HCl and subjected to HPLC purification to afford 7.6 mg 13.....................................
Step 6. Preparation of Final ProductThe Acid 11 (30 mg, 0.045 mmol) in 1.0 mL DMF is stirred with pepridine (0.27 mmol, 6 eq), HATU (34 mg, 0.09 mmol, 2.0 eq), and DIEA (0.14 mmol, 3.0 eq) for 2 hrs. The mixture is subjected to HPLC purification to afford 21.2 mg 12 (65% yield), which is hydrolyzed in methanol with 2N NaOH for 6 hrs. The mixture is acidified with 6N HCl and subjected to HPLC purification to afford 7.6 mg 13.....................................
- ..................................................................................................
- 5 VEDROPREVIR
- GS 9451, GS-9451, 1098189-15-1 USAN ZZ-81VEDROPREVIR THERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES 1. Cyclopropanecarboxylic acid, N-[[(1α,3β,5α)-bicyclo[3.1.0]hex-3- yloxy]carbonyl]-3-methyl-L-valyl-(4R)-4-[[8-chloro-2-[2-[(1-methylethyl)amino]- 4-thiazolyl]-7-[2-(4-morpholinyl)ethoxy]-4-quinolinyl]oxy]-L-prolyl-1-amino-2- ethyl-, (1R,2R)-2. N-{[(1R,3r,5S)-bicyclo[3.1.0]hex-3-yloxy]carbonyl}-3-methyl-L-valyl-(4R)-4-[(8- chloro-2-{2-[(1-methylethyl)amino]thiazol-4-yl}-7-[2-(morpholin-4- yl)ethoxy]quinolin-4-yl)oxy]-L-prolyl-(1R,2R)-1-amino-2- ethylcyclopropanecarboxylic acidMOLECULAR FORMULA C45H60ClN7O9SMOLECULAR WEIGHT 910.5 daltonsSPONSOR Gilead Sciences, Inc.CODE DESIGNATION GS-9451CAS REGISTRY NUMBER1098189-15-1WHO NUMBER9745GS-9451 is a NS3 protease inhibitor in phase II clinical trials at Gilead for the oral treatment of hepatitis C.....................................................................................Discovery of GS-9451: An acid inhibitor of the hepatitis C virus NS3/4A protease Bioorg Med Chem Lett 2012, 22(7): 2629 .....................................................................PATENTS WO 2012087596 WO 2009005676 WO 2013106631 WO2013101550
............................
WO2012087596A1 Compound 3 can be prepared using synthetic methods and intermediates like those described in USSN 12/215,605 (US 20090257978 A1). Compound 3 can also be prepared described in the following Example. Example 3: Preparation of Compound 3 Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows.
Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows. 301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).
301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).

 c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig's base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig's base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat'd NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat'd NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig's base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig's base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat'd NH4CI (500mL), 1 N HCI (aq) (500mL), sat'd NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat'd aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.
c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig's base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig's base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat'd NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat'd NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig's base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig's base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat'd NH4CI (500mL), 1 N HCI (aq) (500mL), sat'd NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat'd aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.

 h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give - 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )Preparation of 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylic acid 215. To a solution of methyl 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylate 214 (36.5g, 0.145 mol) in a mixture of 1 :1 of MeOH: THF (160 mL total) was added a solution of LiOH (30.5 g, 0.725 mol) in H20 (80 mL). The mixture was stirred at room temperature for an hour when LCMS analysis showed complete conversion to the carboxylic acid. The reaction was worked up by removal of the volatiles and adjusting the pH of the solution to 6 using aqueous 6N HCI. The resulted gummy residue was filtered and dried on the lyophilizer for 2 days to provide 34.4 g (99.6 %) of compound 215 as a white solid. El MS (mlz) 253.9 [M+H]. j. Preparation of 2-(2-diazo-l-oxo)-8-chloro-7-methoxyquinolin-4-yl isobutyl carbonate 216. To a solution of 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylic acid 215 (10.2 g, 0.04 mol) in THF (400 mL) was added triethyl amine (12.3 mL, 0.088 mol) and /-Butylchloroformate (11.6 mL, 0.088 mol) at 0°C under an argon atmosphere. The mixture was stirred at 0°C for 1 hour when LCMS analysis demonstrated completion of the reaction to provide the desired mixed anhydride. El MS (mlz) 454.0 [M+H]. To the reaction mixture of the anhydride was added a 1 M solution of diazomethane (121 mL, 0.121 mol) in diethyl ether via a plastic funnel at 0°C. This mixture was allowed to stir while warming up to room temperature for additional 2 hours. Analysis of the mixture by LCMS demonstrated completion of the reaction. The septum was removed and the reaction was stirred for additional 20 minutes before removal of the solvent. The residue was dried further under high vacuum to provide compound 216, which was carried on to the next step. El MS (m/z) 377.9 [M+H]. k. Preparation of 8-chloro-2-(2-(isopropylamino)thiazol-4-yl)-7-methoxyquinoiin-4-ol 207. To a cooled solution of 2-(2-diazo-l-oxo)-8-chloro-7-methoxyquinolin-4-yl isobutyl carbonate 216 (15.2 g, 0.040 mol) at 0°C in THF (268 mL) was added 48% HBr (23 mL, 0.201 mol) slowly over 15 minutes. The solution was stirred at 0°C for an additional 40 minutes when LCMS analysis demonstrated complete reaction. The reaction was worked up by addition of aqueous 1 N NaOH (180 mL) at 0° C to adjust the pH of the aqueous layer to 9. The layers were separated and the aqueous layer was washed with EtOAc (2 x 200 mL). Combined organic extracts were washed with brine and dried over MgS04. The solvent was removed in vacuo to provide 17.7 g of a yellow solid. El MS (m/z) 431.9 [M+H]. The solution of the bromoketone obtained from the previous reaction was suspended in i-propanol (270 mL) and isopropylisourea (9.4 g, 0.080 mol). The reaction mixture was heated at 72 °C for 32 hours. LCMS analysis of the reaction demonstrated complete conversion to the desired- product. The reaction was allowed to cool to room temperature to allow for the product to precipitate out of the solution. The reaction was further cooled to 0°C for 12 hours before filtration. The filtrate was washed with ether and dried on lyopholizer to provide 8.03 g of compound 207 as an orange solid. 1H NMR (500 MHz, CDCI3): δ 8.21 (d, J= 9 Hz, 1 H), 7.74 (s, 1 H), 7.44 (d, J= 10Hz), 1 H), 7.07 (s, 1 H), 4.05 (s, 3H), 3.92 (pentet, J=6 Hz, 1 H), 1.25 (d, J= 7 Hz, 6H): El MS (m/z) 350.0 [M+H].
h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give - 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )Preparation of 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylic acid 215. To a solution of methyl 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylate 214 (36.5g, 0.145 mol) in a mixture of 1 :1 of MeOH: THF (160 mL total) was added a solution of LiOH (30.5 g, 0.725 mol) in H20 (80 mL). The mixture was stirred at room temperature for an hour when LCMS analysis showed complete conversion to the carboxylic acid. The reaction was worked up by removal of the volatiles and adjusting the pH of the solution to 6 using aqueous 6N HCI. The resulted gummy residue was filtered and dried on the lyophilizer for 2 days to provide 34.4 g (99.6 %) of compound 215 as a white solid. El MS (mlz) 253.9 [M+H]. j. Preparation of 2-(2-diazo-l-oxo)-8-chloro-7-methoxyquinolin-4-yl isobutyl carbonate 216. To a solution of 8-chloro-4-hydroxy-7-methoxyquinoline-2-carboxylic acid 215 (10.2 g, 0.04 mol) in THF (400 mL) was added triethyl amine (12.3 mL, 0.088 mol) and /-Butylchloroformate (11.6 mL, 0.088 mol) at 0°C under an argon atmosphere. The mixture was stirred at 0°C for 1 hour when LCMS analysis demonstrated completion of the reaction to provide the desired mixed anhydride. El MS (mlz) 454.0 [M+H]. To the reaction mixture of the anhydride was added a 1 M solution of diazomethane (121 mL, 0.121 mol) in diethyl ether via a plastic funnel at 0°C. This mixture was allowed to stir while warming up to room temperature for additional 2 hours. Analysis of the mixture by LCMS demonstrated completion of the reaction. The septum was removed and the reaction was stirred for additional 20 minutes before removal of the solvent. The residue was dried further under high vacuum to provide compound 216, which was carried on to the next step. El MS (m/z) 377.9 [M+H]. k. Preparation of 8-chloro-2-(2-(isopropylamino)thiazol-4-yl)-7-methoxyquinoiin-4-ol 207. To a cooled solution of 2-(2-diazo-l-oxo)-8-chloro-7-methoxyquinolin-4-yl isobutyl carbonate 216 (15.2 g, 0.040 mol) at 0°C in THF (268 mL) was added 48% HBr (23 mL, 0.201 mol) slowly over 15 minutes. The solution was stirred at 0°C for an additional 40 minutes when LCMS analysis demonstrated complete reaction. The reaction was worked up by addition of aqueous 1 N NaOH (180 mL) at 0° C to adjust the pH of the aqueous layer to 9. The layers were separated and the aqueous layer was washed with EtOAc (2 x 200 mL). Combined organic extracts were washed with brine and dried over MgS04. The solvent was removed in vacuo to provide 17.7 g of a yellow solid. El MS (m/z) 431.9 [M+H]. The solution of the bromoketone obtained from the previous reaction was suspended in i-propanol (270 mL) and isopropylisourea (9.4 g, 0.080 mol). The reaction mixture was heated at 72 °C for 32 hours. LCMS analysis of the reaction demonstrated complete conversion to the desired- product. The reaction was allowed to cool to room temperature to allow for the product to precipitate out of the solution. The reaction was further cooled to 0°C for 12 hours before filtration. The filtrate was washed with ether and dried on lyopholizer to provide 8.03 g of compound 207 as an orange solid. 1H NMR (500 MHz, CDCI3): δ 8.21 (d, J= 9 Hz, 1 H), 7.74 (s, 1 H), 7.44 (d, J= 10Hz), 1 H), 7.07 (s, 1 H), 4.05 (s, 3H), 3.92 (pentet, J=6 Hz, 1 H), 1.25 (d, J= 7 Hz, 6H): El MS (m/z) 350.0 [M+H]. - .....................................................................................................................................



- ....................................................................................................................
- 6 VANIPREVIR
 (1R,21S,24S)-21-tert-butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-16,16-dimethyl-3,19,22-trioxo-2,18-dioxa-4,20,23-triazatetracyclo[21.2.1.14,7.06,11]-heptacosa-6,8,10-triene-24-carboxamide923590-37-8 cas no
(1R,21S,24S)-21-tert-butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-16,16-dimethyl-3,19,22-trioxo-2,18-dioxa-4,20,23-triazatetracyclo[21.2.1.14,7.06,11]-heptacosa-6,8,10-triene-24-carboxamide923590-37-8 cas noMolecular formula C38H53N5O9S Molar mass 755.92 g mol−1 vaniprevir (MK-7009) is a macrocyclic hepatitis C virus NS3/4a protease inhibitor, is active against both the genotype 1 and genotype 2 NS3/4a protease enzymes. vaniprevir (MK-7009) has good plasma exposure and excellent liver exposure in multiple species.Vaniprevir (MK-7009) is a macrocyclic Hepatitis C virus (HCV) NS3/4a protease inhibitor, developed by Merck & Co., which is currently in clinical testing.[1]- McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ (March 2010). "Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor". J. Med. Chem. 53 (6): 2443–63.doi:10.1021/jm9015526. PMID 20163176.
Song ZJ, Tellers DM, Journet M, Kuethe JT, Lieberman D, Humphrey G, Zhang F, Peng Z, Waters MS, Zewge D, Nolting A, Zhao D, Reamer RA, Dormer PG, Belyk KM, Davies IW, Devine PN, Tschaen DM.Synthesis of vaniprevir (MK-7009): lactamization to prepare a 20-membered [corrected] macrocycle.J Org Chem. 2011 Oct 7;76(19):7804-15. Epub 2011 Aug 31.
Abstract
Development of a practical synthesis of MK-7009, a 20-membered [corrected] macrocycle, is described. A variety of ring-closing strategies were evaluated, including ring-closing metathesis, intermolecular palladium-catalyzed cross-couplings, and macrolactamization. Ring closure via macrolactamization was found to give the highest yields under relatively high reaction concentrations. Optimization of the ring formation step and the synthesis of key intermediates en route to MK-7009 are reported.........................................Kong J, * Chen C.-y, * Balsells-Padros J, Cao Y, Dunn RF, Dolman SJ, Janey J, Li H, Zacuto MJ. Merck Research Laboratory, Rahway, USA
Synthesis of the HCV Protease Inhibitor Vaniprevir (MK-7009) Using Ring-Closing Metathesis Strategy.J. Org. Chem. 2012; 77: 3820-3828
The key step in this synthesis of vaniprevir is the construction of the macrocycle (91% yield) via ring-closing metathesis (RCM). By using simultaneous slow addition of the substrate and the catalyst D (0.2 mol%), the RCM reaction could be conducted at high concentration (0.13 M) on a 100 g scale.
2,6-Dichloro-1,4-benzoquinone was added to suppress isomerization of the allyl alkene in the isoindoline unit in C and consequent competing formation of a 19-membered ring by-product. An important contributor to the success of the RCM reaction was the high purity of crystalline B......................................... J. Org. Chem., 2012, 77 (8), pp 3820–3828DOI: 10.1021/jo3001595nmrSynthesis of the HCV protease inhibitor vaniprevir (MK-7009) using ring-closing metathesis strategy
J. Org. Chem., 2012, 77 (8), pp 3820–3828DOI: 10.1021/jo3001595nmrSynthesis of the HCV protease inhibitor vaniprevir (MK-7009) using ring-closing metathesis strategy
J Org Chem 2012, 77(8): 3820
Song, Z.G.J.; Tellers, D.M.; Journet, M.; et al.
Synthesis of vaniprevir (MK-7009): Lactamization to prepare a 22-membered macrocycle
J Org Chem 2011, 76(19): 9553WO 2013106631WO 2013101550WO 2007015787
WO 2007015855WO 2013066753WO 2012082672WO 2011025849WO 2003099274WO 2007016441........................................................................................EXAMPLE 46 (5R,7S,10S)-10-tert-Butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-15,15-dimethyl-3,9,12-trioxo-6,7,9,10,11,12,14,15,16,17,18,19-dodecahydro-1H,5H-2,23-ethano-5,8-methano-4,13,2,8,11-benzodioxatriazacyclohenicosine-7-carboxamide (III-231)
 Step 1: 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide
Step 1: 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide A mixture of 8-methoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride [Tetrahedron Letters, 1991, 32(17), 1965.] (3.0 g 15.0 mmol) and 45 mL of 48% aqueous HBr was heated for 18 h at 120° C. The resulting brown suspension was filtered and dried to provide 8-hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide (2.8 g, 81% yield). LRMS (ESI) m/z 150.1 [(M+H)+; calcd for C9H1NO: 150.2].Step 2: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate:
A mixture of 8-methoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride [Tetrahedron Letters, 1991, 32(17), 1965.] (3.0 g 15.0 mmol) and 45 mL of 48% aqueous HBr was heated for 18 h at 120° C. The resulting brown suspension was filtered and dried to provide 8-hydroxy-1,2,3,4-tetrahydroisoquinoline hydrobromide (2.8 g, 81% yield). LRMS (ESI) m/z 150.1 [(M+H)+; calcd for C9H1NO: 150.2].Step 2: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate: Carbonyldiimidazole (0.176 g, 1.086 mmol) was added to a stirred, room temperature solution of DMF (5 mL) and N-Boc-trans-4-hydroxy-L-proline methyl ester (0.21 g, 0.87 mmol) and the mixture was stirred 45 min. 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline (0.20 g, 0.87 mmol) and Et3N (0.18 g, 1.74 mmol) were added and the resulting solution was heated at 50° C. for 2 h. The reaction mixture was poured into aqueous saturated NH4Cl and extracted with EtOAc, dried over Na2SO4and concentrated to an oil. The residue was purified by column chromatography on silica gel (gradient elution, 10 to 80% EtOAc in hexanes) to give 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (0.25 g, 0.60 mmol, 69% yield) as a colorless foam after evaporation of solvent. LRMS (ESI) m/z 321.3 [((M-Boc)+H)+; calcd for C16H21N2O5: 321.4].Step 3: 1-tert-Butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate
Carbonyldiimidazole (0.176 g, 1.086 mmol) was added to a stirred, room temperature solution of DMF (5 mL) and N-Boc-trans-4-hydroxy-L-proline methyl ester (0.21 g, 0.87 mmol) and the mixture was stirred 45 min. 8-Hydroxy-1,2,3,4-tetrahydroisoquinoline (0.20 g, 0.87 mmol) and Et3N (0.18 g, 1.74 mmol) were added and the resulting solution was heated at 50° C. for 2 h. The reaction mixture was poured into aqueous saturated NH4Cl and extracted with EtOAc, dried over Na2SO4and concentrated to an oil. The residue was purified by column chromatography on silica gel (gradient elution, 10 to 80% EtOAc in hexanes) to give 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (0.25 g, 0.60 mmol, 69% yield) as a colorless foam after evaporation of solvent. LRMS (ESI) m/z 321.3 [((M-Boc)+H)+; calcd for C16H21N2O5: 321.4].Step 3: 1-tert-Butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate Trifluoromethanesulfonic anhydride (1.76 g, 6.24 mmol) was added to a stirred, 0° C. mixture of 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.81 g, 4.30 mmol) and Et3N (1.31 g, 12.90 mmol) in DCM (20 mL) and stirred for 18 h. The resulting mixture was poured into saturated aqueous NaHCO3 and extracted into dichloromethane. The organic layer was washed with 10% citric acid solution, dried over Na2SO4and concentrated to red oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 70% EtOAc in hexanes) to give a yellow oil, 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoro methyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.65 g, 69.4% yield). LRMS (ESI) m/z 453.2 [((M-Boc)+H)+; calcd for C17H20F3N2O7S: 453.4].Step 4: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate
Trifluoromethanesulfonic anhydride (1.76 g, 6.24 mmol) was added to a stirred, 0° C. mixture of 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-hydroxy-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.81 g, 4.30 mmol) and Et3N (1.31 g, 12.90 mmol) in DCM (20 mL) and stirred for 18 h. The resulting mixture was poured into saturated aqueous NaHCO3 and extracted into dichloromethane. The organic layer was washed with 10% citric acid solution, dried over Na2SO4and concentrated to red oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 70% EtOAc in hexanes) to give a yellow oil, 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoro methyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.65 g, 69.4% yield). LRMS (ESI) m/z 453.2 [((M-Boc)+H)+; calcd for C17H20F3N2O7S: 453.4].Step 4: 1-tert-Butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate A solution of 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.74 g, 3.15 mmol), tri-n-butyl vinyl tin (1.10 g, 1.46 mmol) and lithium chloride (0.40 g, 9.45 mmol) in 25 mL DMF was purged with nitrogen for 10 min. Then bis(triphenylphosphine)palladium (II) chloride (0.22 g, 0.32 mmol) was added, and the mixture stirred at 25° C. under nitrogen for 18 h. The mixture was partitioned between EtOAc and saturated NaHCO3, the organic layer separated and washed with water then brine, dried over anhydrous sodium sulfate and concentrated to an oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 65% EtOAc in hexanes) to give a colorless oil, 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.00 g, 74% yield). LRMS (ESI) m/z 453.2 [(M+Na)+; calcd for C23H30N2O6Na: 453.5].Step 5: (5R,7S,10S)-10-tert-Butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-15,15-dimethyl-3,9,12-trioxo-6,7,9,10,11,12,14,15,16,17,18,19-dodecahydro-1H,5H-2,23-ethano-5.8-methano-4,13,2,8,11-benzodioxatriazacyclohenicosine-7-carboxamide (III-231)Synthesis of the HCV protease inhibitor Vaniprevir (MK-7009) using ring-closing metathesis strategy.Kong J, Chen CY, Balsells-Padros J, Cao Y, Dunn RF, Dolman SJ, Janey J, Li H, Zacuto MJ.J Org Chem. 2012 Apr 20;77(8):3820-8. doi: 10.1021/jo3001595. Epub 2012 Apr 10.Song ZJ, Tellers DM, Journet M, Kuethe JT, Lieberman D, Humphrey G, Zhang F, Peng Z, Waters MS, Zewge D, Nolting A, Zhao D, Reamer RA, Dormer PG, Belyk KM, Davies IW, Devine PN, Tschaen DM.J Org Chem. 2011 Oct 7;76(19):7804-15. doi: 10.1021/jo2011494. Epub 2011 Aug 31. Erratum in: J Org Chem. 2011 Nov 18;76(22):9553.2.McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ.J Med Chem. 2010 Mar 25;53(6):2443-63. doi: 10.1021/jm9015526.Pompei M, Di Francesco ME, Pesci S, Koch U, Vignetti SE, Veneziano M, Pace P, Summa V.Bioorg Med Chem Lett. 2010 Jan 1;20(1):168-74. doi: 10.1016/j.bmcl.2009.11.005. Epub 2009 Nov 10.
A solution of 1-tert-butyl 2-methyl (2S,4R)-4-({[8-{[(trifluoromethyl)sulfonyl]oxy}-3,4-dihydroisoquinolin-2(1H)-yl]carbonyl}oxy)pyrrolidine-1,2-dicarboxylate (1.74 g, 3.15 mmol), tri-n-butyl vinyl tin (1.10 g, 1.46 mmol) and lithium chloride (0.40 g, 9.45 mmol) in 25 mL DMF was purged with nitrogen for 10 min. Then bis(triphenylphosphine)palladium (II) chloride (0.22 g, 0.32 mmol) was added, and the mixture stirred at 25° C. under nitrogen for 18 h. The mixture was partitioned between EtOAc and saturated NaHCO3, the organic layer separated and washed with water then brine, dried over anhydrous sodium sulfate and concentrated to an oil. The oil was purified by column chromatography on silica gel (gradient elution, 10 to 65% EtOAc in hexanes) to give a colorless oil, 1-tert-butyl 2-methyl (2S,4R)-4-{[(8-vinyl-3,4-dihydroisoquinolin-2(1H)-yl)carbonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.00 g, 74% yield). LRMS (ESI) m/z 453.2 [(M+Na)+; calcd for C23H30N2O6Na: 453.5].Step 5: (5R,7S,10S)-10-tert-Butyl-N-((1R,2R)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-ethylcyclopropyl)-15,15-dimethyl-3,9,12-trioxo-6,7,9,10,11,12,14,15,16,17,18,19-dodecahydro-1H,5H-2,23-ethano-5.8-methano-4,13,2,8,11-benzodioxatriazacyclohenicosine-7-carboxamide (III-231)Synthesis of the HCV protease inhibitor Vaniprevir (MK-7009) using ring-closing metathesis strategy.Kong J, Chen CY, Balsells-Padros J, Cao Y, Dunn RF, Dolman SJ, Janey J, Li H, Zacuto MJ.J Org Chem. 2012 Apr 20;77(8):3820-8. doi: 10.1021/jo3001595. Epub 2012 Apr 10.Song ZJ, Tellers DM, Journet M, Kuethe JT, Lieberman D, Humphrey G, Zhang F, Peng Z, Waters MS, Zewge D, Nolting A, Zhao D, Reamer RA, Dormer PG, Belyk KM, Davies IW, Devine PN, Tschaen DM.J Org Chem. 2011 Oct 7;76(19):7804-15. doi: 10.1021/jo2011494. Epub 2011 Aug 31. Erratum in: J Org Chem. 2011 Nov 18;76(22):9553.2.McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ.J Med Chem. 2010 Mar 25;53(6):2443-63. doi: 10.1021/jm9015526.Pompei M, Di Francesco ME, Pesci S, Koch U, Vignetti SE, Veneziano M, Pace P, Summa V.Bioorg Med Chem Lett. 2010 Jan 1;20(1):168-74. doi: 10.1016/j.bmcl.2009.11.005. Epub 2009 Nov 10.- ......................................................................
- 7 NARLAPREVIR
- NARLAPREVIRAn antiviral agent that inhibits hepatitis C virus NS3 protease.M.Wt: 707.96
Formula: C36H61N5O7SCAS No.: 865466-24-6SCH 900518;SCH900518;SCH-9005183-Azabicyclo[3.1.0]hexane-2-carboxamide, N-[(1S)-1-[2-(cyclopropylamino)-2-
oxoacetyl]pentyl]-3-[(2S)-2-[[[[1-[[(1,1-dimethylethyl)sulfonyl]methyl]cyclohexyl]
amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-, (1R,2S,5S)-2. (1R,2S,5S)-N-{(1S)-1-[2-(cyclopropylamino)-2-oxoacetyl]pentyl}-3-[(2S)-2-{[(1-{[(1,1-
dimethylethyl)sulfonyl]methyl}cyclohexyl)carbamoyl]amino}-3,3-dimethylbutanoyl]-6,6-
dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide3. (1R,2S,5S)-3-{N-[({1-[(tert-butylsulfonyl)methyl]cyclohexyl}amino)carbonyl]-3-methyl-L-
valyl}-N-{(1S)-1-[(cyclopropylamino)(oxo)acetyl]pentyl}-6,6-dimethyl-3-
azabicyco[3.1.0]hexane-2-carboxamideNarlaprevir is a potent, Second Generation HCV NS3 Serine Protease Inhibitor.Narlaprevir is useful for AntiviralMerck & Co. (Originator)SCH-900518 had been in phase II clinical trials by Merck & Co. for the treatment of genotype 1 chronic hepatitis C; however, no recent development has been reported for this indication.A potent oral inhibitor of HCV NS3 protease, SCH-900518 disrupts hepatitis C virus (HCV) polyprotein processing. When added to the current standard of care (SOC), peginterferon-alfa plus ribavirin, SCH-900518 is likely to increase the proportion of patients achieving undetectable HCV-RNA levels and sustained virologic response (SVR).In 2012, the product was licensed by Merck & Co. to R-Pharm in Russia and the Commonwealth of Independent States (CIS) for the development and commercialization as treatment of hepatitis C (HCV)PATENTSWO 2011014494WO 2010068714(1 R,5S)-N-[1 (S)-[2-(cyclopropylamino)-1 ,2-dioxoethyl]pentyl]-3-[2(S)- [[[[1-[[1.1-dimethylethyl)sulfonyl]methyl]cyclohexyl]amino]carbonyl]amino]-3,3- dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2(S)-carboxamide. Identification of any publication in this section or any section of this application is not an admission that such publication is prior art to the present invention.The compound of Formula I is generically and specifically disclosed inPublished U.S. Patent No.2007/0042968, published February 22, 2007 (the '968 publication), incorporated herein by reference.Processes suitable for making the compound of Formula I are generally described in the '968 publication. In particular, the '968 publication discusses preparing a sulfone carbamate compound, for example, the compound of Formula 837 comprising a cyclic sulfone substituent (paragraphs [0395] through [0403]). The following reaction scheme describes the procedure:
Identification of any publication in this section or any section of this application is not an admission that such publication is prior art to the present invention.The compound of Formula I is generically and specifically disclosed inPublished U.S. Patent No.2007/0042968, published February 22, 2007 (the '968 publication), incorporated herein by reference.Processes suitable for making the compound of Formula I are generally described in the '968 publication. In particular, the '968 publication discusses preparing a sulfone carbamate compound, for example, the compound of Formula 837 comprising a cyclic sulfone substituent (paragraphs [0395] through [0403]). The following reaction scheme describes the procedure: The process disclosed in the '968 publication produces the intermediate alcohol in step S7 as a mixture of diastereomers at the hydroxyl group; while this chiral center is lost in the final step of the disclosed process, the alcohol intermediate as a mixture of isomers cannot be crystallized and required a volumetrically inefficient precipitative isolation that did not remove any impurities,.......................................................................................................................................
The process disclosed in the '968 publication produces the intermediate alcohol in step S7 as a mixture of diastereomers at the hydroxyl group; while this chiral center is lost in the final step of the disclosed process, the alcohol intermediate as a mixture of isomers cannot be crystallized and required a volumetrically inefficient precipitative isolation that did not remove any impurities,....................................................................................................................................... ....................................................................................................................................Preparation of Compound VIJ
....................................................................................................................................Preparation of Compound VIJ LDA was made by slowly charging n-butyl lithium (2.5 M, 159 kg) to diisopropyl amine (60 kg) dissolved in THF (252 kg), keeping the temperature at about −20° C., followed by agitation at this temperature for about 30 min. To this solution was charged cyclohexane carboxylic acid, methyl ester (70 kg), keeping the temperature below −10° C. The mixture was agitated at this temperature for about 2 h. To the resulting enolate was charged TMSCI (64.4 kg). The mixture was agitated at −10 to −20° C. for about 30 min, and then heated to about 25° C. and held at this temperature to allow for conversion to the silylenol ether Compound VIH. The reaction mixture was solvent exchanged to n-heptane under vacuum, keeping the temperature below 50° C., resulting in the precipitation of solids. The solids were filtered and washed with n-heptane, and the wash was combined with the n-heptane reaction mixture. The n-heptane mixture of Compound VIH was concentrated under vacuum and diluted with CH2Cl2.In a separate reactor was charged CH2Cl2 (461 kg) and anhydrous ZnBr2 (14.5 kg). The temperature of the zinc slurry was adjusted to about 20° C. To the zinc slurry was simultaneously charged the solution of Compound VIH and 2-chloromethylsulfanyl-2-methyl-propane (63.1 kg, ref: Bioorg. Med. Chem. Lett, 1996, 6, 2053-2058), keeping the temperature below 45° C. After complete addition, the mixture was agitated for about 1.5 h at 35 to 45° C., after which the reaction mixture was cooled to 10 to 15° C. A solution of dilute aqueous HCl was then charged, keeping the temperature between 0 and 15° C., followed by a separation of the aqueous and organic layers (desired compound in organic layer). The organic layer was washed with aqueous NaHCO3 and water. The organic layer was solvent exchanged to methanol by vacuum distillation, keeping the temperature below 35° C., and kept as a solution in methanol for further processing to Compound VIK. Active Yield of Compound VIJ=69.7 kg (molar yield=57.9%).Preparation of Compound VIK
LDA was made by slowly charging n-butyl lithium (2.5 M, 159 kg) to diisopropyl amine (60 kg) dissolved in THF (252 kg), keeping the temperature at about −20° C., followed by agitation at this temperature for about 30 min. To this solution was charged cyclohexane carboxylic acid, methyl ester (70 kg), keeping the temperature below −10° C. The mixture was agitated at this temperature for about 2 h. To the resulting enolate was charged TMSCI (64.4 kg). The mixture was agitated at −10 to −20° C. for about 30 min, and then heated to about 25° C. and held at this temperature to allow for conversion to the silylenol ether Compound VIH. The reaction mixture was solvent exchanged to n-heptane under vacuum, keeping the temperature below 50° C., resulting in the precipitation of solids. The solids were filtered and washed with n-heptane, and the wash was combined with the n-heptane reaction mixture. The n-heptane mixture of Compound VIH was concentrated under vacuum and diluted with CH2Cl2.In a separate reactor was charged CH2Cl2 (461 kg) and anhydrous ZnBr2 (14.5 kg). The temperature of the zinc slurry was adjusted to about 20° C. To the zinc slurry was simultaneously charged the solution of Compound VIH and 2-chloromethylsulfanyl-2-methyl-propane (63.1 kg, ref: Bioorg. Med. Chem. Lett, 1996, 6, 2053-2058), keeping the temperature below 45° C. After complete addition, the mixture was agitated for about 1.5 h at 35 to 45° C., after which the reaction mixture was cooled to 10 to 15° C. A solution of dilute aqueous HCl was then charged, keeping the temperature between 0 and 15° C., followed by a separation of the aqueous and organic layers (desired compound in organic layer). The organic layer was washed with aqueous NaHCO3 and water. The organic layer was solvent exchanged to methanol by vacuum distillation, keeping the temperature below 35° C., and kept as a solution in methanol for further processing to Compound VIK. Active Yield of Compound VIJ=69.7 kg (molar yield=57.9%).Preparation of Compound VIK To a fresh reactor was charged Compound VIJ (99.8 kg active in a methanol solution), water (270 kg), NaOH (70 kg), and methanol (603 kg). The mixture was heated to −70° C. and agitated at this temperature for about 16 h. Upon conversion to the sodium salt of Compound VIK, the reaction mixture was concentrated under vacuum, keeping the temperature below 55° C., and then cooled to about 25° C. Water and MTBE were then charged, agitiated, and the layers were separated (product in the aqueous layer). The product-containing aqueous layer was further washed with MTBE.CH2Cl2 was charged to the aqueous layer and the temperature was adjusted to ˜10° C. The resultant mixture was acidified to a pH of about 1.5 with HCl, agitated, settled, and separated (the compound was in the organic layer). The aqueous layer was extracted with CH2Cl2, and the combined organic layers were stored as a CH2Cl2 solution for further processing to Compound VID. Active yield of Compound VIK=92.7 kg (molar yield=98.5 kg). MS Calculated: 230.13; MS Found (ES−, M−H): 229.11.Preparation of Compound VID
To a fresh reactor was charged Compound VIJ (99.8 kg active in a methanol solution), water (270 kg), NaOH (70 kg), and methanol (603 kg). The mixture was heated to −70° C. and agitated at this temperature for about 16 h. Upon conversion to the sodium salt of Compound VIK, the reaction mixture was concentrated under vacuum, keeping the temperature below 55° C., and then cooled to about 25° C. Water and MTBE were then charged, agitiated, and the layers were separated (product in the aqueous layer). The product-containing aqueous layer was further washed with MTBE.CH2Cl2 was charged to the aqueous layer and the temperature was adjusted to ˜10° C. The resultant mixture was acidified to a pH of about 1.5 with HCl, agitated, settled, and separated (the compound was in the organic layer). The aqueous layer was extracted with CH2Cl2, and the combined organic layers were stored as a CH2Cl2 solution for further processing to Compound VID. Active yield of Compound VIK=92.7 kg (molar yield=98.5 kg). MS Calculated: 230.13; MS Found (ES−, M−H): 229.11.Preparation of Compound VID To a reactor was charged water (952 kg), Oxone® (92.7 kg), and Compound VIK (92.7 kg active as a solution in CH2Cl2). The reaction mixture was agitated for about 24 h at a temperature of about 15° C., during which time Compound VIK oxidized to sulfone Compound VID. The excess Oxone® was quenched with aqueous Na2S2O5, the reaction mixture was settled and the layers separated; the aqueous layer was back-extracted with CH2Cl2, and the combined product-containing organic layers were washed with water.The resultant solution was then concentrated under vacuum. To precipitate Compound VID, n-heptane was charged, and the resulting slurry was agitated for about 60 min at a temperature of about 30° C. The reaction mixture was filtered, and the wet cake was washed with n-heptane. The wet cake was redissolved in CH2Cl2, followed by the addition of n-heptane. The resultant solution was then concentrated under vacuum, keeping the temperature below 35° C., to allow for product precipitation. The resultant solution was cooled to about 0° C. and agitated at this temperature for about 1 h. The solution was filtered, the wet cake was washed with n-heptane, and dried under vacuum at about 45° C. to yield 68.7 kg Compound VID (molar yield=65.7%). MS Calculated: 262.37; MS Found (ES−, M−H): 261.09Preparation of Compound VI
To a reactor was charged water (952 kg), Oxone® (92.7 kg), and Compound VIK (92.7 kg active as a solution in CH2Cl2). The reaction mixture was agitated for about 24 h at a temperature of about 15° C., during which time Compound VIK oxidized to sulfone Compound VID. The excess Oxone® was quenched with aqueous Na2S2O5, the reaction mixture was settled and the layers separated; the aqueous layer was back-extracted with CH2Cl2, and the combined product-containing organic layers were washed with water.The resultant solution was then concentrated under vacuum. To precipitate Compound VID, n-heptane was charged, and the resulting slurry was agitated for about 60 min at a temperature of about 30° C. The reaction mixture was filtered, and the wet cake was washed with n-heptane. The wet cake was redissolved in CH2Cl2, followed by the addition of n-heptane. The resultant solution was then concentrated under vacuum, keeping the temperature below 35° C., to allow for product precipitation. The resultant solution was cooled to about 0° C. and agitated at this temperature for about 1 h. The solution was filtered, the wet cake was washed with n-heptane, and dried under vacuum at about 45° C. to yield 68.7 kg Compound VID (molar yield=65.7%). MS Calculated: 262.37; MS Found (ES−, M−H): 261.09Preparation of Compound VI To a reactor was charged Compound VID (68.4 kg), toluene (531 kg), and Et3N (31 kg). The reaction mixture was atmospherically refluxed under Dean-Stark conditions to remove water (target KF <0.05%). The reaction temperature was adjusted to 80° C., DPPA (73.4 kg) was charged over 7 h, and the mixture was agitated for an additional 2 h. After conversion to isocyanate Compound VIE via the azide, the reaction mixture was cooled to about 0 to 5° C. and quenched with aqueous NaHCO3. The resultant mixture was agitated, settled and the layers were separated. The aqueous layer was extracted with toluene, and the combined isocyante Compound VIE organic layers were washed with water.In a separate vessel was charged L-tert- Leucine (L-Tle, 30.8 kg), water (270 kg), and Et3N (60 kg). While keeping the temperature at about 5° C., the toluene solution of Compound VIE was transferred to the solution of L-Tle. The reaction mixture was stirred at 0 to 5° C. for about 5 h, at which time the mixture was heated to 15 to 20° C. and agitated at this temperature for 2 h to allow for conversion to urea Compound VI.The reaction was quenched by the addition of aqueous NaOH, keeping the temperature between 0 and 25° C. The reaction mixture was separated, and the organic layer was extracted with water. The combined Compound VI-containing aqueous layers were washed with toluene, and acidified to pH 2 by the addition of HCl, at which time the product precipitated from solution. The reaction mixture was filtered, washed with water and dried under vacuum at 65 to 70° C. to yield 79.7 kg crude Compound VI (molar yield 52.7%). MS Calculated: 390.54; MS Found (ES−, M−H): 389.20.Compound VI is further purified by slurrying in CH3CN at reflux (about 80° C.), followed by cooling to RT. Typical recovery is 94%, with an increase in purity from about 80% to 99%.Preparation of Compound Va
To a reactor was charged Compound VID (68.4 kg), toluene (531 kg), and Et3N (31 kg). The reaction mixture was atmospherically refluxed under Dean-Stark conditions to remove water (target KF <0.05%). The reaction temperature was adjusted to 80° C., DPPA (73.4 kg) was charged over 7 h, and the mixture was agitated for an additional 2 h. After conversion to isocyanate Compound VIE via the azide, the reaction mixture was cooled to about 0 to 5° C. and quenched with aqueous NaHCO3. The resultant mixture was agitated, settled and the layers were separated. The aqueous layer was extracted with toluene, and the combined isocyante Compound VIE organic layers were washed with water.In a separate vessel was charged L-tert- Leucine (L-Tle, 30.8 kg), water (270 kg), and Et3N (60 kg). While keeping the temperature at about 5° C., the toluene solution of Compound VIE was transferred to the solution of L-Tle. The reaction mixture was stirred at 0 to 5° C. for about 5 h, at which time the mixture was heated to 15 to 20° C. and agitated at this temperature for 2 h to allow for conversion to urea Compound VI.The reaction was quenched by the addition of aqueous NaOH, keeping the temperature between 0 and 25° C. The reaction mixture was separated, and the organic layer was extracted with water. The combined Compound VI-containing aqueous layers were washed with toluene, and acidified to pH 2 by the addition of HCl, at which time the product precipitated from solution. The reaction mixture was filtered, washed with water and dried under vacuum at 65 to 70° C. to yield 79.7 kg crude Compound VI (molar yield 52.7%). MS Calculated: 390.54; MS Found (ES−, M−H): 389.20.Compound VI is further purified by slurrying in CH3CN at reflux (about 80° C.), followed by cooling to RT. Typical recovery is 94%, with an increase in purity from about 80% to 99%.Preparation of Compound Va To a reactor was charged Compound VI (87.6 kg), Compound VII-1 (48.2 kg), HOBt (6 kg) and CH3CN (615 kg). The reaction mixture was cooled to about 5° C., and NMM (35 kg) and EDCi (53.4 kg) were charged. The reaction was heated to 20 to 25° C. for about 1 h, and then to 35 to 40° C., at which time water was charged to crystallize Compound Va. The reaction mixture was cooled to 5° C. and held at this temperature for about 4 h. Compound Va was filtered and washed with water. XRD data for the hydrated polymorph of Va is as follows:The Compound Va wet cake was charged to a fresh vessel and was dissolved in ethyl acetate at 25 to 30° C. The solution was washed with an aqueous HCl solution, aqueous K2CO3 solution, and brine. The solution was then concentrated under vacuum, keeping the temperature between 35 to 50° C. Additional ethyl acetate was charged, and the solution was heated to 65 to 70° C. While keeping the temperature at 65 to 70° C., n-heptane was charged, followed by cooling the resultant solution to 0 to 5° C. Compound Va was filtered and washed with an ethyl acetate/n-heptane mix.The wet cake was dried under vacuum between 55 to 60° C. to yield 96.6 kg crystalline Compound Va (molar yield 79.2%). MS Calculated: 541.32; MS Found (ES+, M+H): 542.35.Preparation of Compound IUB
To a reactor was charged Compound VI (87.6 kg), Compound VII-1 (48.2 kg), HOBt (6 kg) and CH3CN (615 kg). The reaction mixture was cooled to about 5° C., and NMM (35 kg) and EDCi (53.4 kg) were charged. The reaction was heated to 20 to 25° C. for about 1 h, and then to 35 to 40° C., at which time water was charged to crystallize Compound Va. The reaction mixture was cooled to 5° C. and held at this temperature for about 4 h. Compound Va was filtered and washed with water. XRD data for the hydrated polymorph of Va is as follows:The Compound Va wet cake was charged to a fresh vessel and was dissolved in ethyl acetate at 25 to 30° C. The solution was washed with an aqueous HCl solution, aqueous K2CO3 solution, and brine. The solution was then concentrated under vacuum, keeping the temperature between 35 to 50° C. Additional ethyl acetate was charged, and the solution was heated to 65 to 70° C. While keeping the temperature at 65 to 70° C., n-heptane was charged, followed by cooling the resultant solution to 0 to 5° C. Compound Va was filtered and washed with an ethyl acetate/n-heptane mix.The wet cake was dried under vacuum between 55 to 60° C. to yield 96.6 kg crystalline Compound Va (molar yield 79.2%). MS Calculated: 541.32; MS Found (ES+, M+H): 542.35.Preparation of Compound IUB Pyridine (92 L) was charged to the reactor and was cooled to 5° C. To the cooled pyridine was slowly charged malonic acid (48.5 kg) and valeraldehyde (59 L), keeping the temperature below 25° C. The reaction was stirred between 25 to 35° C. for at least 60 h. After this time, H2SO4 was charged to acidify, keeping the temperature below 30° C. The reaction mixture was then extracted into MTBE. The organic layer was washed with water. In a separate reactor was charged water and NaOH. The MTBE solution was charged to the NaOH solution, keeping the temperature below 25° C., and the desired material was extracted into the basic layer. The basic layer was separated and the organic layer was discarded. MTBE was charged, the mixture was agitated, settled, and separated, and the organic layer was discarded. To the resultant solution (aqueous layer) was charged water and H2SO4 to acidify, keeping the temperature between 10 to 15° C. To the acidified mixture was charged MTBE, keeping the temperature below 25° C. The resultant solution was agitated, settled, and separated, and the aqueous layer was discarded. The product-containing organic layer was washed with water and was concentrated under vacuum, keeping the temperature below 70° C., to yield 45.4 kg Compound IIIB (molar yield=76.2%) as an oil. Compound Reference: Concellon, J. M.; Concellon, C J. Org. Chem., 2006, 71, 1728-1731Preparation of Compound IIIC
Pyridine (92 L) was charged to the reactor and was cooled to 5° C. To the cooled pyridine was slowly charged malonic acid (48.5 kg) and valeraldehyde (59 L), keeping the temperature below 25° C. The reaction was stirred between 25 to 35° C. for at least 60 h. After this time, H2SO4 was charged to acidify, keeping the temperature below 30° C. The reaction mixture was then extracted into MTBE. The organic layer was washed with water. In a separate reactor was charged water and NaOH. The MTBE solution was charged to the NaOH solution, keeping the temperature below 25° C., and the desired material was extracted into the basic layer. The basic layer was separated and the organic layer was discarded. MTBE was charged, the mixture was agitated, settled, and separated, and the organic layer was discarded. To the resultant solution (aqueous layer) was charged water and H2SO4 to acidify, keeping the temperature between 10 to 15° C. To the acidified mixture was charged MTBE, keeping the temperature below 25° C. The resultant solution was agitated, settled, and separated, and the aqueous layer was discarded. The product-containing organic layer was washed with water and was concentrated under vacuum, keeping the temperature below 70° C., to yield 45.4 kg Compound IIIB (molar yield=76.2%) as an oil. Compound Reference: Concellon, J. M.; Concellon, C J. Org. Chem., 2006, 71, 1728-1731Preparation of Compound IIIC To a pressure vessel was charged Compound IIIB (9.1 kg), heptane (9 L), and H2SO4 (0.5 kg). The pressure vessel was sealed and isobutylene (13.7 kg) was charged, keeping the temperature between 19 to 25° C. The reaction mixture was agitated at this temperature for about 18 h. The pressure was released, and a solution of K2CO3 was charged to the reaction mixture, which was agitated and settled, and the bottom aqueous layer was then separated. The resultant organic solution was washed with water and distilled under vacuum (temp below 45° C.) to yield 13.5 kg Compound IIIC (molar yield=88.3%) as a yellow oil.Preparation of Compound IIID
To a pressure vessel was charged Compound IIIB (9.1 kg), heptane (9 L), and H2SO4 (0.5 kg). The pressure vessel was sealed and isobutylene (13.7 kg) was charged, keeping the temperature between 19 to 25° C. The reaction mixture was agitated at this temperature for about 18 h. The pressure was released, and a solution of K2CO3 was charged to the reaction mixture, which was agitated and settled, and the bottom aqueous layer was then separated. The resultant organic solution was washed with water and distilled under vacuum (temp below 45° C.) to yield 13.5 kg Compound IIIC (molar yield=88.3%) as a yellow oil.Preparation of Compound IIID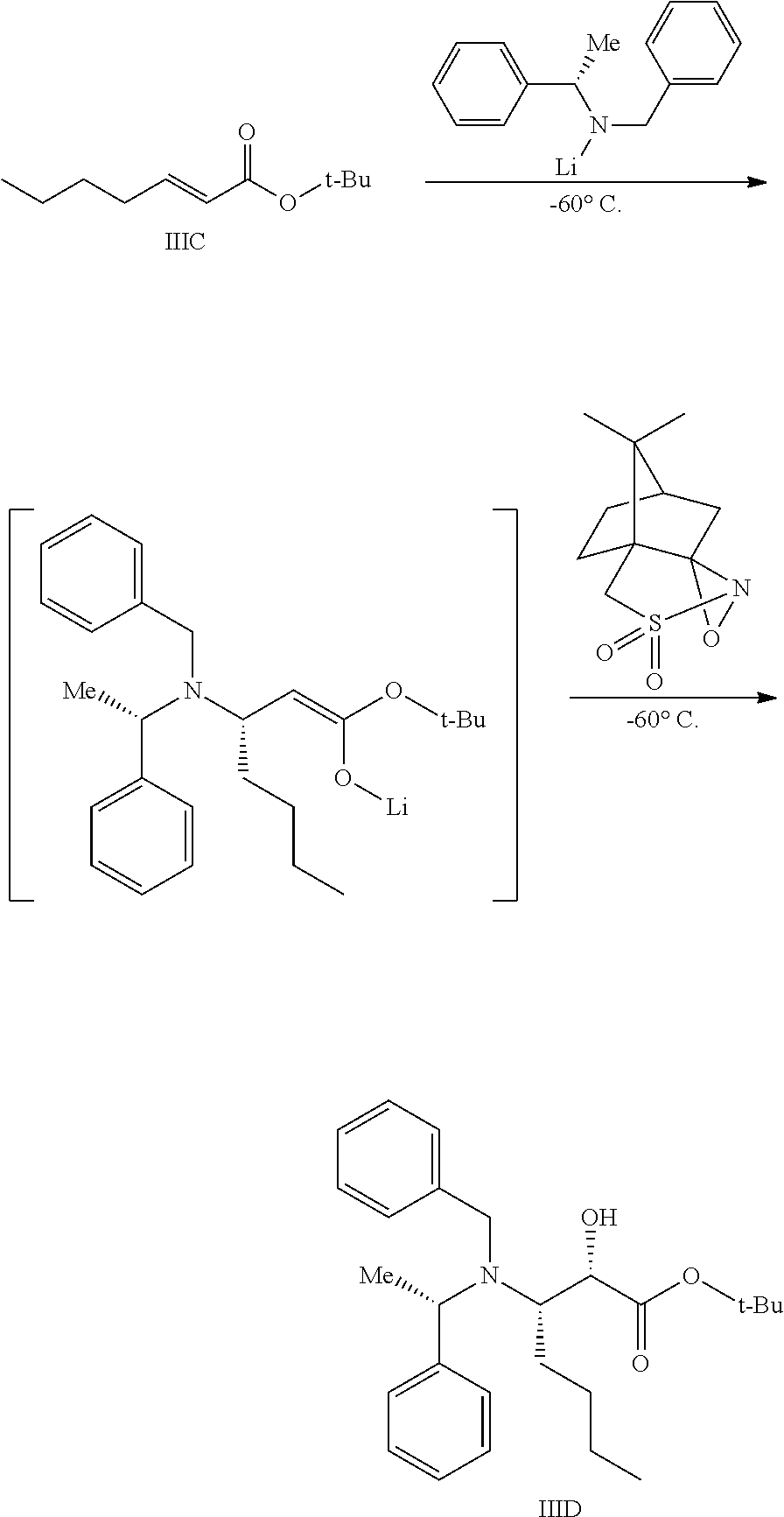 To a reactor capable of maintaining a temperature of −60° C. was charged (S)-benzyl-1-phenyl ethylamine (18 kg) and THF (75 L). The reaction mixture was cooled to −60° C. To the mixture was charged n-hexyl lithium (42 L of 2.3 M in heptane) while maintaining a temperature of −65 to −55° C., followed by a 30 min agitation within this temperature range. To the in situ-formed lithium amide was charged Compound IIIC over 1 h, keeping the temperature between −65 to −55° C. . The reaction mixture was agitated at this temperature for 30 min to allow for conversion to the enolate intermediate. To the resultant reaction mixture was charged (+)-camphorsulfonyl oxaziridine (24 kg) as a solid, over a period of 2 h, keeping the temperature between −65 to −55° C. . The mixture was agitated at this temperature for 4 h.The resultant reaction mixture was quenched by the addition of acetic acid (8 kg), keeping the temperature between −60 to −40° C. The mixture was warmed to 20 to 25° C., then charged into a separate reactor containing heptane. The resultant mixture was concentrated under vacuum, keeping the temperature below 35° C. Heptane and water were charged to the reaction mixture, and the precipitated solids were removed by filtration (the desired compound is in the supernatant). The cake was washed with heptane and this wash was combined with the supernatant. The heptane/water solution was agitated, settled, and separated to remove the aqueous layer. An aqueous solution of H2SO4 was charged, and the mixture was agitated, settled, and separated. The heptane layer was washed with a solution of K2CO3.The heptane layer was concentrated under reduced pressure, keeping the temperature below 45° C., and the resulting oil was diluted in toluene, yielding 27.1 kg (active) of Compound IIID (molar yield=81.0%). MS Calculated: 411.28; MS Found (ES+, M+H): 412.22.A similar procedure for this step was reported in: Beevers, R, et al, Bioorg. Med. Chem. Lett. 2002, 12, 641-643.Preparation of Compound IDE
To a reactor capable of maintaining a temperature of −60° C. was charged (S)-benzyl-1-phenyl ethylamine (18 kg) and THF (75 L). The reaction mixture was cooled to −60° C. To the mixture was charged n-hexyl lithium (42 L of 2.3 M in heptane) while maintaining a temperature of −65 to −55° C., followed by a 30 min agitation within this temperature range. To the in situ-formed lithium amide was charged Compound IIIC over 1 h, keeping the temperature between −65 to −55° C. . The reaction mixture was agitated at this temperature for 30 min to allow for conversion to the enolate intermediate. To the resultant reaction mixture was charged (+)-camphorsulfonyl oxaziridine (24 kg) as a solid, over a period of 2 h, keeping the temperature between −65 to −55° C. . The mixture was agitated at this temperature for 4 h.The resultant reaction mixture was quenched by the addition of acetic acid (8 kg), keeping the temperature between −60 to −40° C. The mixture was warmed to 20 to 25° C., then charged into a separate reactor containing heptane. The resultant mixture was concentrated under vacuum, keeping the temperature below 35° C. Heptane and water were charged to the reaction mixture, and the precipitated solids were removed by filtration (the desired compound is in the supernatant). The cake was washed with heptane and this wash was combined with the supernatant. The heptane/water solution was agitated, settled, and separated to remove the aqueous layer. An aqueous solution of H2SO4 was charged, and the mixture was agitated, settled, and separated. The heptane layer was washed with a solution of K2CO3.The heptane layer was concentrated under reduced pressure, keeping the temperature below 45° C., and the resulting oil was diluted in toluene, yielding 27.1 kg (active) of Compound IIID (molar yield=81.0%). MS Calculated: 411.28; MS Found (ES+, M+H): 412.22.A similar procedure for this step was reported in: Beevers, R, et al, Bioorg. Med. Chem. Lett. 2002, 12, 641-643.Preparation of Compound IDE Toluene (324 L) and a toluene solution of Compound IIID (54.2 kg active) was charged to the reactor. TFA (86.8 kg) was charged over about 1.5 h, keeping the temperature below 50° C. The reaction mixture was agitated for 24 h at 50° C. The reaction mixture was cooled to 15° C. and water was charged. NaOH was slowly charged, keeping the temperature below 20° C., to adjust the batch to a pH between 5.0 and 6.0. The reaction mixture was agitated, settled, and separated; the aqueous layer was discarded. The organic layer was concentrated under vacuum, keeping the temperature below 40° C., and the resulting acid intermediate (an oil), was dissolved in 2-MeTHF.In a separate reactor, 2-MeTHF (250 L), HOBt (35.2 kg), and EDCi-HCl (38.0 kg) were charged and the mixture was adjusted to a temperature between 0 to 10° C. DIPEA (27.2 kg) was charged, keeping the mixture within this temperature range. The mixture was agitated for 5 min, followed by the addition of cyclopropyl amine (11.4 kg), keeping the temperature between 0 to 10° C.To this solution was charged the 2-MeTHF/ acid intermediate solution, keeping the resultant solution between 0 to 10° C. The resultant mixture was heated to 25 to 35° C., and was agitated at this temperature for about 4 h. The reaction mixture was cooled to about 20° C., and was washed with aqueous citric acid, aqueous K2CO3, and water. The solvent was exchanged to n-heptane, and the desired compound was crystallized from a mix of n-heptane and toluene by cooling to 0° C. The crystalline product was filtered, washed with n-heptane, and dried to yield 37.1 kg Compound IIIE (molar yield=70.7%). MS Calculated: 394.26; MS Found (ES+, M+H): 395.22.Preparation of Compound III
Toluene (324 L) and a toluene solution of Compound IIID (54.2 kg active) was charged to the reactor. TFA (86.8 kg) was charged over about 1.5 h, keeping the temperature below 50° C. The reaction mixture was agitated for 24 h at 50° C. The reaction mixture was cooled to 15° C. and water was charged. NaOH was slowly charged, keeping the temperature below 20° C., to adjust the batch to a pH between 5.0 and 6.0. The reaction mixture was agitated, settled, and separated; the aqueous layer was discarded. The organic layer was concentrated under vacuum, keeping the temperature below 40° C., and the resulting acid intermediate (an oil), was dissolved in 2-MeTHF.In a separate reactor, 2-MeTHF (250 L), HOBt (35.2 kg), and EDCi-HCl (38.0 kg) were charged and the mixture was adjusted to a temperature between 0 to 10° C. DIPEA (27.2 kg) was charged, keeping the mixture within this temperature range. The mixture was agitated for 5 min, followed by the addition of cyclopropyl amine (11.4 kg), keeping the temperature between 0 to 10° C.To this solution was charged the 2-MeTHF/ acid intermediate solution, keeping the resultant solution between 0 to 10° C. The resultant mixture was heated to 25 to 35° C., and was agitated at this temperature for about 4 h. The reaction mixture was cooled to about 20° C., and was washed with aqueous citric acid, aqueous K2CO3, and water. The solvent was exchanged to n-heptane, and the desired compound was crystallized from a mix of n-heptane and toluene by cooling to 0° C. The crystalline product was filtered, washed with n-heptane, and dried to yield 37.1 kg Compound IIIE (molar yield=70.7%). MS Calculated: 394.26; MS Found (ES+, M+H): 395.22.Preparation of Compound III To a pressure reactor was charged acetic acid (1.1 kg), methanol (55 kg), and Compound IIIE (10.9 kg). In a separate vessel, Pd/C (50% water wet, 0.5 kg) was suspended in methanol (5 kg). The Pd/C suspension was transferred to the solution containing Compound IIIE. The resultant mixture was pressurized to 80 psi with hydrogen, and agitated at 60° C. for 7 h. The reaction mixture was then purged with nitrogen, and the Pd/C catalyst was filtered off. The resultant solution was concentrated under vacuum and adjusted to about 20° C. MTBE was charged, and the resultant solution was brought to reflux. Concentrated HCl (3 L) was charged and the product was crystallized by cooling the reaction mixture to about 3° C. The desired compound was filtered, washed with MTBE, and dried under vacuum, keeping the temperature below 40° C. to yield 5.5 kg Compound III (molar yield=83.0%). MS Calculated (free base): 200.15; MS Found (ES+, M+H): 201.12.Preparation of Compound II
To a pressure reactor was charged acetic acid (1.1 kg), methanol (55 kg), and Compound IIIE (10.9 kg). In a separate vessel, Pd/C (50% water wet, 0.5 kg) was suspended in methanol (5 kg). The Pd/C suspension was transferred to the solution containing Compound IIIE. The resultant mixture was pressurized to 80 psi with hydrogen, and agitated at 60° C. for 7 h. The reaction mixture was then purged with nitrogen, and the Pd/C catalyst was filtered off. The resultant solution was concentrated under vacuum and adjusted to about 20° C. MTBE was charged, and the resultant solution was brought to reflux. Concentrated HCl (3 L) was charged and the product was crystallized by cooling the reaction mixture to about 3° C. The desired compound was filtered, washed with MTBE, and dried under vacuum, keeping the temperature below 40° C. to yield 5.5 kg Compound III (molar yield=83.0%). MS Calculated (free base): 200.15; MS Found (ES+, M+H): 201.12.Preparation of Compound II Compound Va (119.3 kg) was dissolved in 2-MeTHF (720 kg) and water (180 kg). To this solution was charged 50% NaOH (21.4 kg) while maintaining a temperature between 20 and 30° C. The reaction mixture was then agitated for about 7 h at a temperature between 50 and 60° C. The reaction mixture was cooled to a temperature between 20 and 30° C.The pH of the reaction mixture was adjusted to 1.5-3.0 with dilute phosphoric acid, maintaining a temperature between 20 and 30° C. The resultant mixture was agitated for 10 min, settled for 30 min, and the bottom aqueous layer was separated and removed. The top organic layer was washed with water, followed by concentration by atmospheric distillation.The concentrated solution was solvent exchanged to CH3CN by continuous atmospheric distillation, and crystallized by cooling to 0° C. The crystalline product was filtered, washed with CH3CN, and dried under vacuum at a temperature between 45 and 55° C. to yield 97.9 kg Compound II (molar yield=83.7%). MS Calculated: 527.30; MS Found (ES+, M+H): 528.29.Preparation of Compound IV
Compound Va (119.3 kg) was dissolved in 2-MeTHF (720 kg) and water (180 kg). To this solution was charged 50% NaOH (21.4 kg) while maintaining a temperature between 20 and 30° C. The reaction mixture was then agitated for about 7 h at a temperature between 50 and 60° C. The reaction mixture was cooled to a temperature between 20 and 30° C.The pH of the reaction mixture was adjusted to 1.5-3.0 with dilute phosphoric acid, maintaining a temperature between 20 and 30° C. The resultant mixture was agitated for 10 min, settled for 30 min, and the bottom aqueous layer was separated and removed. The top organic layer was washed with water, followed by concentration by atmospheric distillation.The concentrated solution was solvent exchanged to CH3CN by continuous atmospheric distillation, and crystallized by cooling to 0° C. The crystalline product was filtered, washed with CH3CN, and dried under vacuum at a temperature between 45 and 55° C. to yield 97.9 kg Compound II (molar yield=83.7%). MS Calculated: 527.30; MS Found (ES+, M+H): 528.29.Preparation of Compound IV Compound II (21.1 kg), Compound III (9.9 kg), HOBt (3.2 kg) and EDCi (11.2 kg) were charged to the vessel, followed by CH3CN (63 kg), ethyl acetate (20 kg) and water (1.5 kg). The reaction mixture was agitated and the heterogeneous mixture was cooled to −5 to +5° C. DIPEA (11.2 kg) was charged to the reaction mixture, maintaining a temperature between −5 to +5° C. and the mixture was agitated at a temperature of −5 to +5° C. for 1 h. The resultant reaction mixture was warmed to 20 to 30° C. and agitated for 2 to 3 h.The resultant product was extracted with aqueous HCl, aqueous K2CO3, and water.The desired product was crystallized from ethyl acetate by cooling from reflux (78° C.) to about 0° C. The crystalline product was filtered and dried at 30° C. under vacuum to yield 23.1 kg Compound IV (molar yield=81.3%). MS Calculated: 709.44; MS Found (ES+, M+H): 710.47.Preparation of Compound I
Compound II (21.1 kg), Compound III (9.9 kg), HOBt (3.2 kg) and EDCi (11.2 kg) were charged to the vessel, followed by CH3CN (63 kg), ethyl acetate (20 kg) and water (1.5 kg). The reaction mixture was agitated and the heterogeneous mixture was cooled to −5 to +5° C. DIPEA (11.2 kg) was charged to the reaction mixture, maintaining a temperature between −5 to +5° C. and the mixture was agitated at a temperature of −5 to +5° C. for 1 h. The resultant reaction mixture was warmed to 20 to 30° C. and agitated for 2 to 3 h.The resultant product was extracted with aqueous HCl, aqueous K2CO3, and water.The desired product was crystallized from ethyl acetate by cooling from reflux (78° C.) to about 0° C. The crystalline product was filtered and dried at 30° C. under vacuum to yield 23.1 kg Compound IV (molar yield=81.3%). MS Calculated: 709.44; MS Found (ES+, M+H): 710.47.Preparation of Compound I Compound IV (22.5 kg), TEMPO (5 kg), NaOAc (45 kg), methyl acetate (68 L), MTBE (158 L), water (23 L) and acetic acid (22.5 L) were charged to the reactor. The reaction mixture was stirred at 20-30° C. to allow for dissolution of the solids, and was then cooled to 5-15° C. NaOCl solution (1.4 molar equivalents) was charged to the reaction mixture, keeping the temperature at about 10° C. After complete addition of NaOCl, the reaction mixture was agitated at 10° C. for 2 h.The reaction was quenched by washing with a buffered sodium ascorbate/HCl aqueous solution, followed by a water wash.The reaction mixture was solvent exchanged to acetone under vacuum, keeping the temperature below 20° C.; the desired product was crystallized by the addition of water, and dried under vacuum, keeping the temperature below 40° C. to yield 18.6 kg Compound I (molar yield=82.7%). MS Calculated: 707.43: MS Found (ES+, M+H): 708.44.
Compound IV (22.5 kg), TEMPO (5 kg), NaOAc (45 kg), methyl acetate (68 L), MTBE (158 L), water (23 L) and acetic acid (22.5 L) were charged to the reactor. The reaction mixture was stirred at 20-30° C. to allow for dissolution of the solids, and was then cooled to 5-15° C. NaOCl solution (1.4 molar equivalents) was charged to the reaction mixture, keeping the temperature at about 10° C. After complete addition of NaOCl, the reaction mixture was agitated at 10° C. for 2 h.The reaction was quenched by washing with a buffered sodium ascorbate/HCl aqueous solution, followed by a water wash.The reaction mixture was solvent exchanged to acetone under vacuum, keeping the temperature below 20° C.; the desired product was crystallized by the addition of water, and dried under vacuum, keeping the temperature below 40° C. to yield 18.6 kg Compound I (molar yield=82.7%). MS Calculated: 707.43: MS Found (ES+, M+H): 708.44.
- .....................................................................
- 8 DELDEPREVIR OR NECEPREVIR
 ACH-0142684, ACH-2684HCV NS3 PRUSAN (YY-152) DELDEPREVIRTHERAPEUTIC CLAIM Treatment of Hepatitis C
ACH-0142684, ACH-2684HCV NS3 PRUSAN (YY-152) DELDEPREVIRTHERAPEUTIC CLAIM Treatment of Hepatitis C
CHEMICAL NAMES
1. Cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-carboxamide, N-
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, (2R,6R,12Z,13aS,14aR,16aS)-
2. (2R,6R,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6-[2-(3,3-difluoropiperidin-1-yl)-
2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-yl}oxy)-
5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16atetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-
carboxamideMOLECULAR FORMULA C45H56F2N6O8S2
MOLECULAR WEIGHT 911.1
SPONSOR Achillion Pharmaceuticals, Inc.
CODE DESIGNATION ACH-0142684, ACH-2684
CAS REGISTRY NUMBER 1229626-28-1
WHO NUMBER 9600
NOTE: This adoption statement replaces adoption N12/17 and the name neceprevir is hereby rescinded.......................................................................................................................DELDEPREVIR SODIUMUSAN (yy-153) DELDEPREVIR SODIUMTHERAPEUTIC CLAIM Treatment of Hepatitis CCHEMICAL NAMES1. Cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-carboxamide, N-
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, sodium salt (1:1),
(2R,6R,12Z,13aS,14aR,16aS)-2. Sodium (cyclopropylsulfonyl){[(2R,6R,12Z,13aS,14aR,16aS)-6-[2-(3,3-difluoropiperidin-
1-yl)-2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-
yl}oxy)-5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-
tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-14a(5H)-
yl]formyl]azanideMOLECULAR FORMULA C45H55F2N6NaO8S2MOLECULAR WEIGHT 933.1SPONSOR Achillion Pharmaceuticals, Inc.CODE DESIGNATION ACH-0142684.Na, ACH-2684.NaCAS REGISTRY NUMBER 1298053-61-8NOTE: This adoption statement replaces adoption N12/18 and the name neceprevir sodiumis hereby rescinded.ACH-2684 is a HCV NS3 protease inhibitor in phase I clinical development at Achillion for the oral treatment of chronic hepatitis C genotype 1 and 3.WO 2010068761 US 2010152103 COMPD 133(2R,6R,14aR,16aS,Z)- N-(cyclopropylsulfonyl)- 6-(2-(3,3-difluoropiperidin- 1-yl)-2-oxoethyl)-2- (2-(2-isopropylthiazol- 4-yl)-7-methoxy-8- methylquinolin-4- yloxy)-5,16-dioxo- 1,2,3,5,6,7,8,9,10,11, 13a,14,14a,15,16,16a- hexadecahydrocyclopropa [e]pyrrolo[1,2- a][1,4] diazacyclopentadecine- 14a-carboxamide- ........................................................................................................
- 9 FALDAPREVIR
 FALDAPREVIR801283-95-4(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acidBoehringer Ingelheim (Originator)BI-201335 is an HCV NS3 protease inhibitor awaiting approval in the E.U. by Boehringer Ingelheim for the treatment of chronic hepatitis C, in combination with pegylated Interferon and ribavirin.Faldaprevir (formerly BI 201335) is an experimental drug for the treatment of hepatitis C. It is being developed by Boehringer-Ingelheim and is currently in Phase III trials.[1]Faldaprevir is a hepatitis C virus protease inhibitor.Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.
FALDAPREVIR801283-95-4(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acidBoehringer Ingelheim (Originator)BI-201335 is an HCV NS3 protease inhibitor awaiting approval in the E.U. by Boehringer Ingelheim for the treatment of chronic hepatitis C, in combination with pegylated Interferon and ribavirin.Faldaprevir (formerly BI 201335) is an experimental drug for the treatment of hepatitis C. It is being developed by Boehringer-Ingelheim and is currently in Phase III trials.[1]Faldaprevir is a hepatitis C virus protease inhibitor.Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.- Efficacy and Safety of BI 201335 (Faldaprevir) in Combination With Pegylated Interferon-alpha and Ribavirin in Treatment-naïve Genotype 1 Hepatitis C Infected Patients (STARTverso 1). Cliicaltrials.gov. March 6, 2013.
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
Phase II clinical trials are also ongoing for the treatment of patients with chronic genotype-1a hepatitis C virus (HCV) infection, in combination with PPI-668 and BI-207127.In 2007, fast track designation was assigned to the compound in the U.S. for the treatment of chronic genotype-1 hepatitis C (HCV).Protease inhibitors that are active against NS3/4a are a fertile area of research, not least because of the early promise shown by the two already-approved agents FaldaprevirProtease inhibitors that are active against NS3/4a are a fertile area of research. Boehringer Ingelheim’s compound faldaprevir is currently in Phase III trials.1 In one 24-week trial in 429 treatment-naïve patients with genotype-1 hepatitis C infection, subjects were given standard peg-interferon and ritonavir therapy plus placebo, or standard therapy plus either 120mg or 240mg of faldaprevir either with or without a three day lead-in of standard therapy alone, or standard therapy plus the higher dose of faldaprevir.ADDN LITDiscovery of a potent and selective noncovalent linear inhibitor of the hepatitis C virus NS3 protease (BI 201335)
FaldaprevirProtease inhibitors that are active against NS3/4a are a fertile area of research. Boehringer Ingelheim’s compound faldaprevir is currently in Phase III trials.1 In one 24-week trial in 429 treatment-naïve patients with genotype-1 hepatitis C infection, subjects were given standard peg-interferon and ritonavir therapy plus placebo, or standard therapy plus either 120mg or 240mg of faldaprevir either with or without a three day lead-in of standard therapy alone, or standard therapy plus the higher dose of faldaprevir.ADDN LITDiscovery of a potent and selective noncovalent linear inhibitor of the hepatitis C virus NS3 protease (BI 201335)
J Med Chem 2010, 53(17): 6466WO 2010033444WO 2004103996US6323180 Aug 5, 1999 Nov 27, 2001 Boehringer Ingelheim (Canada) Ltd Hepatitis C inhibitor tri-peptides US7514557 * May 23, 2005 Apr 7, 2009 Boehringer Ingelheim International Gmbh Process for preparing acyclic HCV protease inhibitors US7585845 * May 20, 2004 Sep 8, 2009 Boehringer Ingelheim International Gmbh Hepatitis C inhibitor compounds US20050020503 * May 20, 2004 Jan 27, 2005 Boehringer Ingelheim International Gmbh Hepatitis C inhibitor compounds US20120059033 Mar 9, 2011 Mar 8, 2012 Boehringer Ingelheim International Gmbh Crystalline Salts of a Potent HCV Inhibitor USRE40525 Sep 30, 2005 Sep 30, 2008 Boehringer Ingelheim (Canada) Ltd. Hepatitis C inhibitor tri-peptides WO2000009543A2 Aug 9, 1999 Feb 24, 2000 Boehringer Ingelheim Ca Ltd Hepatitis c inhibitor tri-peptides WO2004087741A1 Mar 25, 2004 Oct 14, 2004 Boehringer Ingelheim Int Crystalline phases of a potent hcv inhibitor WO2004103996A1 May 19, 2004 Dec 2, 2004 Boehringer Ingelheim Int Hepatitis c inhibitor compounds WO2011112761A1 Mar 10, 2011 Sep 15, 2011 Boehringer Ingelheim International Gmbh Crystalline salts of a potent hcv inhibitor ................................................. EXAMPLES Example 1 Preparation of Quinoline Starting Material Compound 11
EXAMPLES Example 1 Preparation of Quinoline Starting Material Compound 11 Step 1The dianion of amide 1 (prepared exactly as described above, from 1.00 g amide 1) was cooled to −78° C., then 2.19 mL perfluorooctyl bromide (8.46 mmol, 1.75 eq.) was added dropwise via syringe over 5 minutes. The dark-colored reaction mixture was then placed in a −10° C. bath. After two hours, 10 mL 1N HCl was cautiously added, and the mixture extracted with EtOAc (2×25 mL), dried (MgSO4), and the solvents removed in vacuo. The residue was then chromatographed on silica gel eluting with 4:1 Hexane:EtOAc to give 1.13 g bromoamide 5 (81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.12 (br s, 1H), 8.04 (dd, J=1.3, 8.4 Hz, 1H), 7.24 (t, J=8.3 Hz, 1H), 6.63 (dd, J=1.3, 8.3 Hz, 1H), 3.87 (s, 3H), 1.33 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 176.57 (s), 155.74 (s), 136.98 (s), 128.34 (d), 113.63 (d), 106.86 (d), 103.07 (s), 56.26 (q), 40.20 (s), 27.45 (q).Step 20.25 g bromoamide 5 (0.87 mmol, 1 eq.), 2.0 mL con. HCl (24 mmol, 28 eq.), and 1.0 mL diglyme were heated at 100° C. for 24 hours. The mixture was then cooled and filtered (product). The filtrate was evaporated in vacuo using H2O to azeotropically remove all solvents. The residue was triturated with EtOAc to cause precipitation of additional product, which was also filtered. The combined solids were dried to give 0.16 g (77%) of bromoaniline 6.HCl as a light brown solid. 1H NMR (400 MHz, CDCl3) δ: 7.09 (t, J=8.1 Hz, 1H), 6.61 (d, J=8.0 Hz, 1H), 6.47 (d, J=8.1 Hz, 1H), 3.84 (br s, 2H), 3.77 (s, 3H).Step 3Bromoanisidine.HCl (5.73 g, 24.0 mmol), Aluminumtrichloride (3.52 g) and chlorobenzene (15.0 mL) are charged into an oven dried 100 mL three necked flask at rt (temperature rise to 30° C.). The resulting mixture is then stirred for 10 min then cooled to 0-5° C. followed by slow addition of acetonitrile (1.89 mL, 36.0 mmol) followed by addition of BCl3 (2.82 g), transferred as gas (or liquid) into the reaction mixture, keeping the temperature below 5° C. The resulting mixture is then stirred at rt for 20 min then heated to 85-100° C. for 16 h. HPLC indicate completion of the reaction (SM<0.5% at 220 nm). The mixture is cooled down to 50° C. then Toluene (15 mL) was added followed by slow addition of IPA (11.1 mL) then slow addition of water (32 mL) at 50° C. The resulting mixture stirred for additional 2 h at this temperature then 3 g Celite was added and the stirred mixture cooled to rt. Filtration then wash of the organic fraction with water 1×15 mL, 2×15 m: 5% NaHCO3, 1×15 mL water then concentration under reduced pressure provided 3.92-4.4 g of the desired product in 68-72% isolated yield. 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=9.0 Hz, 1H), 7.1 (br s, 2H), 6.28 (d, J=9.1 Hz, 1H), 3.94 (s, 3H), 2.55 (s, 3H).Step 4Oxalyl chloride (8.15 mL) is added dropwise to the cold mixture (10±5° C.) of Thiazole acid 8 (20.18 g) is dissolved in THF (300 mL) and DMF (300 μL) over a period of ˜5 min keeping the internal temperature at 10±5° C. The reaction mixture becomes yellow and homogenous. The cooling bath is removed and the mixture is allowed to reach ambient temperature over a period of ˜30 min. Gas evolution is observed. The mixture is stirred at ambient temperature for 30 min to 1 hour. A solution of aniline 7 (19.8 g), DMAP (140 mg) and THF (35 mL) was added at 10±5° C. Et3N (13.2 mL) was added in portions at 10±5° C. over a period of 10 min. The ice bath was removed and mixture was heated to 65±2° C. and stirred overnight (18 h). The mixture was allowed to reach ambient temperature, diluted with EtOAc (150 mL) and washed with water (150 mL). NaHCO3(5%, 225 mL) was added to the organic portion and the mixture was stirred at ambient temperature for 30 min. The organic portion was concentrated under reduced pressure at approx. 40° C. EtOAc (150 mL) was added to the resulting material and the residual water was removed and the mixture was concentrated under reduced pressure at approx. 40° C. (to azeotrope water). EtOAc (94 mL) was added and the resulting slurry was stirred for 2-6 h and filtered. The solid was washed with EtOAc (30 mL) followed by heptane (30 mL) and air dried for 1 h to give the desired product in 70% yield.1H NMR (400 MHz, CDCl3) δ: 1.32 (d, 6H, J=7.8 Hz), 2.58 (s, 3H), 2.65-2.72 (m, 1H), 3.98 (s, 3H), 6.83 (d, 1H, J=8.7 Hz), 7.70 (d, 1H, J=8.7 Hz), 7.86 (s, 1H), 8.98 (bs, 1H), 10.13 (bs, 1H).Step 5In a 2 L flask was placed potassium t-butoxide (112 g). Dry DME was added at room temperature (exothermic: temperature went up to 35° C.). The resultant solution was heated to ca. 80° C., and amide (88 g) was added in 10 portions slowly so temperature was kept between 80-85° C. Upon completion, reaction mixture was stirred at 85° C. for 2 hours. Solid precipitated during the reaction. HPLC analysis indicated that the reaction was completed at this point (conversion: 100%). The reaction mixture was cooled to room temperature and then to 10° C. with a cool bath. Aqueous 2N HCl solution (ca. 500 ml) was added slowly so temperature was kept under 25° C. to quench the reaction mixture. pH was adjusted to 4-5. About 100 ml of water was added (Note: amount of water may need adjustment to facilitate filtration), and the resulting suspension was stirred at room temperature for 5-10 hours. Product was isolated by filtration, washing with THF and drying under vacuum. Yield: 81 g, 96% yield.1H-NMR (400 M Hz, DMSO-d6): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.48 (1H, hept., J=6.8 Hz, i-Pr), 3.99 (3H, s, MeO), 6.75 (1H, s, H-3), 7.24 (1H, d, J=8.5 Hz, H-6), 8.10 (1H, d, J=8.5 Hz, H5), 8.22 (1H, s, H-5′), 9.87 (1H, s, OH), 12.40 (1H, s, amide NH).Step 6In a 100 ml flask was placed starting material quinoline (4.22 g) and dioxane (40 ml). POCl3 (4.6 g) was added, and the mixture was heated to 75° C. After 2 hours, HPLC showed the reaction finished (99.7% conversion). Reaction mixture was cooled to room temperature, and then poured to 100 ml saturated NaHCO3 solution and 20 ml EtOAc. The resulting suspension was stirred for 3 hours. Product was isolated by filtration, washing with EtOAc and drying under vacuum. Yield: 4.0 g, 90.9%.1H-NMR (400 M Hz, CDCl3): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.76 (1H, hept., J=6.8 Hz, i-Pr), 4.05 (3H, s, MeO), 7.68 (1H, d, J=8.5 Hz, H-6), 8.07 (1H, s, H-3), 8.13 (1H, s, H-5′), 8.20 (1H, d, J=8.5 Hz, H5), 12.30 (1H, s, amide NH).Example 2 Preparation of Dipeptide Acid Compound 13 Starting Material
Step 1The dianion of amide 1 (prepared exactly as described above, from 1.00 g amide 1) was cooled to −78° C., then 2.19 mL perfluorooctyl bromide (8.46 mmol, 1.75 eq.) was added dropwise via syringe over 5 minutes. The dark-colored reaction mixture was then placed in a −10° C. bath. After two hours, 10 mL 1N HCl was cautiously added, and the mixture extracted with EtOAc (2×25 mL), dried (MgSO4), and the solvents removed in vacuo. The residue was then chromatographed on silica gel eluting with 4:1 Hexane:EtOAc to give 1.13 g bromoamide 5 (81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.12 (br s, 1H), 8.04 (dd, J=1.3, 8.4 Hz, 1H), 7.24 (t, J=8.3 Hz, 1H), 6.63 (dd, J=1.3, 8.3 Hz, 1H), 3.87 (s, 3H), 1.33 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 176.57 (s), 155.74 (s), 136.98 (s), 128.34 (d), 113.63 (d), 106.86 (d), 103.07 (s), 56.26 (q), 40.20 (s), 27.45 (q).Step 20.25 g bromoamide 5 (0.87 mmol, 1 eq.), 2.0 mL con. HCl (24 mmol, 28 eq.), and 1.0 mL diglyme were heated at 100° C. for 24 hours. The mixture was then cooled and filtered (product). The filtrate was evaporated in vacuo using H2O to azeotropically remove all solvents. The residue was triturated with EtOAc to cause precipitation of additional product, which was also filtered. The combined solids were dried to give 0.16 g (77%) of bromoaniline 6.HCl as a light brown solid. 1H NMR (400 MHz, CDCl3) δ: 7.09 (t, J=8.1 Hz, 1H), 6.61 (d, J=8.0 Hz, 1H), 6.47 (d, J=8.1 Hz, 1H), 3.84 (br s, 2H), 3.77 (s, 3H).Step 3Bromoanisidine.HCl (5.73 g, 24.0 mmol), Aluminumtrichloride (3.52 g) and chlorobenzene (15.0 mL) are charged into an oven dried 100 mL three necked flask at rt (temperature rise to 30° C.). The resulting mixture is then stirred for 10 min then cooled to 0-5° C. followed by slow addition of acetonitrile (1.89 mL, 36.0 mmol) followed by addition of BCl3 (2.82 g), transferred as gas (or liquid) into the reaction mixture, keeping the temperature below 5° C. The resulting mixture is then stirred at rt for 20 min then heated to 85-100° C. for 16 h. HPLC indicate completion of the reaction (SM<0.5% at 220 nm). The mixture is cooled down to 50° C. then Toluene (15 mL) was added followed by slow addition of IPA (11.1 mL) then slow addition of water (32 mL) at 50° C. The resulting mixture stirred for additional 2 h at this temperature then 3 g Celite was added and the stirred mixture cooled to rt. Filtration then wash of the organic fraction with water 1×15 mL, 2×15 m: 5% NaHCO3, 1×15 mL water then concentration under reduced pressure provided 3.92-4.4 g of the desired product in 68-72% isolated yield. 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=9.0 Hz, 1H), 7.1 (br s, 2H), 6.28 (d, J=9.1 Hz, 1H), 3.94 (s, 3H), 2.55 (s, 3H).Step 4Oxalyl chloride (8.15 mL) is added dropwise to the cold mixture (10±5° C.) of Thiazole acid 8 (20.18 g) is dissolved in THF (300 mL) and DMF (300 μL) over a period of ˜5 min keeping the internal temperature at 10±5° C. The reaction mixture becomes yellow and homogenous. The cooling bath is removed and the mixture is allowed to reach ambient temperature over a period of ˜30 min. Gas evolution is observed. The mixture is stirred at ambient temperature for 30 min to 1 hour. A solution of aniline 7 (19.8 g), DMAP (140 mg) and THF (35 mL) was added at 10±5° C. Et3N (13.2 mL) was added in portions at 10±5° C. over a period of 10 min. The ice bath was removed and mixture was heated to 65±2° C. and stirred overnight (18 h). The mixture was allowed to reach ambient temperature, diluted with EtOAc (150 mL) and washed with water (150 mL). NaHCO3(5%, 225 mL) was added to the organic portion and the mixture was stirred at ambient temperature for 30 min. The organic portion was concentrated under reduced pressure at approx. 40° C. EtOAc (150 mL) was added to the resulting material and the residual water was removed and the mixture was concentrated under reduced pressure at approx. 40° C. (to azeotrope water). EtOAc (94 mL) was added and the resulting slurry was stirred for 2-6 h and filtered. The solid was washed with EtOAc (30 mL) followed by heptane (30 mL) and air dried for 1 h to give the desired product in 70% yield.1H NMR (400 MHz, CDCl3) δ: 1.32 (d, 6H, J=7.8 Hz), 2.58 (s, 3H), 2.65-2.72 (m, 1H), 3.98 (s, 3H), 6.83 (d, 1H, J=8.7 Hz), 7.70 (d, 1H, J=8.7 Hz), 7.86 (s, 1H), 8.98 (bs, 1H), 10.13 (bs, 1H).Step 5In a 2 L flask was placed potassium t-butoxide (112 g). Dry DME was added at room temperature (exothermic: temperature went up to 35° C.). The resultant solution was heated to ca. 80° C., and amide (88 g) was added in 10 portions slowly so temperature was kept between 80-85° C. Upon completion, reaction mixture was stirred at 85° C. for 2 hours. Solid precipitated during the reaction. HPLC analysis indicated that the reaction was completed at this point (conversion: 100%). The reaction mixture was cooled to room temperature and then to 10° C. with a cool bath. Aqueous 2N HCl solution (ca. 500 ml) was added slowly so temperature was kept under 25° C. to quench the reaction mixture. pH was adjusted to 4-5. About 100 ml of water was added (Note: amount of water may need adjustment to facilitate filtration), and the resulting suspension was stirred at room temperature for 5-10 hours. Product was isolated by filtration, washing with THF and drying under vacuum. Yield: 81 g, 96% yield.1H-NMR (400 M Hz, DMSO-d6): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.48 (1H, hept., J=6.8 Hz, i-Pr), 3.99 (3H, s, MeO), 6.75 (1H, s, H-3), 7.24 (1H, d, J=8.5 Hz, H-6), 8.10 (1H, d, J=8.5 Hz, H5), 8.22 (1H, s, H-5′), 9.87 (1H, s, OH), 12.40 (1H, s, amide NH).Step 6In a 100 ml flask was placed starting material quinoline (4.22 g) and dioxane (40 ml). POCl3 (4.6 g) was added, and the mixture was heated to 75° C. After 2 hours, HPLC showed the reaction finished (99.7% conversion). Reaction mixture was cooled to room temperature, and then poured to 100 ml saturated NaHCO3 solution and 20 ml EtOAc. The resulting suspension was stirred for 3 hours. Product was isolated by filtration, washing with EtOAc and drying under vacuum. Yield: 4.0 g, 90.9%.1H-NMR (400 M Hz, CDCl3): 1.14 (6H, d, J=6.8 Hz, i-Pr), 2.76 (1H, hept., J=6.8 Hz, i-Pr), 4.05 (3H, s, MeO), 7.68 (1H, d, J=8.5 Hz, H-6), 8.07 (1H, s, H-3), 8.13 (1H, s, H-5′), 8.20 (1H, d, J=8.5 Hz, H5), 12.30 (1H, s, amide NH).Example 2 Preparation of Dipeptide Acid Compound 13 Starting Material A 250 mL 3-neck flask with a thermocouple, nitrogen inlet, and magnetic stir bar was charged with N-cyclopentyloxy carbonyl-tert-L-leucine (20.0 g, 82.2 mmol, 1.0 eq.), 1-hydroxy-benzotriazole (12.73 g, 90.42 mmol, 1.1 eq), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (17.33 g, 90.42 mmol, 1.1 eq.) The flask was purged with nitrogen, and the stiffing started. Anhydrous DMF (62 mL) was added to the flask and the mixture was stirred for 20 minutes at room temperature (about 24° C.). The reaction was mildly exothermic, the internal temperature rose to 29° C. Solid trans-4-hydroxyproline methyl ester HCl (14.93 g, 82.2 mmol, 1.0 eq) was added to the reaction in one portion. Using a syringe, diisopropyl ethyl amine (14.36 mL, 82.2 mmol, 1.0 eq) was added to the reaction dropwise over 25 min. The internal temperature rose to 34.5° C. from 29° C. The reaction was stirred for 1.75 h, forming 12. The reaction was then quenched with 0.1 M HCl (100 mL), the internal temperature rose to 34° C. The reaction was extracted three times with 75 mL of ethyl acetate, and the organic layers were combined. The organic layer was washed with 75 mL H2O, and 2×75 mL of sat. NaHCO3. The organic layer (about 235 mL) was transferred to a 500 mL flask fitted with a mechanical stirrer, shortpath distillation head, internal and external thermocouples, and distilled to minimal stirrable volume under house vacuum (˜110 mm Hg) below 35° C. internal temperature with an oil bath temperature of 40° C. To this crude mixture of 12 was then added tetrahydrofuran (150 mL) and it was distilled to minimum stirrable volume. Tetrahydrofuran (100 mL) was added to the flask, and it was again distilled to minimum stirrable volume. The distillation head was replaced with an addition funnel. Tetrahydrofuran, (100 mL) and methanol (50 mL) were added to the flask, and the solution stirred for about 15 minutes. A 3.2 M solution of LiOH (77 mL, 246.6 mmol, 3 eq.) was charged to the addition funnel, and added over 45 minutes. The temperature rose from 22° C. to 29° C., and the reaction mixture became slightly cloudy. The mixture was cooled in a cold water bath, then the reaction was quenched by slow (45 min.) addition of 4 M HCl (58-65 mL) to adjust the pH to 3.5, causing a slight increase in temperature to 27° C. The flask was fitted with a distillation head, and the methanol and tetrahydrofuran were removed by distillation at reduced pressure, with a bath temperature of 40° C., internal temperature below 30° C. The mixture was extracted twice with 150 mL of MTBE. The MTBE solution was concentrated at reduced pressure, (350 mmHg) to minimum stirrable volume. 50 mL of MTBE was added, it was removed by distillation, internal temp below 35° C. The reaction was a clear viscous liquid, 20 mL of MTBE was added, the mixture was heated to 50° C., solution was clear, the oil bath was turned off, and the solution cooled to rt, ˜24° C. over 1.5 h. To the resultant slurry was then added 60 mL MTBE, stirred 2 h, then the slurry was filtered, using ˜20 mL MTBE to transfer the mixture. The solid was then dried under vacuum at 35° C. to constant weight, 16.4 g (52%), to give the ⅓ MTBE solvate compound 13 as a colorless solid, m.p. 117-124° C.; αD=−58.6 (c 2.17, MeOH); 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 6.76 (d, J=9.3 Hz, 1H), 5.15 (s, 1H), 4.92 (m, 1H), 4.31 (br s, 1H), 4.26 (t, J=8.3 Hz, 1H), 4.19 (d, J=9.3 Hz, 1H), 3.63 (m, 2H), 3.06 (s, 1H, (MTBE)), 2.08 (m, 1H), 1.87-1.48 (m, 9H), 1.09 (s, 3H, (MTBE)), 0.92 (s, 9H).Example 3 Preparation of Tripeptide Acid Compound 16 Starting Material
A 250 mL 3-neck flask with a thermocouple, nitrogen inlet, and magnetic stir bar was charged with N-cyclopentyloxy carbonyl-tert-L-leucine (20.0 g, 82.2 mmol, 1.0 eq.), 1-hydroxy-benzotriazole (12.73 g, 90.42 mmol, 1.1 eq), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (17.33 g, 90.42 mmol, 1.1 eq.) The flask was purged with nitrogen, and the stiffing started. Anhydrous DMF (62 mL) was added to the flask and the mixture was stirred for 20 minutes at room temperature (about 24° C.). The reaction was mildly exothermic, the internal temperature rose to 29° C. Solid trans-4-hydroxyproline methyl ester HCl (14.93 g, 82.2 mmol, 1.0 eq) was added to the reaction in one portion. Using a syringe, diisopropyl ethyl amine (14.36 mL, 82.2 mmol, 1.0 eq) was added to the reaction dropwise over 25 min. The internal temperature rose to 34.5° C. from 29° C. The reaction was stirred for 1.75 h, forming 12. The reaction was then quenched with 0.1 M HCl (100 mL), the internal temperature rose to 34° C. The reaction was extracted three times with 75 mL of ethyl acetate, and the organic layers were combined. The organic layer was washed with 75 mL H2O, and 2×75 mL of sat. NaHCO3. The organic layer (about 235 mL) was transferred to a 500 mL flask fitted with a mechanical stirrer, shortpath distillation head, internal and external thermocouples, and distilled to minimal stirrable volume under house vacuum (˜110 mm Hg) below 35° C. internal temperature with an oil bath temperature of 40° C. To this crude mixture of 12 was then added tetrahydrofuran (150 mL) and it was distilled to minimum stirrable volume. Tetrahydrofuran (100 mL) was added to the flask, and it was again distilled to minimum stirrable volume. The distillation head was replaced with an addition funnel. Tetrahydrofuran, (100 mL) and methanol (50 mL) were added to the flask, and the solution stirred for about 15 minutes. A 3.2 M solution of LiOH (77 mL, 246.6 mmol, 3 eq.) was charged to the addition funnel, and added over 45 minutes. The temperature rose from 22° C. to 29° C., and the reaction mixture became slightly cloudy. The mixture was cooled in a cold water bath, then the reaction was quenched by slow (45 min.) addition of 4 M HCl (58-65 mL) to adjust the pH to 3.5, causing a slight increase in temperature to 27° C. The flask was fitted with a distillation head, and the methanol and tetrahydrofuran were removed by distillation at reduced pressure, with a bath temperature of 40° C., internal temperature below 30° C. The mixture was extracted twice with 150 mL of MTBE. The MTBE solution was concentrated at reduced pressure, (350 mmHg) to minimum stirrable volume. 50 mL of MTBE was added, it was removed by distillation, internal temp below 35° C. The reaction was a clear viscous liquid, 20 mL of MTBE was added, the mixture was heated to 50° C., solution was clear, the oil bath was turned off, and the solution cooled to rt, ˜24° C. over 1.5 h. To the resultant slurry was then added 60 mL MTBE, stirred 2 h, then the slurry was filtered, using ˜20 mL MTBE to transfer the mixture. The solid was then dried under vacuum at 35° C. to constant weight, 16.4 g (52%), to give the ⅓ MTBE solvate compound 13 as a colorless solid, m.p. 117-124° C.; αD=−58.6 (c 2.17, MeOH); 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 6.76 (d, J=9.3 Hz, 1H), 5.15 (s, 1H), 4.92 (m, 1H), 4.31 (br s, 1H), 4.26 (t, J=8.3 Hz, 1H), 4.19 (d, J=9.3 Hz, 1H), 3.63 (m, 2H), 3.06 (s, 1H, (MTBE)), 2.08 (m, 1H), 1.87-1.48 (m, 9H), 1.09 (s, 3H, (MTBE)), 0.92 (s, 9H).Example 3 Preparation of Tripeptide Acid Compound 16 Starting Material In a 25 ml flask 14 was dissolved in 3 ml DMF. HOBt (149 mg, 1.1 mmol), EDC (211 mg, 1.1 mmol), 13 (290 mg, 1.0 mmol) and i-Pr2NEt (129 mg, 1.0 mmol) were added in the given order at room temperature. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into 15 ml aqueous NaHCO3 and extracted with ethyl acetate (20 ml). The organic layer was washed with HCl (0.5 N, 2×10 ml) and saturated aqueous NaHCO3 (10 ml). After removal of solvent by rotary evaporation, 15 was obtained as a white solid. 0.46 g (95% yield). 1H-NMR (400 M Hz, CDCl3): 0.96 (s, 9H), 1.35 (1H, dd, J=3.0, 4.5 Hz), 1.45-1.90 (m, 9H), 1.77 (1H, dd, J=3.0, 4.0 Hz), 2.00-2.09 (1H, m), 2.45-2.52 (1H, m), 3.02 (1H, br), 3.50 (1H, dd, J=11.0, 3.0 Hz), 3.58 (3H, s), 3.99 (1H, d, J=11.0 Hz), 4.18 (1H, d, J=9.0 Hz), 4.43 (1H, br), Hz), 4.63 (1H, t, J=8.0 Hz), 4.93-5.00 (1H, m), 5.04 (1H, dd, J=10.5, 2.0 Hz), 5.20 (1H, d, J=18.0 Hz), 5.20-5.25 (1H, m), 5.65-5.77 (1H, ddd, J=18.0, 10.5, 2.0 Hz), 7.78 (1H, br) ppm.320 mg ester 15 (0.667 mmol, 1 eq.) was dissolved in 6.7 mL THF+3.4 mL MeOH at ambient temperature under N2. To this solution was then added 3.34 mL 1.6 M LiOH (5.34 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 hours, the solvents were removed in vacuo, and the residue diluted with 15 mL EtOAc+10 mL sat'd NaCl, then 1N HCl was added until pH 3.45 was reached. The phases were separated and the aqueous phase reextracted with 15 mL EtOAc. The combined EtOAc layers were washed with H2O (1×50 mL), dried (MgSO4), and the solvents removed in vacuo to give an oil. The oil was azeotroped with MTBE (1×15 mL), and the residue dried under high vacuum to give 320 mg of 16 (100%) as a colorless foam. Exact mass calc'd for C23H35N3O7: 465.25. Found (ES−): 464.29; 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.40 (br s, 1H), 8.49 (s, 1H), 6.77 (d, J=8.2 Hz, 1H), 5.71 (m, 1H), 5.22-4.85 (m, 4H), 4.36-4.10 (m, 3H), 3.80-3.21 (m, 4H), 2.00-1.42 (m, 11H), 0.92 (s, 9H).Example 4 Dipeptide SNAr Approach to Amorphous Compound (1)
In a 25 ml flask 14 was dissolved in 3 ml DMF. HOBt (149 mg, 1.1 mmol), EDC (211 mg, 1.1 mmol), 13 (290 mg, 1.0 mmol) and i-Pr2NEt (129 mg, 1.0 mmol) were added in the given order at room temperature. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was poured into 15 ml aqueous NaHCO3 and extracted with ethyl acetate (20 ml). The organic layer was washed with HCl (0.5 N, 2×10 ml) and saturated aqueous NaHCO3 (10 ml). After removal of solvent by rotary evaporation, 15 was obtained as a white solid. 0.46 g (95% yield). 1H-NMR (400 M Hz, CDCl3): 0.96 (s, 9H), 1.35 (1H, dd, J=3.0, 4.5 Hz), 1.45-1.90 (m, 9H), 1.77 (1H, dd, J=3.0, 4.0 Hz), 2.00-2.09 (1H, m), 2.45-2.52 (1H, m), 3.02 (1H, br), 3.50 (1H, dd, J=11.0, 3.0 Hz), 3.58 (3H, s), 3.99 (1H, d, J=11.0 Hz), 4.18 (1H, d, J=9.0 Hz), 4.43 (1H, br), Hz), 4.63 (1H, t, J=8.0 Hz), 4.93-5.00 (1H, m), 5.04 (1H, dd, J=10.5, 2.0 Hz), 5.20 (1H, d, J=18.0 Hz), 5.20-5.25 (1H, m), 5.65-5.77 (1H, ddd, J=18.0, 10.5, 2.0 Hz), 7.78 (1H, br) ppm.320 mg ester 15 (0.667 mmol, 1 eq.) was dissolved in 6.7 mL THF+3.4 mL MeOH at ambient temperature under N2. To this solution was then added 3.34 mL 1.6 M LiOH (5.34 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 hours, the solvents were removed in vacuo, and the residue diluted with 15 mL EtOAc+10 mL sat'd NaCl, then 1N HCl was added until pH 3.45 was reached. The phases were separated and the aqueous phase reextracted with 15 mL EtOAc. The combined EtOAc layers were washed with H2O (1×50 mL), dried (MgSO4), and the solvents removed in vacuo to give an oil. The oil was azeotroped with MTBE (1×15 mL), and the residue dried under high vacuum to give 320 mg of 16 (100%) as a colorless foam. Exact mass calc'd for C23H35N3O7: 465.25. Found (ES−): 464.29; 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.40 (br s, 1H), 8.49 (s, 1H), 6.77 (d, J=8.2 Hz, 1H), 5.71 (m, 1H), 5.22-4.85 (m, 4H), 4.36-4.10 (m, 3H), 3.80-3.21 (m, 4H), 2.00-1.42 (m, 11H), 0.92 (s, 9H).Example 4 Dipeptide SNAr Approach to Amorphous Compound (1) SNAr Protocol 1: A 100 mL 3-neck round bottom flask was charged with 1.93 g 13 (5.00 mmol, 1 eq.), then evacuated/Ar filled (3×), then 17.0 mL DMSO was added via syringe to give a clear, colorless solution. The flask was again evacuated/Ar filled (3×), then 2.53 g t-BuOK (22.5 mmol, 4.5 eq.) was added neat, at once. An exotherm to a maximum of 31.5° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum (˜60 mm) for one hour, and some foaming (-t-BuOH) was observed. The vacuum was relieved to Ar, then 2.20 g 11 (5.00 mmol, 1 eq.) was added neat, at once. An exotherm to 28.6° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum protected from light at ambient temperature. After 6.5 h the vacuum was relieved to Ar and a sample removed for HPLC, which showed <2% unreacted 11. The flask was then cooled in a cold water bath to 18° C., and 1.72 mL glacial HOAc (30 mmol, 6 eq.) was then added via syringe over ˜10 minutes. An exotherm to 20.5° C. was observed. The mixture was stirred for 10 minutes, then added dropwise over 15 minutes into a second flask containing a well-stirred solution of 30 mL pH 3.5H2O (˜0.001M HCl) at 18° C., causing a precipitate to form immediately, and giving an exotherm to 21.0° C. 2.0 mL DMSO was used to wash the residue into the aqueous mixture, followed by a wash of 5.0 mL ˜0.001M HCl. The resulting suspension was stirred for 15 minutes, then 30 mL of a 1:1 mixture of EtOAc:MTBE was added, and the mixture agitated vigorously for 15 minutes. Agitation was stopped and the phases were allowed to separate. Rapid phase separation and formation of 2 clear phases with no rag layer was seen. The lower aqueous phase was then reextracted with 30 mL of 1:1 EtOAc:MTBE (same fast separation), and the organic extracts were combined and saved. The aqueous phase was discarded as waste.The organic solution was then washed with H2O (3×30 mL), again all extractions gave rapid separation of phases and no rag layer, then the EtOAc was distilled to minimal stirrable volume. The residue was then azeotroped with 30 mL THF (2×), again distilling to minimal stirrable volume. The resultant slurry of crude 18 was used immediately in the peptide coupling. Exact mass calc'd for C34H42BrN5O8S: 759.19. Found (MS−): 757.92.SNAr Protocol 2: 1.00 g 13 (2.59 mmol, 1 eq.) and 1.35 g 11 (2.59 mmol, 1 eq.) were charged to a dry flask. The flask was then evacuated/Ar filled (3×), then 10 mL dry DMSO was added via syringe. The flask was again evacuated/Ar filled (3×), then cooled to 19° C. with a cold water bath. To this mixture was then added a 2M solution of KDMO/heptane (5.71 mL, 11.7 mmol, 4.5 eq.) dropwise over 30 minutes. After six hours, HPLC showed the reaction as complete. The reaction was quenched with 0.89 mL HOAc (6 eq.), and added slowly to 25 mL stirring H2O, causing a precipitate to form. The mixture was then extracted with IPAc (2×25 mL). The combined IPAc phases were washed with H2O (1×25 mL), dried (MgSO4), and the solvents removed in vacuo to give a solid, which was azeotroped with MeCN (1×25 mL), and then diluted with heptane to give a slurry. The slurry was filtered and dried to give 1.80 g 18 (91%).Peptide Coupling Protocol 1: To the THF slurry of crude 18 from SNAr Protocol 1 (taken as 5.00 mmol, 1 eq.) under Ar at ambient temperature in a flask protected from light was added 1.72 g 14 (5.5 mmol, 1.1 eq.) and 25 mL THF. The solution was then cooled to 5° C. under Ar, then 0.958 mL DIEA (5.50 mmol, 1.1 eq.) was added dropwise via syringe over 5 minutes. 5 minutes after the DIEA addition was completed, 0.85 g HOBT hydrate (6.00 mmol, 1.2 eq.), and 1.05 g EDC (5.50 mmol, 1.1 eq.) was then added neat, at once. The flask was then removed from the cold bath and the resultant mixture was then stirred at ambient temperature under Ar for 4 hours. A sample was withdrawn for HPLC which showed <2% unreacted 18 remained. The mixture was cooled to 5° C., then 40 mL 0.1N HCl was added dropwise via addition funnel over 5 minutes, followed by 40 mL EtOAc. The mixture was well agitated for 15 minutes, then agitation was stopped and the phases were allowed to separate. The lower aqueous phase was then reextracted with 40 mL EtOAc and the organic phases were combined and saved. The aqueous phase was discarded as waste. The organic solution was then washed with H2O (1×40 mL), sat'd NaHCO3 (2×40 mL), and again H2O (1×40 mL), then distilled to minimal stirrable volume. The residue was then azeotroped with MTBE (2×40 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 4.70 g of crude 19 as an orange solid, with HPLC purity of 78.3%. This material was then chromatographed on silica gel eluting with 2:1 EtOAc:Hexane to give 3.01 g (68% over 2 steps) pure 19 as a yellow powder. Exact mass calc'd for C41H51BrN6O9S: 882.26, MS+: 883.30. 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.32 (s, 1H), 8.69 (s, 1H), 8.14 (d, J=9.2 Hz, 1H), 8.03 (s, 1H), 7.45 (s, 1H), 7.33 (d, J=9.4 Hz, 1H), 6.97 (d, J=8.6 Hz, 1H), 5.65 (m, 1H), 5.40 (s, 1H), 5.20 (dd, J=1.5, 17 Hz, 1H), 5.06 (dd, J=1.6, 10.2 Hz, 1H), 5.56 (s, 1H), 4.46 (m, 1H), 4.37 (d, J=9 Hz, 1H), 4.08 (m, 1H), 3.99 (s, 3H), 3.90 (m, 1H), 3.56 (s, 3H), 2.81 (m, 1H), 2.51 (m, 1H), 2.25 (m, 1H), 2.07 (m, 1H), 1.70-1.32 (m, 7H), 1.30 (m, 3H), 1.15 (d, J=8.1 Hz, 6H), 0.95 (s, 9H).Peptide Coupling Protocol 2: A 5 L 4-neck RBF fitted with mech. stirrer, addition funnel, and thermocouple was charged with 69.57 g 14 (222 mmol, 1.3 eq.), then evacuated/Ar filled (3×). To this was then added a 200 mL THF solution of 18 (contains 129.85 g 171 mmol, 1 eq.), then 523 mL THF was charged to bring the final THF volume to 1 L. The mixture was then cooled to 4.0° C. under Ar. 38.67 mL DIEA (222 mmol, 1.3 eq.) was then added dropwise via addition funnel over 10 minutes, as the internal temperature fell to 2.4° C. The mixture was aged 5 minutes, then 29.98 g HOBT H2O (222 mmol, 1.3 eq.) was added, followed by 42.57 g EDC (222 mmol, 1.3 eq.). The internal temperature was then 3.6° C. The bath was then removed. The internal temperature rose to 20.5° C. over 90 minutes. 4 h after the EDC addition was completed, HPLC showed the reaction was complete. The mixture was cooled to 4.0° C., then 750 mL 0.1N HCl was added over 30 minutes via addition funnel, giving an exotherm to 9.5° C. To this mixture was then added 250 mL sat'd NaCl, followed by 1 L IPAc. After 5 min. vigorous stirring, the mixture was added to a separatory funnel, and the phases were separated. The lower aq. phase was then reextracted with 500 mL IPAc, and the IPAc phases combined. These were then washed successively with H2O (1×1 L), sat'd NaHCO3 (1×1 L), and then H2O (1×1 L). The mixture was then mech. stirred for 12 h to precipitate quinoline 7. The mixture was then filtered through a medium-fritted funnel, and the filtrate distilled until minimal stirrable volume was reached. The residue was then azeotroped with MTBE (2×400 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 128 g of 19 as a yellow solid, with HPLC purity of 89%.140 mg 19 (0.158 mmol, 1 eq.) was dissolved in 1.6 mL THF+0.80 mL MeOH at ambient temperature under N2. To this solution was then added 0.79 mL 1.6 M LiOH (1.27 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 h, the organic solvents were removed in vacuo, and the residue diluted with 10 mL EtOAc+10 mL sat'd NaCl. The pH was then adjusted to 5.75 with 1N HCl. The mixture was agitated vigorously for one hour, then the phases were separated. The aqueous phase was reextracted with 10 mL EtOAc. The combined EtOAc phases were then washed with H2O (2×25 mL), dried (MgSO4, and the solvents removed in vacuo to give 125 mg of Compound (1) (91%) as an amorphous yellow powder.Example 5 Tripeptide SNAr Approach to Amorphous Compound (1)
SNAr Protocol 1: A 100 mL 3-neck round bottom flask was charged with 1.93 g 13 (5.00 mmol, 1 eq.), then evacuated/Ar filled (3×), then 17.0 mL DMSO was added via syringe to give a clear, colorless solution. The flask was again evacuated/Ar filled (3×), then 2.53 g t-BuOK (22.5 mmol, 4.5 eq.) was added neat, at once. An exotherm to a maximum of 31.5° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum (˜60 mm) for one hour, and some foaming (-t-BuOH) was observed. The vacuum was relieved to Ar, then 2.20 g 11 (5.00 mmol, 1 eq.) was added neat, at once. An exotherm to 28.6° C. was observed. The flask was evacuated/Ar filled (3×), then stirred under house vacuum protected from light at ambient temperature. After 6.5 h the vacuum was relieved to Ar and a sample removed for HPLC, which showed <2% unreacted 11. The flask was then cooled in a cold water bath to 18° C., and 1.72 mL glacial HOAc (30 mmol, 6 eq.) was then added via syringe over ˜10 minutes. An exotherm to 20.5° C. was observed. The mixture was stirred for 10 minutes, then added dropwise over 15 minutes into a second flask containing a well-stirred solution of 30 mL pH 3.5H2O (˜0.001M HCl) at 18° C., causing a precipitate to form immediately, and giving an exotherm to 21.0° C. 2.0 mL DMSO was used to wash the residue into the aqueous mixture, followed by a wash of 5.0 mL ˜0.001M HCl. The resulting suspension was stirred for 15 minutes, then 30 mL of a 1:1 mixture of EtOAc:MTBE was added, and the mixture agitated vigorously for 15 minutes. Agitation was stopped and the phases were allowed to separate. Rapid phase separation and formation of 2 clear phases with no rag layer was seen. The lower aqueous phase was then reextracted with 30 mL of 1:1 EtOAc:MTBE (same fast separation), and the organic extracts were combined and saved. The aqueous phase was discarded as waste.The organic solution was then washed with H2O (3×30 mL), again all extractions gave rapid separation of phases and no rag layer, then the EtOAc was distilled to minimal stirrable volume. The residue was then azeotroped with 30 mL THF (2×), again distilling to minimal stirrable volume. The resultant slurry of crude 18 was used immediately in the peptide coupling. Exact mass calc'd for C34H42BrN5O8S: 759.19. Found (MS−): 757.92.SNAr Protocol 2: 1.00 g 13 (2.59 mmol, 1 eq.) and 1.35 g 11 (2.59 mmol, 1 eq.) were charged to a dry flask. The flask was then evacuated/Ar filled (3×), then 10 mL dry DMSO was added via syringe. The flask was again evacuated/Ar filled (3×), then cooled to 19° C. with a cold water bath. To this mixture was then added a 2M solution of KDMO/heptane (5.71 mL, 11.7 mmol, 4.5 eq.) dropwise over 30 minutes. After six hours, HPLC showed the reaction as complete. The reaction was quenched with 0.89 mL HOAc (6 eq.), and added slowly to 25 mL stirring H2O, causing a precipitate to form. The mixture was then extracted with IPAc (2×25 mL). The combined IPAc phases were washed with H2O (1×25 mL), dried (MgSO4), and the solvents removed in vacuo to give a solid, which was azeotroped with MeCN (1×25 mL), and then diluted with heptane to give a slurry. The slurry was filtered and dried to give 1.80 g 18 (91%).Peptide Coupling Protocol 1: To the THF slurry of crude 18 from SNAr Protocol 1 (taken as 5.00 mmol, 1 eq.) under Ar at ambient temperature in a flask protected from light was added 1.72 g 14 (5.5 mmol, 1.1 eq.) and 25 mL THF. The solution was then cooled to 5° C. under Ar, then 0.958 mL DIEA (5.50 mmol, 1.1 eq.) was added dropwise via syringe over 5 minutes. 5 minutes after the DIEA addition was completed, 0.85 g HOBT hydrate (6.00 mmol, 1.2 eq.), and 1.05 g EDC (5.50 mmol, 1.1 eq.) was then added neat, at once. The flask was then removed from the cold bath and the resultant mixture was then stirred at ambient temperature under Ar for 4 hours. A sample was withdrawn for HPLC which showed <2% unreacted 18 remained. The mixture was cooled to 5° C., then 40 mL 0.1N HCl was added dropwise via addition funnel over 5 minutes, followed by 40 mL EtOAc. The mixture was well agitated for 15 minutes, then agitation was stopped and the phases were allowed to separate. The lower aqueous phase was then reextracted with 40 mL EtOAc and the organic phases were combined and saved. The aqueous phase was discarded as waste. The organic solution was then washed with H2O (1×40 mL), sat'd NaHCO3 (2×40 mL), and again H2O (1×40 mL), then distilled to minimal stirrable volume. The residue was then azeotroped with MTBE (2×40 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 4.70 g of crude 19 as an orange solid, with HPLC purity of 78.3%. This material was then chromatographed on silica gel eluting with 2:1 EtOAc:Hexane to give 3.01 g (68% over 2 steps) pure 19 as a yellow powder. Exact mass calc'd for C41H51BrN6O9S: 882.26, MS+: 883.30. 1H NMR (400 MHz, DMSO, major rotamer reported) δ: 12.32 (s, 1H), 8.69 (s, 1H), 8.14 (d, J=9.2 Hz, 1H), 8.03 (s, 1H), 7.45 (s, 1H), 7.33 (d, J=9.4 Hz, 1H), 6.97 (d, J=8.6 Hz, 1H), 5.65 (m, 1H), 5.40 (s, 1H), 5.20 (dd, J=1.5, 17 Hz, 1H), 5.06 (dd, J=1.6, 10.2 Hz, 1H), 5.56 (s, 1H), 4.46 (m, 1H), 4.37 (d, J=9 Hz, 1H), 4.08 (m, 1H), 3.99 (s, 3H), 3.90 (m, 1H), 3.56 (s, 3H), 2.81 (m, 1H), 2.51 (m, 1H), 2.25 (m, 1H), 2.07 (m, 1H), 1.70-1.32 (m, 7H), 1.30 (m, 3H), 1.15 (d, J=8.1 Hz, 6H), 0.95 (s, 9H).Peptide Coupling Protocol 2: A 5 L 4-neck RBF fitted with mech. stirrer, addition funnel, and thermocouple was charged with 69.57 g 14 (222 mmol, 1.3 eq.), then evacuated/Ar filled (3×). To this was then added a 200 mL THF solution of 18 (contains 129.85 g 171 mmol, 1 eq.), then 523 mL THF was charged to bring the final THF volume to 1 L. The mixture was then cooled to 4.0° C. under Ar. 38.67 mL DIEA (222 mmol, 1.3 eq.) was then added dropwise via addition funnel over 10 minutes, as the internal temperature fell to 2.4° C. The mixture was aged 5 minutes, then 29.98 g HOBT H2O (222 mmol, 1.3 eq.) was added, followed by 42.57 g EDC (222 mmol, 1.3 eq.). The internal temperature was then 3.6° C. The bath was then removed. The internal temperature rose to 20.5° C. over 90 minutes. 4 h after the EDC addition was completed, HPLC showed the reaction was complete. The mixture was cooled to 4.0° C., then 750 mL 0.1N HCl was added over 30 minutes via addition funnel, giving an exotherm to 9.5° C. To this mixture was then added 250 mL sat'd NaCl, followed by 1 L IPAc. After 5 min. vigorous stirring, the mixture was added to a separatory funnel, and the phases were separated. The lower aq. phase was then reextracted with 500 mL IPAc, and the IPAc phases combined. These were then washed successively with H2O (1×1 L), sat'd NaHCO3 (1×1 L), and then H2O (1×1 L). The mixture was then mech. stirred for 12 h to precipitate quinoline 7. The mixture was then filtered through a medium-fritted funnel, and the filtrate distilled until minimal stirrable volume was reached. The residue was then azeotroped with MTBE (2×400 mL), and again distilled to minimal stirrable volume. The residue was dried under high vacuum to give 128 g of 19 as a yellow solid, with HPLC purity of 89%.140 mg 19 (0.158 mmol, 1 eq.) was dissolved in 1.6 mL THF+0.80 mL MeOH at ambient temperature under N2. To this solution was then added 0.79 mL 1.6 M LiOH (1.27 mmol, 8 eq.) dropwise over 5 minutes. After 1.5 h, the organic solvents were removed in vacuo, and the residue diluted with 10 mL EtOAc+10 mL sat'd NaCl. The pH was then adjusted to 5.75 with 1N HCl. The mixture was agitated vigorously for one hour, then the phases were separated. The aqueous phase was reextracted with 10 mL EtOAc. The combined EtOAc phases were then washed with H2O (2×25 mL), dried (MgSO4, and the solvents removed in vacuo to give 125 mg of Compound (1) (91%) as an amorphous yellow powder.Example 5 Tripeptide SNAr Approach to Amorphous Compound (1) 233 mg tripeptide acid 16 (0.50 mmol) was charged to a flask, then the flask was evacuated/Ar filled (3×). 1.7 mL DMSO was then added, and the mixture evacuated/Ar filled (3×). The mixture was then cooled in a cold water bath, then 317 mg t-BuOK (2.82 mmol, 5.63 eq.) were added. The flask was again evacuated/Ar filled (3×), then stirred under 60 mm vacuum for one hour. 220 mg quinoline 11 (0.50 mmol, 1 eq.) was then added, and the flask evacuated/Ar filled (3×), then stirred under 60 mm vacuum in the dark at ambient temperature for 3 hours. 0.30 mL HOAc was then added, then the resulting solution was added to 25 mL 0.001 M HCl, causing a precipitate to form. The slurry was filtered, washing the solids with 25 mL H2O. The solid was dried under N2 for 2 hours, then chromatographed on silica gel eluting with EtOAc to give 226 mg (52%) of Compound (1) as an amorphous yellow solid.Additional methods for preparing amorphous Compound (1) can be found in U.S. Pat. Nos. 6,323,180, 7,514,557 and 7,585,845, which are herein incorporated by reference.Example 6 Preparation of Type A of Compound (1)Amorphous Compound (1) (Batch 7, 13.80 g) was added to a 1000 ml three neck flask. Absolute ethanol (248.9 g) was added to the flask. While stirring, the contents of the flask were heated at 60 degrees C./hr to ˜74 degrees C. (Solids do not dissolve at 74 degrees C.). Water (257.4 g) was then added linearly over 4 hr to the resulting slurry while stirring and maintaining the temperature at 74 degrees C. After the water addition was complete, the temperature was reduced linearly to ambient temperature at 8 degrees C./hr and then held at ambient temperature for 6 hrs while stiffing. The resulting solids were collected by filtration and washed with 50 ml of 1/1 (w/w) EtOH/Water. The wet solids were dried on the funnel for 30 minutes by sucking N2 through the cake. (XRPD analysis on this sample indicates that the pattern is similar to the EtOH solvate). The solids were then dried at 65-70 degrees C. under vacuum (P=25 in Hg) and a nitrogen bleed for 1.5 hr. The resulting solids (12.6 g, 95.5% corrected yield) were confirmed by XRPD as being Type A Compound (1).The unique XRPD pattern and DSC curve of Type A Compound (1) is shown in FIGS. 1 and 2.Example 7 Preparation of the Sodium Salt of Compound (1)—Method 12.1 g of amorphous sodium salt of Compound (1) and 8.90 g of acetone was added to a vial and stirred at ambient temperature for 3 hr. The slurry was filtered off mother liquors and the resulting solids were dried for 20 minutes under nitrogen flow for 20 minutes. 1.51 g of crystalline sodium salt of Compound (1) as solids was collected.Example 8 Preparation of the Sodium Salt of Compound (1)—Method 215.6 g of Type A of Compound (1), 175 ml of acetone and 3.6 ml of water was added to a 250 ml reactor and heated to 53 degrees C. to dissolve the solids. 900 ul of 10.0 N NaOH was added to reactor and the solution was seeded with Type A. The seeded solution was stirred at 53 degrees C. for 10 minutes. A second 900 ul portion of 10.0 N NaOH was added and the system was stirred at 53 degrees C. for 30 minutes over which a slurry developed. The slurry was cooled to 19 degrees C. at a cooling rate of 15 degrees C. per hour and held overnight at 19 degrees C. The final resulting slurry was filtered and the wet solids were washed with 15 ml of acetone. Dried solids for 1 hr at 52 degrees C. under vacuum with a nitrogen flow and then exposed the solids to lab air for one hour. Collected 12.1 g of Compound (1) crystalline sodium salt solids.Example 11 Preparation of the Sodium Salt of Compound (1)—Method 5At room temperature a solution of sodium ethoxide in ethanol (21 weight %; 306 ml) was added to a solution of Compound (1) (745 g) in THF (2000 ml) and water (76.5 ml) while stiffing. After stiffing for 30 minutes, the mixture was filtered and the filter was washed with THF (85 ml). The resulting solution was warmed to 65° C. and treated with filtered butyl acetate (6640 ml, optionally pre-warmed to 65° C.) within 30 minutes. Seeding crystals (0.50 g) were added, and the mixture was stirred at 65° C. for 2 hours, while crystallization starts after about 30 minutes. The suspension was cooled to 50° C. within 1 hour and stirred at this temperature for an additional hour. The title compound was isolated by filtration, washed with filtered butyl acetate (765 ml, optionally pre-warmed to 50° C.) and dried at 65° C. for about 16 h giving Compound (1) crystalline sodium salt (˜725 g).
233 mg tripeptide acid 16 (0.50 mmol) was charged to a flask, then the flask was evacuated/Ar filled (3×). 1.7 mL DMSO was then added, and the mixture evacuated/Ar filled (3×). The mixture was then cooled in a cold water bath, then 317 mg t-BuOK (2.82 mmol, 5.63 eq.) were added. The flask was again evacuated/Ar filled (3×), then stirred under 60 mm vacuum for one hour. 220 mg quinoline 11 (0.50 mmol, 1 eq.) was then added, and the flask evacuated/Ar filled (3×), then stirred under 60 mm vacuum in the dark at ambient temperature for 3 hours. 0.30 mL HOAc was then added, then the resulting solution was added to 25 mL 0.001 M HCl, causing a precipitate to form. The slurry was filtered, washing the solids with 25 mL H2O. The solid was dried under N2 for 2 hours, then chromatographed on silica gel eluting with EtOAc to give 226 mg (52%) of Compound (1) as an amorphous yellow solid.Additional methods for preparing amorphous Compound (1) can be found in U.S. Pat. Nos. 6,323,180, 7,514,557 and 7,585,845, which are herein incorporated by reference.Example 6 Preparation of Type A of Compound (1)Amorphous Compound (1) (Batch 7, 13.80 g) was added to a 1000 ml three neck flask. Absolute ethanol (248.9 g) was added to the flask. While stirring, the contents of the flask were heated at 60 degrees C./hr to ˜74 degrees C. (Solids do not dissolve at 74 degrees C.). Water (257.4 g) was then added linearly over 4 hr to the resulting slurry while stirring and maintaining the temperature at 74 degrees C. After the water addition was complete, the temperature was reduced linearly to ambient temperature at 8 degrees C./hr and then held at ambient temperature for 6 hrs while stiffing. The resulting solids were collected by filtration and washed with 50 ml of 1/1 (w/w) EtOH/Water. The wet solids were dried on the funnel for 30 minutes by sucking N2 through the cake. (XRPD analysis on this sample indicates that the pattern is similar to the EtOH solvate). The solids were then dried at 65-70 degrees C. under vacuum (P=25 in Hg) and a nitrogen bleed for 1.5 hr. The resulting solids (12.6 g, 95.5% corrected yield) were confirmed by XRPD as being Type A Compound (1).The unique XRPD pattern and DSC curve of Type A Compound (1) is shown in FIGS. 1 and 2.Example 7 Preparation of the Sodium Salt of Compound (1)—Method 12.1 g of amorphous sodium salt of Compound (1) and 8.90 g of acetone was added to a vial and stirred at ambient temperature for 3 hr. The slurry was filtered off mother liquors and the resulting solids were dried for 20 minutes under nitrogen flow for 20 minutes. 1.51 g of crystalline sodium salt of Compound (1) as solids was collected.Example 8 Preparation of the Sodium Salt of Compound (1)—Method 215.6 g of Type A of Compound (1), 175 ml of acetone and 3.6 ml of water was added to a 250 ml reactor and heated to 53 degrees C. to dissolve the solids. 900 ul of 10.0 N NaOH was added to reactor and the solution was seeded with Type A. The seeded solution was stirred at 53 degrees C. for 10 minutes. A second 900 ul portion of 10.0 N NaOH was added and the system was stirred at 53 degrees C. for 30 minutes over which a slurry developed. The slurry was cooled to 19 degrees C. at a cooling rate of 15 degrees C. per hour and held overnight at 19 degrees C. The final resulting slurry was filtered and the wet solids were washed with 15 ml of acetone. Dried solids for 1 hr at 52 degrees C. under vacuum with a nitrogen flow and then exposed the solids to lab air for one hour. Collected 12.1 g of Compound (1) crystalline sodium salt solids.Example 11 Preparation of the Sodium Salt of Compound (1)—Method 5At room temperature a solution of sodium ethoxide in ethanol (21 weight %; 306 ml) was added to a solution of Compound (1) (745 g) in THF (2000 ml) and water (76.5 ml) while stiffing. After stiffing for 30 minutes, the mixture was filtered and the filter was washed with THF (85 ml). The resulting solution was warmed to 65° C. and treated with filtered butyl acetate (6640 ml, optionally pre-warmed to 65° C.) within 30 minutes. Seeding crystals (0.50 g) were added, and the mixture was stirred at 65° C. for 2 hours, while crystallization starts after about 30 minutes. The suspension was cooled to 50° C. within 1 hour and stirred at this temperature for an additional hour. The title compound was isolated by filtration, washed with filtered butyl acetate (765 ml, optionally pre-warmed to 50° C.) and dried at 65° C. for about 16 h giving Compound (1) crystalline sodium salt (˜725 g).Patent Filing date Publication date Applicant Title US8399484 Sep 16, 2009 Mar 19, 2013 Boehringer Ingelheim International Gmbh Combination therapy for treating HCV infection US8530497 Mar 9, 2011 Sep 10, 2013 Boehringer Ingelheim International Gmbh Crystalline salts of a potent HCV inhibitor WO2013144193A1 Mar 27, 2013 Oct 3, 2013 Boehringer Ingelheim International Gmbh Combination therapy for treating hcv infection in specific patient subgenotype sub-population - .....................................................
- 10.. LEDIPASVIR
 LEDIPASVIR, GS 5885CAS 1256388-51-8, PHASE 3Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
LEDIPASVIR, GS 5885CAS 1256388-51-8, PHASE 3Methyl N-[(2S)-1-[(6S)-6-[5-[9,9-Difluoro-7-[2-[(1S,2S,4R)-3-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]-3-azabicyclo[2.2.1]heptan-2-yl]-3H-benzimidazol-5-yl]fluoren-2-yl]-1H-imidazol-2-yl]-5-azaspiro[2.4]heptan-5-yl]-3-methyl-1-oxobutan-2-yl]carbamate
GS-5885 had been in phase III clinical development at Gilead for the oral treatment of chronic genotype 1 hepatitis C virus (HCV) infection, however no recent developments have been reported on this research.Ledipasvir (formerly GS-5885) is an experimental drug for the treatment of hepatitis C being developed by Gilead Sciences.[1] It is currently in Phase III clinical trials.[2] It is being studied in combination with other direct-acting antiviral agents that interfere with HCV replication.Ledipasvir is an inhibitor of the hepatitis C virus HCV NS5A protein.Ledipasvir is being tested in interferon-free regimens with other direct-acting antiviral agents for hepatitis C.Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the nucleotide analogue inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[3][4] Gilead is developing a sofosbuvir + ledipasvir coformulation that is being tested with and without ribavirin.- ^ "Ledipasvir". United States Adopted Name.
- ^ "GS-5885". Gilead Sciences.
- ^ ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- ^ CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
Hepatitis C is recognized as a chronic viral disease of the liver which is characterized by liver disease. Although drugs targeting the liver are in wide use and have showneffectiveness, toxicity and other side effects have limited their usefulness. Inhibitors of hepatitis C virus (HCV) are useful to limit the establishment and progression of infection by HCV as well as in diagnostic assays for HCV.The compound (l-{3-[6-(9,9-dif uoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl- butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-lH-benzoimidazol-2- yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester, also known as ledipasvir, designated herein as Compound I, is known to be an effective anti-HCV agent, as described for example in WO 2010/132601. A synthesis of compound I is disclosed in U.S. Patent No. 8,088,368
 acetone solvate . 1H NMR (400 MHz, DMSO-^, δ): 12.29 (s, 0.1H), 12.19 (d, J=4.0 Hz, 1H), 12.14 (s, 0.2H), 11.85 (s, 1H), 8.10 (s, 0.1H), 8.08 (s, 1H), 8.01 (s, 0.1H), 7.963 (m, 1H), 7.955 (s, 1H), 7.89 (d, J=6.4 Hz, 1H), 7.87 (s, 1H), 7.83 (dd, J=8.4, 2.4 Hz, 1H), 7.79 (dd, J=7.2, 2.8 Hz, 1H), 7.78-7.90 (misc., 0.9H), 7.70 (s, 1H), 7.61 (d, J=8.4 Hz, 1H), 7.55 (s, 1H), 7.51 (dd, J=8.8, 1.6 Hz, 1H), 7.44 (m, 0.1H), 7.31 (d, J=8.4 Hz, 1H), 7.21 (d, J=8.4 Hz, 1H), 6.91 (d, J=8.0 Hz, 0.2H), 6.77 (m, 0.2H), 5.34 (d, J=7.6 Hz, 0.1H), 5.20 (dd, J=8.0, 5.2 Hz, 1H), 5.18 (m, 0.1H), 4.88 (s, 0.1H), 4.67 (d, J=6.4 Hz, 1H), 4.55 (s, 1H), 4.17 (dd, J=8.0, 8.0 Hz, 1H), 4.10 (m, 0.2H), 4.01 (dd, J=8.4, 8.0 Hz, 1H), 3.97 (m, 0.1H), 3.82 (d, J=9.6 Hz, 1H), 3.77 (s, 0.2H), 3.71 (d, J=9.6 Hz, 1H), 3.554 (s, 3H), 3.548 (s, 3H), 3.43 (s, 0.4H), 3.20 (d, J=7.6 Hz, 0.3H), 2.77 (s, 0.1H), 2.66 (s, 1H), 2.41 (d, J=8.8 Hz, 1H), 2.22 (dd, J=12.4, 8.0 Hz, 1H), 2.13 (m, 0.4H), 2.08 (s, 6H), 2.05 (dd, J=13.2, 5.2 Hz, 1H), 1.99 (m, 2H), 1.92 (m, 1H), 1.77 (m, 2H), 1.61 (m, 0.3H), 1.56 (m, 1H), 1.46 (d, J=9.2 Hz, 1H), 1.33 (d, J=10.0 Hz, 0.1H), 0.97 (dd, J=6.4, 2.0 Hz, 3H), 0.93 (d, J=6.8 Hz, 3H), 0.88 (d, J=6.4 Hz, 3H), 0.87 (d, J=6.4 Hz, 3H), 0.80-1.05 (misc., 2H), 0.70 (m, 1H), 0.59 (m, 2H), 0.54 (m, 1H), 0.33 (m, 0.1H). HRMS-ESI+: [M + H]+ calcd for C49H5506N8F2, 889.4207; found, 889.4205.https://www.google.co.in/patents/WO2013184698A1
acetone solvate . 1H NMR (400 MHz, DMSO-^, δ): 12.29 (s, 0.1H), 12.19 (d, J=4.0 Hz, 1H), 12.14 (s, 0.2H), 11.85 (s, 1H), 8.10 (s, 0.1H), 8.08 (s, 1H), 8.01 (s, 0.1H), 7.963 (m, 1H), 7.955 (s, 1H), 7.89 (d, J=6.4 Hz, 1H), 7.87 (s, 1H), 7.83 (dd, J=8.4, 2.4 Hz, 1H), 7.79 (dd, J=7.2, 2.8 Hz, 1H), 7.78-7.90 (misc., 0.9H), 7.70 (s, 1H), 7.61 (d, J=8.4 Hz, 1H), 7.55 (s, 1H), 7.51 (dd, J=8.8, 1.6 Hz, 1H), 7.44 (m, 0.1H), 7.31 (d, J=8.4 Hz, 1H), 7.21 (d, J=8.4 Hz, 1H), 6.91 (d, J=8.0 Hz, 0.2H), 6.77 (m, 0.2H), 5.34 (d, J=7.6 Hz, 0.1H), 5.20 (dd, J=8.0, 5.2 Hz, 1H), 5.18 (m, 0.1H), 4.88 (s, 0.1H), 4.67 (d, J=6.4 Hz, 1H), 4.55 (s, 1H), 4.17 (dd, J=8.0, 8.0 Hz, 1H), 4.10 (m, 0.2H), 4.01 (dd, J=8.4, 8.0 Hz, 1H), 3.97 (m, 0.1H), 3.82 (d, J=9.6 Hz, 1H), 3.77 (s, 0.2H), 3.71 (d, J=9.6 Hz, 1H), 3.554 (s, 3H), 3.548 (s, 3H), 3.43 (s, 0.4H), 3.20 (d, J=7.6 Hz, 0.3H), 2.77 (s, 0.1H), 2.66 (s, 1H), 2.41 (d, J=8.8 Hz, 1H), 2.22 (dd, J=12.4, 8.0 Hz, 1H), 2.13 (m, 0.4H), 2.08 (s, 6H), 2.05 (dd, J=13.2, 5.2 Hz, 1H), 1.99 (m, 2H), 1.92 (m, 1H), 1.77 (m, 2H), 1.61 (m, 0.3H), 1.56 (m, 1H), 1.46 (d, J=9.2 Hz, 1H), 1.33 (d, J=10.0 Hz, 0.1H), 0.97 (dd, J=6.4, 2.0 Hz, 3H), 0.93 (d, J=6.8 Hz, 3H), 0.88 (d, J=6.4 Hz, 3H), 0.87 (d, J=6.4 Hz, 3H), 0.80-1.05 (misc., 2H), 0.70 (m, 1H), 0.59 (m, 2H), 0.54 (m, 1H), 0.33 (m, 0.1H). HRMS-ESI+: [M + H]+ calcd for C49H5506N8F2, 889.4207; found, 889.4205.https://www.google.co.in/patents/WO2013184698A1
..................................................................................................

....................................................................................................




..............................................................
https://www.google.co.in/patents/US8088368
Example ED Preparation of Intermediate 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester

4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester
4-Methylene-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (10.0 g, 44 mmol) was dissolved in MeOH (75 mL) at room temperature and HCl (4M in dioxane, 75 mL) was added. Stirring at room temperature was continued for 4 hours. All volatiles were removed in vacuo and a beige solid was obtained.The crude material was suspended in DCM (100 mL) and N-Methyl morpholine (13.3 g, 132 mmol) was added. The mixture was cooled to 0° C. and benzyl chloroformate (8.26 g, 48.4 mmol) was added while stirring. After 30 minutes, the reaction was warmed to room temperature and the solution was washed with water and aqueous HCl (1M). The solution was dried over sodium sulfate. Filtration and evaporation of solvents gave crude product, which was purified by silica gel chromatography (eluent: EtOAc/hexanes) to yield the product (10.2 g). LCMS-ESI+: calc'd for C15H17NO4: 275.3 (M+). Found: 276.4 (M+H+).5-aza-spiro[2.4]heptanes-5,6-dicarboxylic acid benzyl ester: An oven-dried 3-neck round bottom flask was equipped with a nitrogen inlet adaptor and a 250 mL addition funnel. The third neck was sealed with a septum. The flask was charged with a stir bar, dichlorormethane (120 mL) and diethyl zinc (1.0 M in hexane, 118 mL, 118 mmol) then cooled to 0° C. in an ice bath. The addition funnel was charged with dichloromethane (40 mL) and trifluoroacetic acid (9.1 mL, 118 mmol). After the diethyl zinc solution had cooled to 0° C. (about 25 minutes), the trifluoroacetic acid solution was added dropwise over 20 minutes to the stirred reaction mixture. After stirring for another 20 minutes at 0° C., diiodomethane (9.5 mL, 118 mmol) was added slowly over 4 minutes. After another 20 minutes, 4-methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester (8.10 g, 29.4 mmol) was added in 30 mL dichloromethane by cannula. The flask containing 4-methylene-pyrrolidine-1,2-dicarboxylic acid 1-benzyl ester 2-methyl ester was then rinsed with another 10 mL dichloromethane and this solution was also transferred to the reaction mixture by cannula. The reaction mixture was allowed to warm to RT and stirred for 110 hours (about 5 days) after which the reagents were quenched with saturated aqueous ammonium chloride (˜150 mL). The contents of the flask were slowly poured into a 2 L sep funnel containing saturated aqueous sodium bicarbonate (˜800 mL). The aqueous phase was extracted three times with 300 mL ethyl acetate. The combined organics were dried over magnesium sulfate and concentrated to provide the crude material. The crude material was dissolved in 3:1:1 THF/water/acetone (165 mL) then treated with N-methylmorpholine-N-oxide (3.45 g, 29.4 mmol) and osmium tetroxide (4 wt % in water, 5 mL, 0.818 mmol). After stirring at RT for 7 h, the reagents were quenched with 1 M aqueous sodium thiosulfate (˜100 mL). The contents of the flask were then poured into a 1 L sep funnel containing water (˜300 mL). The aqueous phase was extracted three times with 300 mL dichloromethane. The combined organics were dried over magnesium sulfate and concentrated. The crude residue was purified by silica column chromatography (5% to 45% EtOAc/hexane) to provide 5-aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester as a clear oil (5.54 g, 19.15 mmol, 65%) as a clear oil. 1H NMR (CDCl3) δ 7.36-7.29 (m, 5H), 5.21-5.04 (m, 2H), 4.56-4.47 (m, 1H), 3.75 (s, 1.5H), 3.60 (m, 1.5H), 03.51-3.37 (m, 2H), 2.32-2.25 (m, 1H), 1.87-1.80 (m, 1H), 0.64-0.51 (m, 4H).5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-methyl ester (244 mg, 0.840 mmol) was dissolved in THF (2.0 mL)/MeOH (1.5 mL) An aqueous solution of LiOH (35.5 mg, 0.84 mmol) was added and stirring at room temperature was continued. After 3 hours, the reaction was neutralized with aqueous HCl (1M) and the organic solvents were removed in vacuo. The crude mixture was diluted with water and EtOAc and the organic layer was collected. All volatiles were removed in vacuo and the crude acid was used without further purification. LCMS-ESI+: calc'd for C15H17NO4: 275.3 (M+). Found: 276.3 (M+H+).Example ED′


2,7-Dibromo-9,9-difluoro-9H-fluorene
2,7-Dibromo-fluoren-9-one (4.0 g, 11.8 mmol) was suspended in deoxofluor (12 mL) at room temperature and EtOH (4 drops) was added. The stirred suspension was heated at T=90° C. for 24 hours (CAUTION: Use of deoxofluor at elevated temperatures, as described above, is strongly discouraged as rapid and violent exotherms may occur). The reaction was cooled to room temperature and poured onto ice containing sodium bicarbonate. A solid formed and was collected via filtration. The crude material was taken into EtOAc and was washed with aqueous HCl (1M) and brine. The solution was dried over sodium sulfate. Filtration and evaporation of solvents gave crude product, which was purified by silica gel chromatography (eluent: EtOAc/hexanes) to yield the product 2,7-Dibromo-9,9-difluoro-9H-fluorene (3.2 g). 19F-NMR: 282 MHz, (dmso-d6) δ: −111.6 ppm.Before using the material in the next step, it was exposed as a solution in EtOAc to charcoal.5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester
2,7-Dibromo-9,9-difluoro-9H-fluorene (372 mg, 1.04 mmol), Pd(PPh3)4 (30.0 mg, 0.026 mmol), PdCl2(PPh3)2 (18.2 mg, 0.026 mmol), As(PPh3)3 (5.0 mg) were dissolved in dioxane (10 mL) under an argon atmosphere. Ethoxyvinyl-tributyl tin (376.4 mg, 1.04 mmol) was added. The mixture was heated for 140 minutes at 85° C. (oil bath). The reaction was cooled to room temperature. N-bromo succinimide (177 mg, 1.0 mmol) was added followed by water (2 mL). The reaction was stirred at room temperature for 3 hours, after which the majority of the dioxane was removed in vacuo. The crude reaction mixture was diluted with EtOAc and was washed with water. All volatiles were removed in vacuo. Toluene was added and all volatiles were removed in vacuo for a second time. The crude material was dissolved in DMF/MeCN (2 mL, 1:1) at room temperature. A solution of N-Cbz-4-cyclopropyl (L) Proline (0.84 mmol) and DIEA (268 mg, 2.08 mmol) in MeCN (2 mL) was added and stirring at room temperature was continued. After 14 hours, most of the MeCN was removed in vacuo and the crude reaction mixture was diluted with EtOAc. The mixture was washed with aqueous HCl (1M), aqueous LiCl solution (5%), brine, and was dried over sodium sulfate. Filtration and evaporation of solvents gave the crude reaction product, which was purified via silica gel chromatography (eluent: EtOAc/hexanes) to yield the product 5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester (176 mg). LCMS-ESI+: calc'd for C30H24BrF2NO5: 596.4 (M+). Found: 595.2/597.2 (M+H+).6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester
5-Aza-spiro[2.4]heptane-5,6-dicarboxylic acid 5-benzyl ester 6-[2-(7-bromo-9,9-difluoro-9H-fluoren-2-yl)-2-oxo-ethyl]ester (172 mg, 0.293 mmol) was dissolved in m-xylenes (6.0 mL). Ammonium acetate (226 mg, 2.93 mmol) was added and the reaction was stirred at 140° C. for 60 minutes under microwave conditions. The reaction was cooled to room temperature and all volatiles were removed in vacuo. The crude material was purified via silica gel chromatography (eluent: EtOAc/hexanes) to yield the product 6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester (80.3 mg). LCMS-ESL': calc'd for C30H24BrF2N3O2: 576.4 (M+). Found: 575.2/577.2 (M+H+).(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carboxylic acid benzyl ester (800 mg, 1.38 mmol) was dissolved in DCM (15 mL) and HBr in AcOH (37%, 2 mL) was added and stirring at room temperature was continued. After 180 minutes, the suspension was diluted with hexanes and the solid was collected via filtration and was washed with hexanes and subjected to vacuum. The crude material was used in the next step without further purification. The crude material was dissolved in DMF (4.0 mL) and DIEA (356 mg, 2.76 mmol) was added. A solution of 2-(L)-Methoxycarbonylamino-3-methyl-butyric acid (242 mg, 1.38 mmol), HATU (524 mg, 1.38 mmol) and DIEA (178 mg, 1.38 mmol) in DMF (1 mL) was added. The reaction was stirred at room temperature. After 50 minutes, the reaction was diluted with EtOAc and was washed with aqueous bicarbonate solution, aqueous LiCl solution (5%), brine, and was dried over sodium sulfate. Filtration and removal of solvents in vacuo gave the crude material, which was purified by silica gel chromatography (eluent: EtOAc/hexanes) to yield the slightly impure product (1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (878 mg). LCMS-ESI+: calc'd for C29H29BrF2N4O3: 599.5 (M+); Found: 598.5/600.5 (M+H+).3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester
(1-{6-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-5-aza-spiro[2.4]heptane-5-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (840 mg, 1.4 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (615 mg, 1.4 mmol), Pd(PPh3)4 (161 mg, 0.14 mmol), K2CO3 (579 mg, 4.2 mmol), were dissolved in DME (15 mL)/water (3 mL) under an argon atmosphere. The mixture was heated for 120 minutes at 85-90° C. (oil bath). After 120 minutes additional boronate ester (61 mg, 0.14 mmol) was added and heating was continued. After 3 hours, the reaction was cooled to room temperature. Most of the DME was removed in vacuo and the crude reaction mixture was diluted with EtOAc. The mixture was washed with brine and was dried over sodium sulfate. Filtration and evaporation of solvents gave the crude reaction product, which was purified via silica gel chromatography (eluent: EtOAc/hexanes) to yield the product 3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (878 mg). LCMS-ESI+: calc'd for C47H51F2N7O5: 831.9 (M+). Found: 832.7 (M+H+).(1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (115 mg, 0.138 mmol) was dissolved in DCM (2 mL) and HCl in dioxane (4M, 2 mL) was added and stirring at room temperature was continued. After 20 minutes, all volatiles were removed in vacuo. The crude material was used in the next step without further purification. The crude material was dissolved in DMF (1.5 mL) and DIEA (53.4 mg, 0.414 mmol) was added. A solution of 2-(L) Methoxycarbonylamino-3-methyl-butyric acid (24.2 mg, 0.138 mmol), HATU (52.4 mg, 0.138 mmol) and DIEA (17.8 mg, 0.138 mmol) in DMF (1 mL) was added. The reaction was stirred at room temperature. After 20 minutes, the reaction was diluted with EtOAc and was washed with aqueous bicarbonate solution, aqueous LiCl solution (5%), brine, and was dried over sodium sulfate. Filtration and removal of solvents in vacuo gave the crude material, which was purified by RP-HPLC (eluent: water/MeCN w/0.1% TFA) to yield the product (1-{3-[6-(9,9-Difluoro-7-{2-[5-(2-methoxycarbonylamino-3-methyl-butyryl)-5-aza-spiro[2.4]hept-6-yl]-3H-imidazol-4-yl}-9H-fluoren-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (76 mg). LCMS-ESI+: calc'd for C49H54F2N8O6: 888.9 (M+). Found: 890.0 (M+H+).1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37-7.27 (m, 2H), 5.25 (dd, J=7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m, 1H), 3.87 (m, 1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m, 2H), 2.09-2.04 (m, 2H), 1.88-1.79 (m, 2H), 1.54 (m, 1H), 0.94-0.77 (m, 15H) 0.63 (m, 4H) ppm. 19F-NMR: 282 MHz, (dmso-d6) δ: −109.1 ppm [−74.8 ppm TFA]
https://www.google.co.in/patents/US8088368

2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester: A mixture of 2-[5-(7-Bromo-9,9-difluoro-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester (324 mg, 0.627 mmol), 3-[6-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-benzoimidazol-2-yl]-2-aza-bicyclo[2.2.1]heptane-2-carboxylic acid tert-butyl ester (1.1 eq., 304 mg), [1,1′ bis(diphenylphosphino)ferrocene]dichloropalladium(II)(3%, 15 mg), tetrakis(triphenylphosphine)palladium (3%, 22 mg) and potassium carbonate (3.3 eq., 285 mg) in 10 mL DME and 3 mL water was heated to 90° C. under Argon for 3 hours. The reaction mixture was cooled and diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 20 to 100% ethyl acetate/hexane) to give 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, yield 77%). LCMS-ESI−: calc'd for C43H46F2N6O4: 748.86. Found: 749.2 (M+H+).
(1-{2-[5-(9,9-Difluoro-7-{2-[2-(2-methoxycarbonylamino-3-methyl-butyryl)-2-aza-bicyclo[2.2.1]hept-3-yl]-3H-benzoimidazol-5-yl}-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (Example DK): 4N HCl in dioxane (2 mL) was added to 2-(5-{9,9-Difluoro-7-[2-(2-Boc-2-aza-bicyclo[2.2.1]hept-3-yl)-3H-benzoimidazol-5-yl]-9H-fluoren-2-yl}-1H-imidazol-2-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester (361 mg, 0.482 mmol) and the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was concentrated and dried overnight under vacuum. The residue was dissolved in DMF (5 mL) and to this solution was added 2-Methoxycarbonylamino-3-methyl-butyric acid (2 eq., 169 mg), diisopropyl ethylamine (6 eq., 0.5 mL), followed by HATU (2 eq., 367 mg). Reaction mixture was stirred at 0° C. for 30 minutes. The reaction mixture was dissolved in ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was dried (MgSO4), concentrated and purified by flash column chromatography (silica gel, 0 to 20% MeOH/ethyl acetate), followed by preparative reverse phase HPLC (GEMINI, 5 to 100% ACN/H2O+0.1% TFA). Product was lyophilized to give (1-{2-[5-(9,9-Difluoro-7-{2-[2-(2-methoxycarbonylamino-3-methyl-butyryl)-2-aza-bicyclo [2.2.1]hept-3-yl]-3H-benzoimidazol-5-yl}-9H-fluoren-2-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (285 mg, 59%).1H-NMR: 300 MHz, (CD3OD-d4) δ: 8.05-7.82 (m, 9H), 5.40-5.22 (m, 2H), 4.72 (m, 1H), 4.39 (d, 1H), 4.239d, 1H), 4.17 (m, 1H), 3.91 (m, 2H), 3.62 (d, 6H), 2.98 (m, 1H), 2.58 (m, 1H), 2.37-2.18 (m, 4H), 2.18-1.92 (m, 4H), 1.80 (m, 2H), 1.09-0.85 (m, 12H). 19F-NMR: 300 MHz, (CD3OD-d4) δ: −112.88. LCMS-ESI+: calc'd for C47H52F2N8O6 862.96. Found: 863.5 (M+H+).SEE ................................................................................................................
WO 2010132601
WO 2013040492
WO 2013059630
WO 2013059638
.......................................
The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection
J. Med. Chem., Just Accepted ManuscriptDOI: 10.1021/jm401499gPublication Date (Web): December 9, 2013
http://pubs.acs.org/doi/abs/10.1021/jm401499g?prevSearch=LEDIPASVIR&searchHistoryKey=
http://pubs.acs.org/doi/pdf/10.1021/jm401499g
1H-NMR: 300 MHz, (dmso-d6) δ: 8.20-7.99 (m, 8H), 7.73 (s, 2H), 7.37 – 7.27
(m, 2H), 5.25 (dd, J = 7.2 Hz, 1H), 4.78 (s, 1H) 4.54 (s, 1H), 4.16 (m, 1H), 4.02 (m,
1H), 3.87 (m,1H), 3.74 (m, 1H), 3.55 (s, 3H), 3.53 (s, 3H), 2.75 (m, 1H), 2.25 (m,
2H), 2.09 – 2.04 (m, 2H), 1.88 – 1.79 (m, 2H), 1.54 (m, 1H), 0.94 - 0.77 (m, 15H)
0.63 (m, 4H) ppm. 19F-NMR: 282 MHz, (dmso-d6) δ: -109.1 ppm [-74.8 ppm TFA].
HRMS (ESI-TOF) m/z: [M + H]+
calc’d for C49H55F2N8O6: 889.4207; Found: 889.4214.
methyl [(2S)-1-{(6S)-6-[5-(9,9-difluoro-7-{2-
[(1R,3S,4S)-2-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-2-
azabicyclo[2.2.1]hept-3-yl]-1H-benzimidazol-6-yl}-9H-fluoren-2-yl)-1H-imidazol-2-
yl]-5-azaspiro[2.4]hept-5-yl}-3-methyl-1-oxobutan-2-yl]carbamate (39 NOS IS LEDISPAVIR
...............................................................
1)Link, John O.et al; The Discovery of Ledipasvir (GS-5885), a Potent Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection; Journal of Medicinal Chemistry (2013), Ahead of Print.DOI:10.1021/jm401499g
2)Ray, Adrian S. et al; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., WO2013040492
3) Delaney, William E. et al ; Preparation of pyridazinylmethylimidazopyridine derivatives and analogs for use in the treatment of hepatitis C virus using combination chemotherapy, PCT Int. Appl., wo2012087596
4) Delaney, William E., IV et al; Preparation of quinoline derivatives and analogs for use in the treatment of hepatitis C virus infection in combination with ribavirin; PCT Int. Appl., wo2011156757
5) Guo, Hongyan et al; Preparation of biaryls, arylheteroaryls, heteroaryls, biarylacetylenes and related compounds end-capped with amino acid or peptide derivatives as antiviral agents; PCT Int. Appl., WO2010132601
6)Phase III (Sofosbuvir + Ledipasvir) ION-1 study: (Clinical Trial number: NCT01701401):
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
7) Phase III (Sofosbuvir + Ledipasvir) ION-2 study: (Clinical Trial number: NCT01768286)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/GS-5885 Fixed-Dose Combination ± Ribavirin for 12 and 24 Weeks in Treatment-Experienced Subjects With Chronic Genotype 1 HCV Infection
8) Phase III (Sofosbuvir + Ledipasvir) ION-3 study: (Clinical trial number: NCT01851330)
Title:A Phase 3, Multicenter, Randomized, Open-Label Study to Investigate the Efficacy and Safety of Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) +/- Ribavirin for 8 Weeks and Sofosbuvir/Ledipasvir Fixed-Dose Combination (FDC) for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1 HCV Infection
- ............................................
- 11DACLATASVIR
Daclatasvir
BMS-790052,EBP 883; BMS 790052cas no 1009119-64-5THERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES1. Carbamic acid, N,N'-[[1,1'-biphenyl]-4,4'-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C'-dimethyl ester2. dimethyl N,N'-(biphenyl-4,4'-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamateMOLECULAR FORMULA C40H50N8O6MOLECULAR WEIGHT 738.9SPONSOR Bristol-Myers SquibbCODE DESIGNATION BMS-790052CAS REGISTRY NUMBER 1009119-64-5nmrDaclatasvir dihydrochlorideTHERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES1. Carbamic acid, N,N'-[[1,1'-biphenyl]-4,4'-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C'-dimethyl ester,hydrochloride (1:2)2. dimethyl N,N'-(biphenyl-4,4'-diylbis{1H-imidazole-5,2-diyl-[(2S)-pyrrolidine-2,1-diyl][(1S)-1-(1-methylethyl)-2-oxoethane-2,1-diyl]})dicarbamate dihydrochlorideMOLECULAR FORMULA C40H50N8O6 . 2 HClMOLECULAR WEIGHT 811.8SPONSOR Bristol-Myers SquibbCODE DESIGNATION BMS-790052-05CAS REGISTRY NUMBER 1009119-65-6Hepatitis C virus (HCV) is a major global health problem, with an estimated 150-200 million people infected worldwide, including at least 5 million in Europe (Pawlotsky, Trends Microbiol, 2004, 12: 96-102). According to the World Health Organization, 3 to 4 million new infections occur each year. The infection is often asymptomatic; however, the majority of HCV-infected individuals develop chronic infection (Hoof agle, Hepatology, 2002, 36: S21-S29; Lauer et al, N. Engl. J. Med., 2001, 345: 41-52; Seeff, Semin. Gastrointest., 1995, 6: 20-27). Chronic infection frequently results in serious liver disease, including fibrosis and steatosis (Chisari, Nature, 2005, 435: 930-932). About 20% of patients with chronic HCV infection develop liver cirrhosis, which progresses to hepatocellular carcinoma in 5% of the cases (Hoofnagle, Hepatology, 2002, 36: S21-S29; Blonski et al, Clin. Liver Dis., 2008, 12: 661-674; Jacobson et al, Clin. Gastroenterol. Hepatol, 2010, 8: 924-933; Castello et al., Clin. Immunol, 2010, 134: 237-250; McGivern et al., Oncogene, 2011, 30: 1969-1983).Chronic HCV infection is the leading indication for liver transplantations (Seeff et al., Hepatology, 2002, 36: 1-2). Unfortunately, liver transplantation is not a cure for hepatitis C; viral recurrence being an invariable problem and the leading cause of graft loss (Brown, Nature, 2005, 436: 973-978; Watt et al, Am. J. Transplant, 2009, 9: 1707-1713). No vaccine protecting against HCV is yet available. Current therapies include administration of ribavirin and/or interferon-alpha (IFN-Cc), two non-specific anti-viral agents. Using a combination treatment of pegylated IFN-CC and ribavirin, persistent clearance is achieved in about 50% of patients with genotype 1 chronic hepatitis C. However, a large number of patients have contraindications to one of the components of the combination; cannot tolerate the treatment; do not respond to interferon therapy at all; or experience a relapse when administration is stopped. In addition to limited efficacy and substantial side effects such as neutropenia, haemo lytic anemia and severe depression, current antiviral therapies are also characterized by high cost. To improve efficacy of standard of care (SOC), a large number of direct acting antivirals (DAAs) targeting viral polyprotein processing and replication have been developed (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol., 2011, 8: 257-264). These include small molecule compounds targeting HCV nonstructural proteins including the HCV protease, polymerase and NS5A protein. Although a marked improvement of antiviral response was observed when protease inhibitors were combined with SOC (Hofmann et al, Nat. Rev; Gastroenterol. Hepatol, 2011, 8: 257-264; Bacon et al, New Engl. J. Med., 2011, 364: 1207-1217; McHutchison et al, New Engl. J. Med., 2010, 362: 1292-1303; Poordad et al, New Engl. J. Med., 201 1, 364: 1195-1206; Hezode et al, New Engl. J. Med., 2009, 360: 1839-1850; Kwo et al, Lancet, 2010, 376: 705-716), toxicity of the individual compounds and rapid development of viral resistance in a substantial fraction of patients remain major challenges (Pawlotsky, Hepatology, 2011, 53: 1742-1751; Pereira et al, Nat. Rev. Gastroenterol. Hepatol., 2009, 6: 403-411; Sarrazin et al, Gastroenterol., 2010, 138: 447-462). New therapeutic approaches against HCV are therefore still needed. HCV entry into target cells is a promising target for antiviral preventive and therapeutic strategies since it is essential for initiation, spread, and maintenance of infection (Timpe et al, Gut, 2008, 57: 1728-1737; Zeisel et al, Hepatology, 2008, 48: 299-307). Indeed, HCV initiates infection by attaching to molecules or receptors on the surface of hepatocytes. Current evidence suggests that HCV entry is a multistep process involving several host factors including heparan sulfate (Barth et al, J. Biol. Chem., 2003, 278: 41003-41012), the tetraspanin CD81 (Pileri et al, Science, 1998, 282: 938-941), the scavenger receptor class B type I (SR-BI) (Zeisel et al, Hepatology, 2007, 46: 1722-1731; Bartosch et al, J. Exp. Med., 2003, 197: 633-642; Grove et al, J. Virol, 2007, 81 : 3162-3169; Kapadia et al, J. Virol, 2007, 81 : 374- 383; Scarselli et al, EMBO J., 2002, 21 : 5017-5025), Occludin (Ploss et al, Nature, 2009, 457: 882-886) and Claudin-1 (CLDN1), an integral membrane protein and a component of tight-junction strands (Evans et al, Nature, 2007, 446: 801-805). Furthermore, Niemann-Pick CI -like cholesterol absorption receptor has been identified as a new hepatitis C virus entry factor (Sainz et al, Nature Medicine, 2012, 18: 281-285).Daclatasvir (USAN[1]) (formerly BMS-790052) is an experimental drug candidate for the treatment of hepatitis C. It is being developed by Bristol-Myers Squibb.Daclatasvir's mechanism of action involves inhibition of the HCV nonstructural protein NS5A.[2][3] Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA.[4]Daclatasvir has been tested in combination regimens with pegylated interferon and ribavirin,[5] as well as with other direct-acting antiviral agents including asunaprevir[6][7][8][9] and sofosbuvir.[10] Daclatasvir (BMS-790052; EBP 883) is a first-in-class, highly-selective oral HCV NS5A inhibitor. NS5A is an essential component for hepatitis C virus (HCV) replication complex.Daclatasvir (BMS-790052; EBP 883)has broad genotype coverage and exhibits picomolar in vitro potency against genotypes 1a (EC50 50pm) and 1b (EC50 9pm).Daclatasvir (BMS-790052; EBP 883) produces a robust decline in HCV RNA (-3.6 logs after 48 hours from a single 100 mg) dosefollowing a single dose in patients chronically infected with HCV genotype 1.It may be many years before the symptoms of hepatitis C infection appear. However, once they do, the consequences are significant: patients may have developed fibrosis, cirrhosis or even liver cancer, with the end result being liver failure. Even if diagnosed early, there’s no guarantee of a cure. Only around half of patients respond to the standard therapy of an interferon plus the antiviral drug ribavirin, and while two add-on antiviral therapies were approved in 2011, the treatment period is long with no guarantee of a cure, and for non-responders treatment options remain limited.A new drug with a different mechanism is being developed by Bristol-Myers Squibb, in conjunction with Pharmasset. Daclatasvir targets non-structural protein 5A, which is an important component of the viral replication process, although its precise role in this remains unclear. The drug is active in single oral doses, and may have potential as part of a treatment regimen that avoids the use of interferon, and in patients who do not respond to standard therapy.In an open label Phase IIa study, 10 patients with chronic hepatitis C genotype 1b infection who did not respond to standard therapy were given daclatasvir in once daily 60mg doses, plus another experimental drug, BMS-790052, which is an NSP 3 protease inhibitor, in initial twice-daily 600mg doses, later reduced to 200mg twice a day.2 Nine patients completed 24 weeks of treatment, with the 10th discontinuing after 10 weeks. In those who completed the course, HCV RNA was undetectable at week 8, and remained so until the end of the trial, with all achieving a sustained virologic response. It was also undetectable post-treatment in the patient who discontinued.Daclatasvir has also been investigated as monotherapy in a double blind, placebo-controlled, sequential panel, multiple ascending dose study.3 Thirty patients with chronic geno-type 1 hepatitis C infection were randomised to receive a 14 day course of the drug, in once daily doses of 1, 10, 30, 60 or 100mg, 30mg twice a day, or placebo. There was no evidence of antiviral activity in the placebo group, but the mean maximum decline of 2.8 to 4.1 log IU/ml. Most experienced viral rebound on or before day 7 of treatment, which was associated with viral variants that had previously been implicated in resistance development. It was well tolerated in all dose groups.M. Gao et al. Nature 2010, 465, 96
Daclatasvir (BMS-790052; EBP 883) is a first-in-class, highly-selective oral HCV NS5A inhibitor. NS5A is an essential component for hepatitis C virus (HCV) replication complex.Daclatasvir (BMS-790052; EBP 883)has broad genotype coverage and exhibits picomolar in vitro potency against genotypes 1a (EC50 50pm) and 1b (EC50 9pm).Daclatasvir (BMS-790052; EBP 883) produces a robust decline in HCV RNA (-3.6 logs after 48 hours from a single 100 mg) dosefollowing a single dose in patients chronically infected with HCV genotype 1.It may be many years before the symptoms of hepatitis C infection appear. However, once they do, the consequences are significant: patients may have developed fibrosis, cirrhosis or even liver cancer, with the end result being liver failure. Even if diagnosed early, there’s no guarantee of a cure. Only around half of patients respond to the standard therapy of an interferon plus the antiviral drug ribavirin, and while two add-on antiviral therapies were approved in 2011, the treatment period is long with no guarantee of a cure, and for non-responders treatment options remain limited.A new drug with a different mechanism is being developed by Bristol-Myers Squibb, in conjunction with Pharmasset. Daclatasvir targets non-structural protein 5A, which is an important component of the viral replication process, although its precise role in this remains unclear. The drug is active in single oral doses, and may have potential as part of a treatment regimen that avoids the use of interferon, and in patients who do not respond to standard therapy.In an open label Phase IIa study, 10 patients with chronic hepatitis C genotype 1b infection who did not respond to standard therapy were given daclatasvir in once daily 60mg doses, plus another experimental drug, BMS-790052, which is an NSP 3 protease inhibitor, in initial twice-daily 600mg doses, later reduced to 200mg twice a day.2 Nine patients completed 24 weeks of treatment, with the 10th discontinuing after 10 weeks. In those who completed the course, HCV RNA was undetectable at week 8, and remained so until the end of the trial, with all achieving a sustained virologic response. It was also undetectable post-treatment in the patient who discontinued.Daclatasvir has also been investigated as monotherapy in a double blind, placebo-controlled, sequential panel, multiple ascending dose study.3 Thirty patients with chronic geno-type 1 hepatitis C infection were randomised to receive a 14 day course of the drug, in once daily doses of 1, 10, 30, 60 or 100mg, 30mg twice a day, or placebo. There was no evidence of antiviral activity in the placebo group, but the mean maximum decline of 2.8 to 4.1 log IU/ml. Most experienced viral rebound on or before day 7 of treatment, which was associated with viral variants that had previously been implicated in resistance development. It was well tolerated in all dose groups.M. Gao et al. Nature 2010, 465, 96 22/11/2013
22/11/2013European Medicines Agency advises on compassionate use of daclatasvir
Opinion concerns use in combination with sofosbuvir in patients with chronic hepatitis C in urgent need of therapy to prevent progression of liver diseaseThe European Medicines Agency’s Committee for Medicinal Products for Human Use(CHMP) has given an opinion on the use of daclatasvir in combination with sofosbuvir in the treatment of chronic (long-term) hepatitis C virus (HCV) infection, in a compassionate-use programme.Compassionate-use programmes are set up at the level of individual Member States. They are intended to give patients with a life-threatening, long-lasting or seriously disabling disease with no available treatment options access to treatments that are still under development and that have not yet received amarketing authorisation. In this specific case, Sweden has requested an opinion from the CHMP on the conditions under which early access through compassionate use could be given to daclatasvir, for the use in combination with sofosbuvir, with or without ribavirin, for a specific patient population.The recommended compassionate use is intended for adult patients at a high risk of their liver being no longer able to function normally (decompensation) or death within 12 months if left untreated, and who have a genotype 1 infection. Further, it is recognised that the potential benefit of such combination therapy may extend to patients infected with other HCV genotypes.Daclatasvir and sofosbuvir are both first-in-class anti-viral medicines against HCV. These medicines have been studied in combination, with or without ribavirin, in aclinical trial which included treatment-naive (previously untreated) HCV genotype-1, -2 and -3 infected patients, as well as patients with genotype 1 infection who have previously failed telaprevir or boceprevir treatment. Results from the trial indicate high efficacy, also in those who have failed treatment with these protease inhibitors. Many such patients have very advanced liver disease and are in urgent need of effective therapy in order to cease the progression of liver injury.This is the second opinion provided by the CHMP on compassionate use of medicines in development for the treatment of hepatitis C. Overall, it isthe fourth time compassionate use has been assessed by the CHMP.The aim of the CHMP assessment and opinion on a compassionate-use programme for new medicinal products is to ensure a common approach, whenever possible, regarding the criteria and conditions of use under Member States' legislation. The opinion provides recommendations to the EU Member States that are considering setting up such a programme, and its implementation is not mandatory. In addition to describing which patients may benefit from the medicine, it explains how to use it and gives information on safety.The assessment report and conditions of use of daclatasvir in combination with sofosbuvir with or without ribavirin in this setting will be published shortly on the Agency's website.Notes- The first compassionate-use opinion for a hepatitis C treatment was adopted by the CHMP in October 2013.
- Sofosbuvir, which is part of this compassionate-use opinion, received a positive opinion from the CHMP recommending granting of a marketing authorisation at its November 2013 meeting.
- Daclatasvir is developed by Bristol-Myers Squibb and sofosbuvir is developed by Gilead.
References
- ^ Statement on a Nonproprietary Name Adopted by the USAN Council
- ^ Gao, Min; Nettles, Richard E.; Belema, Makonen; Snyder, Lawrence B.; Nguyen, Van N.; Fridell, Robert A.; Serrano-Wu, Michael H.; Langley, David R.; Sun, Jin-Hua; O'Boyle, Donald R., II; Lemm, Julie A.; Wang, Chunfu; Knipe, Jay O.; Chien, Caly; Colonno, Richard J.; Grasela, Dennis M.; Meanwell, Nicholas A.; Hamann, Lawrence G. (2010). "Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect". Nature 465(7294): 96–100. doi:10.1038/nature08960. PMID 20410884.
- ^ Bell, Thomas W. (2010). "Drugs for hepatitis C: unlocking a new mechanism of action". ChemMedChem 5 (10): 1663–1665.doi:10.1002/cmdc.201000334. PMID 20821796.
- ^ Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Guedj, J et al. Proceedings of the National Academy of Sciences. February 19, 2013.
- ^ AASLD: Daclatasvir with Pegylated Interferon/Ribavirin Produces High Rates of HCV Suppression. Highleyman, L. HIVandHepatitis.com. 6 December 2011.
- ^ Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- ^ "Bristol-Myers' Daclatasvir, Asunaprevir Cured 77%: Study". Bloomberg. Apr 19, 2012.
- ^ AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- ^ High rate of response to BMS HCV drugs in harder-to-treat patients – but interferon-free prospects differ by sub-genotype. Alcorn, K. Aidsmap.com. 12 November 2012.
- ^ AASLD 2012: Sofosbuvir + Daclatasvir Dual Regimen Cures Most Patients with HCV Genotypes 1, 2, or 3. Highleyman, L. HIVandHepatitis.com. 15 November 2012.
https://www.google.co.in/patents/US20090041716?pg=PA1&dq=us+2009041716&hl=en&sa=X&ei=3ki4Uo-jEsTirAfzwoHQBQ&ved=0CD4Q6AEwAQ EXAMPLES  A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C.
A 1 L, 3-neck round bottom flask, fitted with a nitrogen line, overhead stirrer and thermocouple, was charged with 20 g (83.9 mmol, 1 equiv) 1,1′-(biphenyl-4,4′-diyl)diethanone, 200 mL CH2Cl2 and 8.7 mL (27.1 g, 169.3 mmol, 2.02 quiv) bromine. The mixture was allowed to stir under nitrogen for about 20 hours under ambient conditions. The resulting slurry was charged with 200 mL CH2Cl2 and concentrated down to about 150 mL via vacuum distillation. The slurry was then solvent exchanged into THF to a target volume of 200 mL via vacuum distillation. The slurry was cooled to 20-25° C. over 1 hour and allowed to stir at 20-25° C. for an additional hour. The off-white crystalline solids were filtered and washed with 150 mL CH2Cl2. The product was dried under vacuum at 60° C. to yield 27.4 g (69.2 mmol, 82%) of the desired product: 1H NMR (400 MHz, CDCl3) δ 7.95-7.85 (m, 4H), 7.60-7.50 (m, 4H), 4.26 (s, 4H); 13C NMR (100 MHz, CDCl3) 6 191.0, 145.1, 133.8, 129.9, 127.9, 30.8; IR (KBr, cm−1) 3007, 2950, 1691, 1599, 1199; Anal calcd for C16H12Br2O2: C, 48.52; H, 3.05; Br, 40.34. Found: C, 48.53; H, 3.03; Br, 40.53 HRMS calcd for C16H13Br2O2 (M+H; DCI+): 394.9282. Found: 394.9292. mp 224-226° C. A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile.
A 500 mL jacketed flask, fitted with a nitrogen line, thermocouple and overhead stirrer, was charged with 20 g (50.5 mmol, 1 equiv) of Compound 2, 22.8 g (105.9 moles, 2.10 equiv) 1-(tert-butoxycarbonyl)-L-proline and 200 mL acetonitrile. The slurry was cooled to 20° C. followed by the addition of 18.2 mL (13.5 g, 104.4 mmol, 2.07 equiv) DIPEA. The slurry was warmed to 25° C. and allowed to stir for 3 hours. The resulting clear, organic solution was washed with 3×100 mL 13 wt % aqueous NaCl. The rich acetonitrile solution was solvent exchanged into toluene (target volume=215 mL) by vacuum distillation until there was less than 0.5 vol % acetonitrile. The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed).
The toluene solution of Compound 3 was charged with 78 g (1.011 moles, 20 equiv) ammonium acetate and heated to 95-100° C. The mixture was allowed to stir at 95-100° C. for 15 hours. After reaction completion, the mixture was cooled to 70-80° C. and charged with 7 mL acetic acid, 40 mL n-butanol, and 80 mL of 5 vol % aqueous acetic acid. The resulting biphasic solution was split while maintaining a temperature >50° C. The rich organic phase was charged with 80 mL of 5 vol % aqueous acetic acid, 30 mL acetic acid and 20 mL n-butanol while maintaining a temperature >50° C. The resulting biphasic solution was split while maintaining a temperature >50° C. and the rich organic phase was washed with an additional 80 mL of 5 vol % aqueous acetic acid. The rich organic phase was then solvent exchanged into toluene to a target volume of 215 mL by vacuum distillation. While maintaining a temperature >60° C., 64 mL methanol was charged. The resulting slurry was heated to 70-75° C. and aged for 1 hour. The slurry was cooled to 20-25° C. over 1 hour and aged at that temperature for an additional hour. The slurry was filtered and the cake was washed with 200 mL 10:3 toluene:methanol. The product was dried under vacuum at 70° C., resulting in 19.8 g (31.7 mmol, 63%) of the desired product: 1H NMR (400 MHz, DMSO-d6) δ 13.00-11.00 (s, 2H), 7.90-7.75 (m, 4H), 7.75-7.60 (m, 4H), 7.60-7.30 (s, 2H), 4.92-4.72 (m, 2H), 3.65-3.49 (m, 2H), 3.49-3.28 (m, 2H), 2.39-2.1 (m, 2H), 2.10-1.87 (m, 6H), 1.60-1.33 (s, 8H), 1.33-1.07 (s, 10H); 13C NMR (100 MHz, DMSO-d6) δ 154.1, 153.8, 137.5, 126.6, 125.0, 78.9, 78.5, 55.6, 55.0, 47.0, 46.7, 33.7, 32.2, 28.5, 28.2, 24.2, 23.5; IR (KBr, cm−1) 2975, 2876, 1663, 1407, 1156, 1125; HRMS calcd for C36H45N6O4 (M+H; ESI+): 625.3502. Found: 625.3502. mp 190-195° C. (decomposed). To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.
To a 250 mL reactor equipped with a nitrogen line and overhead stirrer, 25.0 g of Compound 4 (40.01 mmol, 1 equiv) was charged followed by 250 mL methanol and 32.85 mL (400.1 mmol, 10 equiv) 6M aqueous HCl. The temperature was increased to 50° C. and agitated at 50° C. for 5 hours. The resulting slurry was cooled to 20-25° C. and held with agitation for about 18 hours. Filtration of the slurry afforded a solid which was washed successively with 100 mL 90% methanol/water (V/V) and 2×100 mL of methanol. The wet cake was dried in a vacuum oven at 50° C. overnight to give 18.12 g (31.8 mmol, 79.4%) of the desired product.Recrystallization of Compound 5 To a 250 mL reactor equipped with a nitrogen line and an overhead stirrer, 17.8 g of Compound 5 from above was charged followed by 72 mL methanol. The resulting slurry was agitated at 50° C. for 4 hours, cooled to 20-25° C. and held with agitation at 20-25° C. for 1 hour. Filtration of the slurry afforded a crystalline solid which was washed with 60 mL methanol. The resulting wet cake was dried in a vacuum oven at 50° C. for 4 days to yield 14.7 g (25.7 mmol, 82.6%) of the purified product: 1H NMR (400 MHz, DMSO-d6) δ 10.5-10.25 (br, 2H), 10.1-9.75 (br, 2H), 8.19 (s, 2H), 7.05 (d, J=8.4, 4H), 7.92 (d, J=8.5, 4H), 5.06 (m, 2H), 3.5-3.35 (m, 4H), 2.6-2.3 (m, 4H), 2.25-2.15 (m, 2H), 2.18-1.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 156.6, 142.5, 139.3, 128.1, 127.5, 126.1, 116.9, 53.2, 45.8, 29.8, 24.3; IR (KBr, cm−1) 3429, 2627, 1636, 1567, 1493, 1428, 1028. Anal calcd for C26H32N6Cl4: C, 54.75; H, 5.65; Cl, 24.86; Adjusted for 1.9% water: C, 53.71; H, 5.76; N, 14.46; Cl, 24.39. Found: C, 53.74; H, 5.72; N, 14.50; Cl, 24.49; KF=1.9. mp 240° C. (decomposed). A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product.
A 1 L jacketed flask equipped with a nitrogen line and an overhead stirrer was sequentially charged with 100 mL acetonitrile, 13.69 g (89.4 mmol, 2.5 equiv) hydroxybenzotriazole hydrate, 15.07 g (86 mmol, 2.4 equiv) N-(methoxycarbonyl)-L-valine, 16.46 g (85.9 mmol, 2.4 equiv) 1-(3-dimethyaminopropyl)-3-ethylcarbodiimide hydrochloride and an additional 100 mL acetonitrile. The resulting solution was agitated at 20° C. for 1 hour and charged with 20.4 g (35.8 mmol, 1 equiv) of purified Compound 5. The slurry was cooled to about 0° C. and 18.47 g (142.9 mmol, 4 equiv) diisopropylethylamine was added over 30 minutes while maintaining a temperature below 10° C. The solution was slowly heated to 15° C. over 3 hours and held at 15° C. for 12 hours. The resulting solution was charged with 120 mL 13 wt % aqueous NaCl and heated to 50° C. for 1 hour. After cooling to 20° C., 100 mL of isopropyl acetate was added. The biphasic solution was filtered through a 0.45 μm filter and the mixture split. The rich organic phase was washed with 2×240 mL of a 0.5 N NaOH solution containing 13 wt % NaCl followed by 120 mL 13 wt % aqueous NaCl. The mixture was then solvent exchanged into isopropyl acetate by vacuum distillation with a target volume of 400 mL. The resulting hazy solution was cooled to 20° C. and filtered through a 0.45 μm filter. The clear solution was then solvent exchanged into ethanol by vacuum distillation with a target volume of 140 mL. While maintaining a temperature of 50° C., 66.4 mL (82.3 mmol, 2.3 equiv) of 1.24M HCl in ethanol was added. The mixture was then charged with 33 mg (0.04 mmol, 0.001 equiv) of seed crystals of Compound (I) (see preparation below) and the resulting slurry was stirred at 50° C. for 3 hours. The mixture was cooled to 20° C. over 1 hour and aged at that temperature for an additional 22 hours. The slurry was filtered and the wet cake was washed with 100 mL of 2:1 acetone:ethanol. The solids were dried in a vacuum oven at 70° C. to give 22.15 g (27.3 mmol, 76.3%) of the desired product. A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I): 1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
A solution of Compound (I) was prepared by dissolving 3.17 g of Compound (I) from above in 22 mL methanol. The solution was passed through a 47 mm Cuno Zeta Carbon® 53SP filter at ˜5 psig at a flow rate of ˜58 mL/min. The carbon filter was rinsed with 32 mL of methanol. The solution was concentrated down to 16 mL by vacuum distillation. While maintaining a temperature of 40-50° C., 15.9 mL acetone and 5 mg of seed crystals of Compound (I) (see procedure below) were added. The resulting slurry was then charged with 32 mL acetone over 30 minutes. The slurry was held at 50° C. for 2 hours, cooled to 20° C. over about 1 hour and held at 20° C. for about 20 hours. The solids were filtered, washed with 16 mL 2:1 acetone:methanol and dried in a vacuum oven at 60° C. to give 2.14 g (67.5%) of purified Compound (I): 1H NMR (400 MHz, DMSO-d6, 80° C.): 8.02 (d, J=8.34 Hz, 4 H), 7.97 (s, 2 H), 7.86 (d, J=8.34 Hz, 4 H), 6.75 (s, 2 H), 5.27 (t, J=6.44 Hz, 2 H), 4.17 (t, J=6.95 Hz, 2 H), 3.97-4.11 (m, 2 H), 3.74-3.90 (m, 2 H), 3.57 (s, 6 H), 2.32-2.46 (m, 2 H), 2.09-2.31 (m, 6 H), 1.91-2.07 (m, 2 H), 0.88 (d, J=6.57 Hz, 6 H), 0.79 (d, J=6.32 Hz, 6 H); 13C NMR (75 MHz, DMSO-d6): δ 170.9, 156.9, 149.3, 139.1, 131.7, 127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; IR (neat, cm−1): 3385, 2971, 2873, 2669, 1731, 1650. Anal. Calcd for C40H52N8O6Cl2: C, 59.18; H, 6.45; N, 13.80; Cl, 8.73. Found C, 59.98; H, 6.80; N, 13.68; Cl, 8.77. mp 267° C. (decomposed).
- https://www.google.co.in/patents/US20090041716?pg=PA1&dq=us+2009041716&hl=en&sa=X&ei=3ki4Uo-jEsTirAfzwoHQBQ&ved=0CD4Q6AEwAQ
- The chemical structures of some of these HCV inhibitors as reported by numerous sources are provided below:























- [0152]BMS-791325 preferably is
As used herein, BMS-791325 may also be
See also publications at http://wwwl.easl.eu/eas12011/program/Posters/Abstract680.htm; and http://clinicaltrials.gov/show/NCT00664625. For GS-5885, see publications at http://www.natap.org/2011/EASL/EASL_68.htm; http://wwwl.easl.eu/eas12011/program/Posters/Abstract1097.htm; and http://clinicaltrials.gov/ct2/show/NCT01353248.
Compound 1(
) or a pharmaceutically acceptable salt thereof. Compound 1 is also known as (2R,6S,13aS,14aR,16aS,Z)-N-(cyclopropylsulfonyl)-6-(5-methylpyrazine-2-carboxamido)-5,16-dioxo-2-(phenanthridin-6-yloxy)-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a-carboxamide. Compound 1 is a potent HCV protease inhibitor. The synthesis and formulation of Compound 1 are described in
U.S. Patent Application Publication No. 2010/0144608 ,U.S. Provisional Application Serial No. 61/339,964 filed on March 10, 2010 U.S. Patent Application Publication No. 2011/0312973 filed on March 8, 2011 Table 1 belows lists additional suitable Hepatitis C therapeutic agents that may be used in combinationTable 1
 ALS-2158 Vertex Nl I n/a
ALS-2158 Vertex Nl I n/a
 ................................
................................- .........................................
- 12 DELEOBUVIR
 DELEOBUVIR(2E)-3-(2-{1-[2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-1-methyl-1H-indole-6-carboxamido]cyclobutyl}-1-methyl-1H-benzimidazol- 6-yl)prop-2-enoic acid1221574-24-8 CAS please check may be sodium salt??cas no as per below ref ……863884-77-9 (free acid)PHASE 3BI-207127NA
DELEOBUVIR(2E)-3-(2-{1-[2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-1-methyl-1H-indole-6-carboxamido]cyclobutyl}-1-methyl-1H-benzimidazol- 6-yl)prop-2-enoic acid1221574-24-8 CAS please check may be sodium salt??cas no as per below ref ……863884-77-9 (free acid)PHASE 3BI-207127NA
BI-207127 (free acid)BI-207127 is a novel HCV RNA polymerase inhibitor in phase III clinical development at Boehringer Ingelheim for the treatment of hepatitis C.Company Boehringer Ingelheim GmbH Description Oral non-structural protein 5B (NS5B) RNA-dependent polymerase inhibitor Molecular Target HCV NS5B polymerase Mechanism of Action Viral polymerase inhibitor Therapeutic Modality Small molecule Latest Stage of Development Phase III Indication Hepatitis C virus (HCV) Partner Deleobuvir (formerly BI 207127) is an experimental drug for the treatment of hepatitis C. It is being developed by Boehringer-Ingelheimand is currently in Phase II trials. It is a non-nucleoside hepatitis C virus NS5B polymerase inhibitor. Deleobuvir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including faldaprevir.Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of deleobuvir, faldaprevir, and ribavirin performed well in HCV genotype 1b patients.[1] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.Deleobuvir (BI 207127) is an investigational oral nonnucleoside inhibitor of hepatitis C virus (HCV) NS5B RNA polymerase. Antiviral activity, virology, pharmacokinetics, and safety were assessed in HCV genotype 1-infected patients receiving 5 days’ deleobuvir monotherapy. In this double-blind phase 1b study, treatment-naive (TN; n = 15) and treatment-experienced (TE; n = 45) patients without cirrhosis received placebo or deleobuvir at 100, 200, 400, 800, or 1,200 mg every 8 h (q8h) for 5 days. Patients with cirrhosis (n = 13) received deleobuvir at 400 or 600 mg q8h for 5 days. Virologic analyses included NS5B genotyping and phenotyping of individual isolates. At day 5, patients without cirrhosis had dose-dependent median HCV RNA reductions of up to 3.8 log10 (with no placebo response); patients with cirrhosis had median HCV RNA reductions of approximately 3.0 log10. Three patients discontinued due to adverse events (AEs). The most common AEs were gastrointestinal, nervous system, and skin/cutaneous tissue disorders. Plasma exposure of deleobuvir was supraproportional at doses ≥ 400 mg q8h and approximately 2-fold higher in patients with cirrhosis than in patients without cirrhosis. No virologic breakthrough was observed. NS5B substitutions associated with deleobuvir resistance in vitro were detected in 9/59 patients; seven encoded P495 substitutions, including P495L, which conferred 120- to 310-fold-decreased sensitivity to deleobuvir. P495 variants did not persist in follow-up without selective drug pressure. Deleobuvir monotherapy was generally well tolerated and demonstrated dose-dependent antiviral activity against HCV genotype 1 over 5 days.These results were confirmed in the SOUND-C3 study, presented at the 2013 APASL Liver Conference, which found that 16 week triple therapy with deleobuvir + faldaprevir + ribavirin gave 95% SVR12 in HCV genotype 1b patients but poor virological response in genotype 1a.[2]- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- S Zeuzem, J-F Dufour, M Buti, V Soriano, R Buynak, P Mantry, J Taunk, JO Stern, R Vinisko, J-P Gallivan, WO Bocher and FJ Mensa.“Interferon-free treatment with faldaprevir, deleobuvir (BI 207127) and ribavirin in SOUND-C3: 95% SVR12 in HCV GT-1b”. 23rd Conference of the Asian Pacific Association for the Study of the Liver (APASL) 6–9 June 2013. Retrieved 12 Sep 2013.
PATENTSWO 2013147750WO 2013147749 WO 2012041771WO 2012044520WO 2012016995WO 2005080388……………………………………………………PATENTPatent Filing date Publication date Applicant Title WO2010059667A1 Nov 18, 2009 May 27, 2010 Boehringer Ingelheim International Gmbh Pharmaceutical composition of a potent hcv inhibitor for oral administration WO2011005646A2 Jul 1, 2010 Jan 13, 2011 Boehringer Ingelheim International Gmbh Pharmaceutical composition for a hepatitis c viral protease inhibitor WO2012041771A1 * Sep 23, 2011 Apr 5, 2012 Boehringer Ingelheim International Gmbh Combination therapy for treating hcv infection US4211771 Feb 13, 1978 Jul 8, 1980 Robins Ronald K Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide US6063772 Jun 15, 1998 May 16, 2000 Icn Pharmaceuticals, Inc. Specific modulation of Th1/Th2 cytokine expression by ribavirin in activated T-lymphocytes US6277830 Jul 7, 1999 Aug 21, 2001 Schering Corporation 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon US6323180 Aug 5, 1999 Nov 27, 2001 Boehringer Ingelheim (Canada) Ltd Hepatitis C inhibitor tri-peptides US6403564 Oct 14, 1999 Jun 11, 2002 Schering Corporation Ribavirin-interferon alfa combination therapy for eradicating detectable HCV-RNA in patients having chronic hepatitis C infection US7141574 Jul 18, 2002 Nov 28, 2006 Boehringer Ingelheim (Canada) Ltd. Viral polymerase inhibitors US7514557 May 23, 2005 Apr 7, 2009 Boehringer Ingelheim International Gmbh Process for preparing acyclic HCV protease inhibitors US7582770 Feb 18, 2005 Sep 1, 2009 Boehringer Ingelheim International Gmbh Viral polymerase inhibitors US7585845 May 20, 2004 Sep 8, 2009 Boehringer Ingelheim International Gmbh Hepatitis C inhibitor compounds US7642352 Feb 10, 2006 Jan 5, 2010 Boehringer Ingelheim International Gmbh Process for preparing 2,3-disubstituted indoles US20090087409 Nov 26, 2008 Apr 2, 2009 Boehringer Ingelheim (Canada) Ltd. Viral Polymerase Inhibitors US20100068182 Sep 16, 2009 Mar 18, 2010 Boehringer Ingelheim International Gmbh Combination therapy for treating hcv infection US20100093792 Sep 15, 2009 Apr 15, 2010 Boehringer Ingelheim International Gmbh Crystalline forms of a potent hcv inhibitor * Cited by examiner
NON-PATENT CITATIONSRef 1 BALAGOPAL GASTROENTEROLOGY vol. 139, 2010, pages 1865 – 1876 2 BERG ET AL. HEPATOL vol. 52, no. S1, 2010, 3 * DOMINIQUE LARREY ET AL: “Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin“, JOURNAL OF HEPATOLOGY, vol. 57, no. 1, 7 March 2012 (2012-03-07), pages 39-46, XP55040240, ISSN: 0168-8278, DOI: 10.1016/j.jhep.2012.02.015 4 G. CAIRNS GENE VARIANT THAT HELPS HEPATITIS C TREATMENT MAY HINDER HIV TREATMENT, [Online] 2011, Retrieved from the Internet: <URL:http://www.bhiva.org/Ncws.aspx?NewsID=a7503829-94b9-4d2f-bd91-ld2fbaad6c8d> 5 GE ET AL. NATURE vol. 461, 2009, pages 399 – 401 6 GHANY; MARC ET AL.: ‘An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases‘ HEPATOLOGY vol. 54, no. 4, 2011, pages 1433 – 44 7 * LIZ HIGHLEYMAN: “AASLD: All-Oral Combination of BI 201335, BI 207127 and Ribavirin Shows Good Efficacy at 12 Weeks“, INTERNET CITATION, [Online] 1 December 2011 (2011-12-01), pages 1-3, XP002684260, Retrieved from the Internet: URL:www.hivandhepatitis.com/hepatitis-c/he patitis-c-topics/hcv-treatment/3371-aasld- all-oral-combination-of-bi-201335-bi-20712 7-and-ribavirin-shows-good-efficacy-at-12- weeks> [retrieved on 2012-09-27] 8 * POL S ET AL: “SVR AND PHARMACOKINETICS OF THE HCV PROTEASE INHIBITOR BI201335 WITH PEGIFN/RBV IN HCV GENOTYPE-1 PATIENTS WITH COMPENSATED LIVER CIRRHOSIS AND NON-RESPONSE TO PREVIOUS PEGIFN/RBV“, JOURNAL OF HEPATOLOGY, vol. 54, no. Suppl. 1, March 2011 (2011-03), page S486, XP55038942, & 46TH ANNUAL MEETING OF THE EUROPEAN-ASSOCIATION-FOR-THE-STUDY-OF-THE- LIVER (EASL); BERLIN, GERMANY; MARCH 30 -APRIL 03, 2011 ISSN: 0168-8278 9 S. M. BIRGE ET AL. J. PHARM. SCI. vol. 66, 1977, pages 1 – 19 10 * STEFAN ZEUZEM ET AL: “Efficacy of the Protease Inhibitor BI 201335, Polymerase Inhibitor BI 207127, and Ribavirin in Patients With Chronic HCV Infection“, GASTROENTEROLOGY, ELSEVIER, PHILADELPHIA, PA, vol. 141, no. 6, 1 December 2011 (2011-12-01), pages 2047-2055, XP002664706, ISSN: 0016-5085, DOI: 10.1053/J.GASTRO.2011.08.051 11 SULKOWSKI MS ET AL. HEPATOL vol. 50, 2009, page 2A 12 SULKOWSKI MS ET AL. J HEPATOL vol. 52, no. 1, 2010, pages S462 – S463 13 WHITE PW ET AL. ANTIMICROB AGENTS CHEMOTHER vol. 54, no. 11, 2010, pages 4611 – 4618 14 WHO COLLABORATIVE STUDY GROUP. VOX SANG vol. 76, 1999, pages 149 – 158 15 * ZEUZEM STEFAN ET AL: “STRONG ANTIVIRAL ACTIVITY AND SAFETY OF IFN-SPARING TREATMENT WITH THE PROTEASE INHIBITOR BI 201335, THE HCV POLYMERASE INHIBITOR BI 207127 AND RIBAVIRIN IN PATIENTS WITH CHRONIC HEPATITIS C“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 52, no. Suppl, 1 October 2010 (2010-10-01), pages 876A-877A, XP009154421, ISSN: 0270-9139 16 * ZEUZEM STEFAN ET AL: “VIROLOGIC RESPONSE TO AN INTERFERON-FREE REGIMEN OF BI201335 AND BI207127, WITH AND WITHOUT RIBAVIRIN, IN TREATMENT-NAIVE PATIENTS WITH CHRONIC GENOTYPE-1 HCV INFECTION: WEEK 12 INTERIM RESULTS OF THE SOUND-C2 STUDY“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 54, no. Suppl. 1, 1 November 2011 (2011-11-01), page 1436A, XP009163087, ISSN: 0270-9139, DOI: 10.1002/HEP.24666 [retrieved on 2011-09-30] …………………………………………………………The following…… having the chemical name: (E)-3-[2-(l-{ [2-(5-Bromo-pyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carbonyl]-amino}-cyclobutyl)-3-methyl-3H-benzimidazol-5-yl]- acrylic acid, is known as a selective and potent inhibitor of the HCV NS5B RNA- dependent RNA polymerase and useful in the treatment of HCV infection. Compound (2) falls within the scope of HCV inhibitors disclosed in U.S. Patents 7,141,574 and7,582,770, and US Application Publication 2009/0087409. Compound (2) is disclosed specifically as Compound # 3085 in U.S. Patent 7,582,770. Compound (2), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (2) include the crystalline forms, in particular the crystalline sodium salt form which is prepared as herein described.It is known in the art that particular HCV subtypes and patient subgenotypes may respond differently to HCV therapy. HCV Genotype la is traditionally more difficult to treat and are less responsive to antiviral therapy than Genotype lb. See, e.g., Ghany, Marc et al. “An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases”, Case No.: 09-0592-PCTHepatology, 54(4): 1433-44 (2011)). In addition, and particularly with interferon-based therapy, specific single nucleotide polymorphisms (SNPs) located on the long arm of chromosome 19 within the gene cluster of IL-28B (Interleukin (IL) 28B, (also called lambda interferon), of the patient undergoing therapy can directly effect theresponsiveness of that patient to the antiviral therapy. In particular, patients having a non- CC genotype of SNP rsl2979860 or a non-TT genotype of rs 8099917 are traditionally more difficult to treat and are less responsive in terms of a sustained virological response (SVR) than patients having the CC or TT genotype.. The SNP that was most strongly associated with SVR in the genome-wide analysis was rs 12979860 followed by rs 8099917. See, e.g., Ge et al., Nature, 461 :399-401 (2009) and Balagopal,Gastroenterology, 139: 1865-1876 (2010). See G. Cairns, “Gene variant that helps hepatitis C treatment may hinder HIV treatment”, 2011, at:http://www.bhiva.org^ Thus, there is a need in the art for therapies that are effective against even the more difficult-to-treat patient subpopulations, particularly those exhibiting HCV subtype la and the non-CC IL28B subgenotype, as well as those exhibiting compensated liver disease.ExamplesI. Methods for Preparing Compound (1)Methods for preparing amorphous Compound (1) and a general description ofpharmaceutically acceptable salt forms can be found in US Patents 6,323,180, 7,514,557 and 7,585,845. Methods for preparing additional forms of Compound (1), in particular the crystalline sodium salt form, can be found in U.S. Patent Application Publication No. 2010/0093792.II. Formulations of Compound (1) Case No. : 09-0592-PCTOne example of a pharmaceutical formulation of Compound (1) include an oral solution formulation as disclosed in WO 2010/059667. Additional examples include capsules containing a lipid-based liquid formulation, as disclosed in WO 201 1/005646. III. Methods for Preparing Compound (2)Methods for preparing amorphous Compound (2) can be found in U.S. Patents 7, 141 ,574 and 7,582,770, and US Application Publication 2009/0087409.The following Example provides the method for preparing an additional form ofCompound (2), the sodium salt form, that may be used in the present invention.Example 1 – Preparation of Compound (2) Sodium SaltStep 1. Synthesis of Isopropyl 3-Cyclopentyl-l-methyl-lH-indole-6-carboxylate
having the chemical name: (E)-3-[2-(l-{ [2-(5-Bromo-pyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carbonyl]-amino}-cyclobutyl)-3-methyl-3H-benzimidazol-5-yl]- acrylic acid, is known as a selective and potent inhibitor of the HCV NS5B RNA- dependent RNA polymerase and useful in the treatment of HCV infection. Compound (2) falls within the scope of HCV inhibitors disclosed in U.S. Patents 7,141,574 and7,582,770, and US Application Publication 2009/0087409. Compound (2) is disclosed specifically as Compound # 3085 in U.S. Patent 7,582,770. Compound (2), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (2) include the crystalline forms, in particular the crystalline sodium salt form which is prepared as herein described.It is known in the art that particular HCV subtypes and patient subgenotypes may respond differently to HCV therapy. HCV Genotype la is traditionally more difficult to treat and are less responsive to antiviral therapy than Genotype lb. See, e.g., Ghany, Marc et al. “An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases”, Case No.: 09-0592-PCTHepatology, 54(4): 1433-44 (2011)). In addition, and particularly with interferon-based therapy, specific single nucleotide polymorphisms (SNPs) located on the long arm of chromosome 19 within the gene cluster of IL-28B (Interleukin (IL) 28B, (also called lambda interferon), of the patient undergoing therapy can directly effect theresponsiveness of that patient to the antiviral therapy. In particular, patients having a non- CC genotype of SNP rsl2979860 or a non-TT genotype of rs 8099917 are traditionally more difficult to treat and are less responsive in terms of a sustained virological response (SVR) than patients having the CC or TT genotype.. The SNP that was most strongly associated with SVR in the genome-wide analysis was rs 12979860 followed by rs 8099917. See, e.g., Ge et al., Nature, 461 :399-401 (2009) and Balagopal,Gastroenterology, 139: 1865-1876 (2010). See G. Cairns, “Gene variant that helps hepatitis C treatment may hinder HIV treatment”, 2011, at:http://www.bhiva.org^ Thus, there is a need in the art for therapies that are effective against even the more difficult-to-treat patient subpopulations, particularly those exhibiting HCV subtype la and the non-CC IL28B subgenotype, as well as those exhibiting compensated liver disease.ExamplesI. Methods for Preparing Compound (1)Methods for preparing amorphous Compound (1) and a general description ofpharmaceutically acceptable salt forms can be found in US Patents 6,323,180, 7,514,557 and 7,585,845. Methods for preparing additional forms of Compound (1), in particular the crystalline sodium salt form, can be found in U.S. Patent Application Publication No. 2010/0093792.II. Formulations of Compound (1) Case No. : 09-0592-PCTOne example of a pharmaceutical formulation of Compound (1) include an oral solution formulation as disclosed in WO 2010/059667. Additional examples include capsules containing a lipid-based liquid formulation, as disclosed in WO 201 1/005646. III. Methods for Preparing Compound (2)Methods for preparing amorphous Compound (2) can be found in U.S. Patents 7, 141 ,574 and 7,582,770, and US Application Publication 2009/0087409.The following Example provides the method for preparing an additional form ofCompound (2), the sodium salt form, that may be used in the present invention.Example 1 – Preparation of Compound (2) Sodium SaltStep 1. Synthesis of Isopropyl 3-Cyclopentyl-l-methyl-lH-indole-6-carboxylate Because of the instability of brominated product, methyl 3 -cyclopentyl- 1 -methyl- 1Η- indole-6-carboxylate needed to be converted into the more stable isopropyl 3-cyclopentyl- l-methyl- lH-indole-6-carboxylate via a simple and high yielding operation. The conversion worked the best with stoichiometric amounts of solid lithium isopropoxide. Use of 0.1 eq lithium isopropoxide led to longer reaction times and as a result to more hydrolysis by-product, while lithium isopropoxide solution in THF caused a problematic isolation and required distillation of THF.Procedure: Case No.: 09-0592-PCTThe mixture of methyl 3 -cyclopentyl- 1 -methyl- lH-indole-6-carboxylate (50.0 g, 0.194 mol) and lithium isopropoxide (16.2 g, 95%, 0.233 mol) in 2-propanol was stirred at 65+5 °C for at least 30 min for complete trans-esterification. The batch was cooled to 40+5 °C and water (600 g) was added at a rate to maintain the batch temperature at 40+5°C. After addition, the mixture was cooled to 20-25 °C over 2+0.5 h and held at 20-25 °C for at least 1 h. The batch was filtered and rinsed with 28 wt% 2-propanol in water (186 g), and water (500 g). The wet cake was dried in vacuo (< 200 Torr) at 40-45 °C until the water content was < 0.5% to give isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (52.7 g, 95% yield) in 99.2 A% (240 nm).The starting material methyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate can be prepared as described in Example 12 of U.S. Patent 7,141,574, and in Example 12 of U.S. Patent 7,642,352, both herein incorporated by reference.Step 2. Synthesis of Isopropyl 2-Bromo-3-cyclopentyl-l-methyl-lH-indole-6- carboxylate
Because of the instability of brominated product, methyl 3 -cyclopentyl- 1 -methyl- 1Η- indole-6-carboxylate needed to be converted into the more stable isopropyl 3-cyclopentyl- l-methyl- lH-indole-6-carboxylate via a simple and high yielding operation. The conversion worked the best with stoichiometric amounts of solid lithium isopropoxide. Use of 0.1 eq lithium isopropoxide led to longer reaction times and as a result to more hydrolysis by-product, while lithium isopropoxide solution in THF caused a problematic isolation and required distillation of THF.Procedure: Case No.: 09-0592-PCTThe mixture of methyl 3 -cyclopentyl- 1 -methyl- lH-indole-6-carboxylate (50.0 g, 0.194 mol) and lithium isopropoxide (16.2 g, 95%, 0.233 mol) in 2-propanol was stirred at 65+5 °C for at least 30 min for complete trans-esterification. The batch was cooled to 40+5 °C and water (600 g) was added at a rate to maintain the batch temperature at 40+5°C. After addition, the mixture was cooled to 20-25 °C over 2+0.5 h and held at 20-25 °C for at least 1 h. The batch was filtered and rinsed with 28 wt% 2-propanol in water (186 g), and water (500 g). The wet cake was dried in vacuo (< 200 Torr) at 40-45 °C until the water content was < 0.5% to give isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (52.7 g, 95% yield) in 99.2 A% (240 nm).The starting material methyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate can be prepared as described in Example 12 of U.S. Patent 7,141,574, and in Example 12 of U.S. Patent 7,642,352, both herein incorporated by reference.Step 2. Synthesis of Isopropyl 2-Bromo-3-cyclopentyl-l-methyl-lH-indole-6- carboxylate This process identified the optimal conditions for the synthesis of 2-bromo-3-cyclopentyl- l-methyl-lH-indole-6-carboxylate via bromination of the corresponding 3 -cyclopentyl- 1- methyl-lH-indole-6-carboxylate with bromine. It’s very important to control the reaction temperature and to quench the reaction mixture with a mixture of aqueous sodium thiosulfate and 4-methylmorpholine to minimize the formation of the dibromo- and 2- indolone impurities. Further neutralization of the crude product with NaOH in isopropanol greatly increases the stability of the isolated product. Case No.: 09-0592-PCTProcedure:The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (50.0 g, 0.175 mol) and acetonitrile (393 g) was cooled to -6+3 °C. Bromine (33.6 g, 0.210 mol) was added while the batch was maintained at -6+3°C. The resulting slurry was stirred at – 6+3°C for at least 30 min. When HPLC showed > 94 % conversion (the HPLC sample must be quenched immediately with aqueous 4-methylmorpholine/sodium thiosulfate solution), the mixture was quenched with a solution of sodium thiosulfate (15.3 g) and 28.4 g 4-methylmorpholine in water (440 g) while the temperature was maintained at -5+5 °C. After it was stirred at 0+5 °C for at least 2 h, the batch was filtered and rinsed with 85 wt methanol/water solution (415 g), followed by water (500 g), and dried until water content is < 30%. The wet cake was suspended in 2-propanol (675 g), and heated to 75+5 °C. The resulting hazy solution was treated with 1.0 M aqueous sodium hydroxide solution (9.1 g) and then with 135.0 g water at a rate to maintain the batch at 75+5°C. The suspension was stirred at 75+5°C for at least 30 min, cooled to 15+2 °C over 30-40 min, and held at 15+2 °C for at least 1 h. The batch was filtered, rinsed with 75 wt% 2-propanol/water solution (161 g), and dried in vacuo (<200 Torr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3-cyclopentyl-l -methyl- lH-indole-6-carboxylate as a solid (55.6 g, 87 % yield ) in 99.5 A% (240 nm) and 97.9 Wt%. Alternative Procedure:The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (84 g, 0.294 mol) and isopropyl acetate (1074 g) was cooled to between -10-0 °C. Bromine (50 g, 0.312 mol) was added while the batch was maintained at -10 – 0 °C. The resulting slurry was stirred at the same temperature for additional 30 min and quenched with a pre-cooled solution of sodium thiosulfate pentahydrate (13 g) and triethylamine (64.5 g) in water (240 g) while the temperature was maintained at 0-10 °C. The mixture was heated to 40 – 50 °C and charged with methanol (664 g). After it was stirred at the same temperature for at least 0.5 h, the batch was cooled to 0 – 10 °C and stirred for another 1 hr. The precipitate was filtered, rinsed with 56 wt% methanol/water solution (322 g), and dried in vacuo (<200 Case No. : 09-0592-PCTTorr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3- cyclopentyl-l -methyl- lH-indole-6-carboxylate as a beige solid (90-95 g, 80-85 % yield ).Step 3a,b. Preparation of compound I by one-pot Pd-catalyzed borylation- Suzuki coupling reaction
This process identified the optimal conditions for the synthesis of 2-bromo-3-cyclopentyl- l-methyl-lH-indole-6-carboxylate via bromination of the corresponding 3 -cyclopentyl- 1- methyl-lH-indole-6-carboxylate with bromine. It’s very important to control the reaction temperature and to quench the reaction mixture with a mixture of aqueous sodium thiosulfate and 4-methylmorpholine to minimize the formation of the dibromo- and 2- indolone impurities. Further neutralization of the crude product with NaOH in isopropanol greatly increases the stability of the isolated product. Case No.: 09-0592-PCTProcedure:The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (50.0 g, 0.175 mol) and acetonitrile (393 g) was cooled to -6+3 °C. Bromine (33.6 g, 0.210 mol) was added while the batch was maintained at -6+3°C. The resulting slurry was stirred at – 6+3°C for at least 30 min. When HPLC showed > 94 % conversion (the HPLC sample must be quenched immediately with aqueous 4-methylmorpholine/sodium thiosulfate solution), the mixture was quenched with a solution of sodium thiosulfate (15.3 g) and 28.4 g 4-methylmorpholine in water (440 g) while the temperature was maintained at -5+5 °C. After it was stirred at 0+5 °C for at least 2 h, the batch was filtered and rinsed with 85 wt methanol/water solution (415 g), followed by water (500 g), and dried until water content is < 30%. The wet cake was suspended in 2-propanol (675 g), and heated to 75+5 °C. The resulting hazy solution was treated with 1.0 M aqueous sodium hydroxide solution (9.1 g) and then with 135.0 g water at a rate to maintain the batch at 75+5°C. The suspension was stirred at 75+5°C for at least 30 min, cooled to 15+2 °C over 30-40 min, and held at 15+2 °C for at least 1 h. The batch was filtered, rinsed with 75 wt% 2-propanol/water solution (161 g), and dried in vacuo (<200 Torr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3-cyclopentyl-l -methyl- lH-indole-6-carboxylate as a solid (55.6 g, 87 % yield ) in 99.5 A% (240 nm) and 97.9 Wt%. Alternative Procedure:The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (84 g, 0.294 mol) and isopropyl acetate (1074 g) was cooled to between -10-0 °C. Bromine (50 g, 0.312 mol) was added while the batch was maintained at -10 – 0 °C. The resulting slurry was stirred at the same temperature for additional 30 min and quenched with a pre-cooled solution of sodium thiosulfate pentahydrate (13 g) and triethylamine (64.5 g) in water (240 g) while the temperature was maintained at 0-10 °C. The mixture was heated to 40 – 50 °C and charged with methanol (664 g). After it was stirred at the same temperature for at least 0.5 h, the batch was cooled to 0 – 10 °C and stirred for another 1 hr. The precipitate was filtered, rinsed with 56 wt% methanol/water solution (322 g), and dried in vacuo (<200 Case No. : 09-0592-PCTTorr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3- cyclopentyl-l -methyl- lH-indole-6-carboxylate as a beige solid (90-95 g, 80-85 % yield ).Step 3a,b. Preparation of compound I by one-pot Pd-catalyzed borylation- Suzuki coupling reaction To a clean and dry reactor containing 20.04 g of isopropyl 2-bromo-3-cyclopentyl- l- methyl- lH-indole-6-carboxylate, 1.06 g of Pd(TFP)2Cl2(3 mol%) and 0.76 g of tri(2- furyl)phosphine (6 mol%) was charged 8.35 g of triethylamine (1.5 equivalent), 39.38 g of CH3CN at 23+10 °C under nitrogen or argon and started agitation for 10 min. 9.24 g of 4,4,5, 5-tetramethyl-l ,3,2-dioxaborolane was charged into the reactor. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for 6h until the reaction completed. The batch was cooled to 30+5 °C and quenched with a mixture of 0.99 g of water in 7.86 g ofCH3CN. 17.24 g of 5-bromo-2-iodopyrimidine and 166.7 g of degassed aqueous potassium phosphate solution (pre-prepared from 46.70 g of K3PO4 and 120 g of H20) was charged subsquently under argon or nitrogen. The content was heated to reflux (ca. 76-77 °C) for 2 h until the reaction completed. 4.5 g of 1-methylimidazole was charged into the reactor at 70 °C. The batch was cooled to 20+3 °C over 0.5h and hold at 20+3 °C for at least lh. The solid was collected by filtration. The wet cake was first rinsed with 62.8 g of 2-propanol, Case No. : 09-0592-PCTfollowed by 200 g of H20. The solid was dried under vacuum at the temperature below 50 °C.Into a dry and clean reactor was charged dried I, 10 wt Norit SX Ultra and 5 V of THF. The content was heated at 60+5 °C for at least 1 h. After the content was cooled to 35+5 °C, the carbon was filtered off and rinsed with 3 V of THF. The filtrate was charged into a clean reactor containing 1-methylimidazole (10 wt % relative to I). After removal of 5 V of THF by distillation, the content was then cooled to 31 ±2 °C. After the agitation rate was adjusted to over 120 rpm, 2.5 V of water was charged over a period of at least 40 minutes while maintaining the content temperature at 31 + 2 °C. After the content was agitated at 31 + 2 °C for additional 20 min, 9.5 V of water was charged into the reactor over a period of at least 30 minutes at 31 + 2 °C. The batch was then cooled to about 25 + 3 °C and stirred for additional 30 minutes. The solid was collected and rinsed with 3 V of water. The wet product I was dried under vacuum at the temperature below 50 °C (19.5 g, 95 wt , 76% yield).Alternative Procedure:To a clean and dry reactor containing 40 g of isopropyl 2-bromo-3-cyclopentyl- l-methyl- lH-indole-6-carboxylate (0.1 10 mol), 0.74 g of Pd(OAc)2 (3.30 mmol, 3 mol% equiv.) and 3.2 g of tri(2-furyl)phosphine (13.78 mmol, 12.5 mol% equiv.) was charged 16.8 g of triethylamine (1.5 equivalent), 100 mL of acetonitrile at 25 °C under nitrogen or argon. 20.8 g of 4,4,5, 5-tetramethyl- l ,3,2-dioxaborolane was charged into the reactor within 30 min. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for over 5 hrs until the reaction completed. The batch was cooled to 20 °C and quenched with a mixture of 2.7 g of water in 50 mL of CH3CN. The batch was warmed to 30 °C, stirred for 1 hr and transferred to a second reactor containing 34.4 g of 5-bromo-2-iodopyrimidine in 100 mL of acetonitrile. The reactor was rinsed with 90 mL of acetonitrile. To the second reactor was charged with degassed aqueous potassium phosphate solution (pre-prepared from 93.2 g of K3PO4 and 100 g of H20) under argon or nitrogen. The content was heated to reflux (ca. 80 °C) for over 3 h until the reaction completed. 9.2 g of 1 -methylimidazole was charged into the reactor at 70 °C and the mixture was stirred for at least 10 min. The aqueous phase was removed after phase separation. 257 g of isopropanol was charged at 70 Case No.: 09-0592-PCT°C. The batch was cooled slowly to 0 °C and hold for at least 1 h. The solid was collected by filtration. The wet cake was rinsed twice with 2-propanol (2 x 164 g) and dried under vacuum at the temperature below 50 °C to give I as a yellow to brown solid (26 g, 75% yield).Step 4. Hydrolysis of I to II
To a clean and dry reactor containing 20.04 g of isopropyl 2-bromo-3-cyclopentyl- l- methyl- lH-indole-6-carboxylate, 1.06 g of Pd(TFP)2Cl2(3 mol%) and 0.76 g of tri(2- furyl)phosphine (6 mol%) was charged 8.35 g of triethylamine (1.5 equivalent), 39.38 g of CH3CN at 23+10 °C under nitrogen or argon and started agitation for 10 min. 9.24 g of 4,4,5, 5-tetramethyl-l ,3,2-dioxaborolane was charged into the reactor. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for 6h until the reaction completed. The batch was cooled to 30+5 °C and quenched with a mixture of 0.99 g of water in 7.86 g ofCH3CN. 17.24 g of 5-bromo-2-iodopyrimidine and 166.7 g of degassed aqueous potassium phosphate solution (pre-prepared from 46.70 g of K3PO4 and 120 g of H20) was charged subsquently under argon or nitrogen. The content was heated to reflux (ca. 76-77 °C) for 2 h until the reaction completed. 4.5 g of 1-methylimidazole was charged into the reactor at 70 °C. The batch was cooled to 20+3 °C over 0.5h and hold at 20+3 °C for at least lh. The solid was collected by filtration. The wet cake was first rinsed with 62.8 g of 2-propanol, Case No. : 09-0592-PCTfollowed by 200 g of H20. The solid was dried under vacuum at the temperature below 50 °C.Into a dry and clean reactor was charged dried I, 10 wt Norit SX Ultra and 5 V of THF. The content was heated at 60+5 °C for at least 1 h. After the content was cooled to 35+5 °C, the carbon was filtered off and rinsed with 3 V of THF. The filtrate was charged into a clean reactor containing 1-methylimidazole (10 wt % relative to I). After removal of 5 V of THF by distillation, the content was then cooled to 31 ±2 °C. After the agitation rate was adjusted to over 120 rpm, 2.5 V of water was charged over a period of at least 40 minutes while maintaining the content temperature at 31 + 2 °C. After the content was agitated at 31 + 2 °C for additional 20 min, 9.5 V of water was charged into the reactor over a period of at least 30 minutes at 31 + 2 °C. The batch was then cooled to about 25 + 3 °C and stirred for additional 30 minutes. The solid was collected and rinsed with 3 V of water. The wet product I was dried under vacuum at the temperature below 50 °C (19.5 g, 95 wt , 76% yield).Alternative Procedure:To a clean and dry reactor containing 40 g of isopropyl 2-bromo-3-cyclopentyl- l-methyl- lH-indole-6-carboxylate (0.1 10 mol), 0.74 g of Pd(OAc)2 (3.30 mmol, 3 mol% equiv.) and 3.2 g of tri(2-furyl)phosphine (13.78 mmol, 12.5 mol% equiv.) was charged 16.8 g of triethylamine (1.5 equivalent), 100 mL of acetonitrile at 25 °C under nitrogen or argon. 20.8 g of 4,4,5, 5-tetramethyl- l ,3,2-dioxaborolane was charged into the reactor within 30 min. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for over 5 hrs until the reaction completed. The batch was cooled to 20 °C and quenched with a mixture of 2.7 g of water in 50 mL of CH3CN. The batch was warmed to 30 °C, stirred for 1 hr and transferred to a second reactor containing 34.4 g of 5-bromo-2-iodopyrimidine in 100 mL of acetonitrile. The reactor was rinsed with 90 mL of acetonitrile. To the second reactor was charged with degassed aqueous potassium phosphate solution (pre-prepared from 93.2 g of K3PO4 and 100 g of H20) under argon or nitrogen. The content was heated to reflux (ca. 80 °C) for over 3 h until the reaction completed. 9.2 g of 1 -methylimidazole was charged into the reactor at 70 °C and the mixture was stirred for at least 10 min. The aqueous phase was removed after phase separation. 257 g of isopropanol was charged at 70 Case No.: 09-0592-PCT°C. The batch was cooled slowly to 0 °C and hold for at least 1 h. The solid was collected by filtration. The wet cake was rinsed twice with 2-propanol (2 x 164 g) and dried under vacuum at the temperature below 50 °C to give I as a yellow to brown solid (26 g, 75% yield).Step 4. Hydrolysis of I to II I (20 g) and l-methyl-2-pyrrolidinone (NMP) (113 g) were charged into a clean reactor under nitrogen. After the batch was heated to 50-53 °C with agitation, premixed aq. NaOH (5.4 g of 50% aq. NaOH and 14.3 g of water) was introduced into the reactor. The resulting mixture was stirred at 50-53 °C for about 10 hrs until the reaction completed. A premixed aq. HOAc (60 g of water and 9.0 g of HOAc) was added over 0.5 h at 45 ±5 °C to reach pH 5.5- 7.5. The batch was cooled to 20+5 °C and then kept for at least 1.0 h. The solid product was collected and rinsed with 80 g of NMP/water (1 :3 volume ratio) and then 60 g of water. The product was dried under vacuum at the temperature below 50 °C to give II as a pale yellow powder (19 -20 g, purity > 99.0 A% and 88.4 wt%, containing 5.4 wt% NMP). The yield is about 93-98%.Notes: The original procedure used for the hydrolysis of I was carried out with aq. NaOH (2.5 eq) in MeOH/THF at 60 °C. Although it has been applied to the preparation of II on several hundred grams scale, one disadvantage of this method is the formation of 5-MeO pyrimidine during hydrolysis (ca. 0.4 A%), which is extremely difficult to remove in the subsequent steps. In addition, careful control has to be exerted during crystallization. Case No.: 09-0592-PCTOtherwise, a thick slurry might form during acidification with HO Ac. The use of NMP as solvent could overcome all aforementioned issues and give the product with desired purity.Alternative ProcessTo a reactor was charged I (71 g), isopropanol (332 g), aqueous NaOH (22 g, 45 wt ) and water (140 g) at ambient temperature. The mixture was heated to reflux (80 °C) and stirred for at least 3 hrs until the reaction completed. The batch was cooled to 70 °C and charged a suspension of charcoal (3.7 g) in isopropanol (31 g). The mixture was stirred at the same temperature for over 10 min and filtered. The residue was rinsed with isopropanol (154 g). Water (40 g) was charged to the filtrate at 70 – 80 °C, followed by slow addition of 36% HC1 solution (20 g) to reach pH 5- 6. The batch was stirred for over 30 min at 70 °C, then cooled to 20 °C over 1 hr and kept for at least 1.0 h. The solid product was collected and rinsed with 407 g of isopropanol/water (229 g IPA, 178 g H20). The product was dried under vacuum at 80 °C for over 5 hrs to give II as a white powder (61 g, 95% yield).Notes on Steps 5 to 8 below:A concise and scalable 4-step process for the preparation of the benzimidazoleintermediate V was developed. The first step was the preparation of 4-chloro-2-(methyl)- aminonitrobenzene starting from 2,4-dichloronitrobenzene using aqueous methyl amine in DMSO at 65 °C. Then, a ligandless Heck reaction with n-butyl acrylate in the presence of Pd(OAc)2, ‘PrzNEt, LiCl, and DMAc at 110 °C was discovered.Step 5: SNAr reaction of (5-chloro-2-nitrophenyl)-methylamine
I (20 g) and l-methyl-2-pyrrolidinone (NMP) (113 g) were charged into a clean reactor under nitrogen. After the batch was heated to 50-53 °C with agitation, premixed aq. NaOH (5.4 g of 50% aq. NaOH and 14.3 g of water) was introduced into the reactor. The resulting mixture was stirred at 50-53 °C for about 10 hrs until the reaction completed. A premixed aq. HOAc (60 g of water and 9.0 g of HOAc) was added over 0.5 h at 45 ±5 °C to reach pH 5.5- 7.5. The batch was cooled to 20+5 °C and then kept for at least 1.0 h. The solid product was collected and rinsed with 80 g of NMP/water (1 :3 volume ratio) and then 60 g of water. The product was dried under vacuum at the temperature below 50 °C to give II as a pale yellow powder (19 -20 g, purity > 99.0 A% and 88.4 wt%, containing 5.4 wt% NMP). The yield is about 93-98%.Notes: The original procedure used for the hydrolysis of I was carried out with aq. NaOH (2.5 eq) in MeOH/THF at 60 °C. Although it has been applied to the preparation of II on several hundred grams scale, one disadvantage of this method is the formation of 5-MeO pyrimidine during hydrolysis (ca. 0.4 A%), which is extremely difficult to remove in the subsequent steps. In addition, careful control has to be exerted during crystallization. Case No.: 09-0592-PCTOtherwise, a thick slurry might form during acidification with HO Ac. The use of NMP as solvent could overcome all aforementioned issues and give the product with desired purity.Alternative ProcessTo a reactor was charged I (71 g), isopropanol (332 g), aqueous NaOH (22 g, 45 wt ) and water (140 g) at ambient temperature. The mixture was heated to reflux (80 °C) and stirred for at least 3 hrs until the reaction completed. The batch was cooled to 70 °C and charged a suspension of charcoal (3.7 g) in isopropanol (31 g). The mixture was stirred at the same temperature for over 10 min and filtered. The residue was rinsed with isopropanol (154 g). Water (40 g) was charged to the filtrate at 70 – 80 °C, followed by slow addition of 36% HC1 solution (20 g) to reach pH 5- 6. The batch was stirred for over 30 min at 70 °C, then cooled to 20 °C over 1 hr and kept for at least 1.0 h. The solid product was collected and rinsed with 407 g of isopropanol/water (229 g IPA, 178 g H20). The product was dried under vacuum at 80 °C for over 5 hrs to give II as a white powder (61 g, 95% yield).Notes on Steps 5 to 8 below:A concise and scalable 4-step process for the preparation of the benzimidazoleintermediate V was developed. The first step was the preparation of 4-chloro-2-(methyl)- aminonitrobenzene starting from 2,4-dichloronitrobenzene using aqueous methyl amine in DMSO at 65 °C. Then, a ligandless Heck reaction with n-butyl acrylate in the presence of Pd(OAc)2, ‘PrzNEt, LiCl, and DMAc at 110 °C was discovered.Step 5: SNAr reaction of (5-chloro-2-nitrophenyl)-methylamine To a solution of (5-chloro-2-nitrophenyl)-methylamine (40 g, 208.3 mmol, 1 equiv) in DMSO (160 mL) was added 40% MeNH2solution in water (100 mL, 1145. 6 mmol, 5.5 eq) slowly keeping the temperature below 35 °C. The reaction was stirred at r.t. until the Case No.: 09-0592-PCTcomplete consumption of the starting material (>10 h). Water (400 mL) was added to the resulting orange slurry and stirred at r.t. for additional 2 h. The solid was filtered, rinsed with water (200 mL) and dried under reduced pressure at 40 °C. (5-chloro-2-nitrophenyl)- methylamine (36.2 g, 93% yield, 94 A% purity) was isolated as a solid.Step 6: Heck Reaction of (5-chloro-2-nitrophenyl)-methylamine
To a solution of (5-chloro-2-nitrophenyl)-methylamine (40 g, 208.3 mmol, 1 equiv) in DMSO (160 mL) was added 40% MeNH2solution in water (100 mL, 1145. 6 mmol, 5.5 eq) slowly keeping the temperature below 35 °C. The reaction was stirred at r.t. until the Case No.: 09-0592-PCTcomplete consumption of the starting material (>10 h). Water (400 mL) was added to the resulting orange slurry and stirred at r.t. for additional 2 h. The solid was filtered, rinsed with water (200 mL) and dried under reduced pressure at 40 °C. (5-chloro-2-nitrophenyl)- methylamine (36.2 g, 93% yield, 94 A% purity) was isolated as a solid.Step 6: Heck Reaction of (5-chloro-2-nitrophenyl)-methylamine DMAc (5 vol), 1 10 °C, 7-22 h To a mixture of 4-chloro-2-methylaminonitrobenzene (50.0 g, 268.0 mmol, 1.0 eq),Pd(OAc)2 (0.30 g, 1.3 mmol, 0.005 eq) and LiCI (11.4 g 268.0 mmol, 1.0 eq) in DMAc (250 mL) was added ‘Pr2NEt (56 mL, 321.5 mmol, 1.2 eq) followed by n-butyl acrylate (40 mL, 281.4 mmol, 1.05 eq) under nitrogen. The reaction mixture was stirred at 110 °C for 12 h, then cooled to 50 °C. 1 -methylimidazole (10.6 mL, 134.0 mmol, 0.5 eq) was added and the mixture was stirred for 30 min before filtering and adding water (250 mL). The resulting mixture was cooled to r.t. over 1 h. The resulting solid was filtered and washed with water and dried to yield n-butyl 3-methylamino-4-nitrocinnamate (71.8 g, 96 %, 99.2 A% purity).Step 7: Reduction of n-butyl (3-methylamino-4-nitro)-cinnamate
DMAc (5 vol), 1 10 °C, 7-22 h To a mixture of 4-chloro-2-methylaminonitrobenzene (50.0 g, 268.0 mmol, 1.0 eq),Pd(OAc)2 (0.30 g, 1.3 mmol, 0.005 eq) and LiCI (11.4 g 268.0 mmol, 1.0 eq) in DMAc (250 mL) was added ‘Pr2NEt (56 mL, 321.5 mmol, 1.2 eq) followed by n-butyl acrylate (40 mL, 281.4 mmol, 1.05 eq) under nitrogen. The reaction mixture was stirred at 110 °C for 12 h, then cooled to 50 °C. 1 -methylimidazole (10.6 mL, 134.0 mmol, 0.5 eq) was added and the mixture was stirred for 30 min before filtering and adding water (250 mL). The resulting mixture was cooled to r.t. over 1 h. The resulting solid was filtered and washed with water and dried to yield n-butyl 3-methylamino-4-nitrocinnamate (71.8 g, 96 %, 99.2 A% purity).Step 7: Reduction of n-butyl (3-methylamino-4-nitro)-cinnamate III Case No.: 09-0592-PCTTo a reactor was charged n-butyl 3-methylamino-4-nitrocinnamate (70.0 g, mmol, 1.0 eq) , Raney Ni (4.9 g, ~20wt% H20), charcoal “Norit SX Ultra” (3.5 g), toluene (476 mL) and MeOH (224 mL). The reactor was charged with hydrogen (4 bar) and the mixture was stirred at 20- 25 °C for about 2 hrs until the reaction was completed. The reaction mixture was filtered and rinsed the filter residue with toluene (70 mL). To the combined filtrates were added “Norit SX Ultra” charcoal (3.5 g). The mixture was stirred at 50 °C for 1.0 hr and filtered. The filtrate was concentrated under reduced pressure to remove solvents to 50% of the original volume. The remained content was heated to 70 °C and charged slowly methyl cyclohexane (335 mL) at the same temperature. The mixture was cooled to about 30 – 40 °C and seeded with III seed crystals, then slowly cooled the suspension to— 10 °C. The solid was filtered and rinsed with methyl cyclohexane in three portions (3 x 46 mL). The wet cake was dried in vacuo at 40 °C to give III (53.3 g, 215 mmol, 86%).Step 8: Preparation of benzimidazole VDCC
III Case No.: 09-0592-PCTTo a reactor was charged n-butyl 3-methylamino-4-nitrocinnamate (70.0 g, mmol, 1.0 eq) , Raney Ni (4.9 g, ~20wt% H20), charcoal “Norit SX Ultra” (3.5 g), toluene (476 mL) and MeOH (224 mL). The reactor was charged with hydrogen (4 bar) and the mixture was stirred at 20- 25 °C for about 2 hrs until the reaction was completed. The reaction mixture was filtered and rinsed the filter residue with toluene (70 mL). To the combined filtrates were added “Norit SX Ultra” charcoal (3.5 g). The mixture was stirred at 50 °C for 1.0 hr and filtered. The filtrate was concentrated under reduced pressure to remove solvents to 50% of the original volume. The remained content was heated to 70 °C and charged slowly methyl cyclohexane (335 mL) at the same temperature. The mixture was cooled to about 30 – 40 °C and seeded with III seed crystals, then slowly cooled the suspension to— 10 °C. The solid was filtered and rinsed with methyl cyclohexane in three portions (3 x 46 mL). The wet cake was dried in vacuo at 40 °C to give III (53.3 g, 215 mmol, 86%).Step 8: Preparation of benzimidazole VDCC To reactor-1 was charged III (35 g, 140.95 mmol) in toluene (140 g). The mixture was heated to 50 °C to obtain a clear solution. To a second reactor was charged IV (36.4 g, 169.10 mmol) and toluene (300 g), followed by addition of a solution of dicyclohexyl carbodimide (11.6 g, in 50% toluene, 28.11 mmol) at 0 – 10 °C. The mixture was stirred at the same temperature for 15 min, then charged parallelly with the content of reactor-1 and the solution of dicyclohexyl carbodimide (52.4 g, in 50% toluene, 126.98 mmol) within 1 hr while maintaining the batch temperature at 0 – 10 °C. The mixture was agitated at the same temperature for 3 hrs, and warmed to 25 °C for another 1 hr. Once III was consumed, toluene (-300 mL) was distilled off under reduced pressure at 70 – 80 °C. n-Butanol (200 g) was added, followed by 3 M HCI solution in n-butanol (188 g) while maintaining the Case No.: 09-0592-PCTtemperature at 70 – 80 °C (Gas evolution, product precipitates). After stirring for over 30 min. at 70 – 80 °C, the mixture was cooled to 20 – 30 °C over 1 hr. The precipitate was filtered and washed with acetone (172 g) and toluene (88 g). The wet cake was dried in vacuo at -60 °C to give V toluene solvate as off white solid (60 – 72 g, 85 – 95% yield). Compound V could be used directly for the next step or basified prior to next step to obtain the free base compound VI used in the next step.Step 9. Synthesis of (E)-Butyl 3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l- hydroxy-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate VII
To reactor-1 was charged III (35 g, 140.95 mmol) in toluene (140 g). The mixture was heated to 50 °C to obtain a clear solution. To a second reactor was charged IV (36.4 g, 169.10 mmol) and toluene (300 g), followed by addition of a solution of dicyclohexyl carbodimide (11.6 g, in 50% toluene, 28.11 mmol) at 0 – 10 °C. The mixture was stirred at the same temperature for 15 min, then charged parallelly with the content of reactor-1 and the solution of dicyclohexyl carbodimide (52.4 g, in 50% toluene, 126.98 mmol) within 1 hr while maintaining the batch temperature at 0 – 10 °C. The mixture was agitated at the same temperature for 3 hrs, and warmed to 25 °C for another 1 hr. Once III was consumed, toluene (-300 mL) was distilled off under reduced pressure at 70 – 80 °C. n-Butanol (200 g) was added, followed by 3 M HCI solution in n-butanol (188 g) while maintaining the Case No.: 09-0592-PCTtemperature at 70 – 80 °C (Gas evolution, product precipitates). After stirring for over 30 min. at 70 – 80 °C, the mixture was cooled to 20 – 30 °C over 1 hr. The precipitate was filtered and washed with acetone (172 g) and toluene (88 g). The wet cake was dried in vacuo at -60 °C to give V toluene solvate as off white solid (60 – 72 g, 85 – 95% yield). Compound V could be used directly for the next step or basified prior to next step to obtain the free base compound VI used in the next step.Step 9. Synthesis of (E)-Butyl 3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l- hydroxy-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate VII 5) MeOH/H20Notes:The conversion of the acid into acid chloride was achieved using inexpensive thionyl chloride in the presence of catalytic amount of NMP or DMF. An efficient crystallization was developed for the isolation of the desired product in high yield and purity.Procedure (using free base VI):To the suspension of 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (see Step 4) (33.36 g, 90.0 wt %, containing -0.2 equiv of NMP from previous step,75.00 mmol) in THF (133.4 g) was added thionyl chloride (10.71 g). The mixture was stirred at 25+5 °C for at least 1 h. After the conversion was completed as determined by HPLC (as derivative of diethylamine), the mixture was cooled to 10+5 °C and N,N-diisopropylethylamine (378.77 g, 300 mmol) below 25 °C. A solution of (E)-butyl 3-(2-(l-aminocyclobutyl)-l-methyl-lH-benzo[if|imidazol-6-yl)acrylate VI (25.86 g, 97.8 Wt%, 77.25 mmol) dissolved in THF (106.7 g) was added at a rate to maintain the Case No.: 09-0592-PCTtemperature of the content < 25 °C. The mixture was stirred at 25+5 °C for at least 30 min for completion of the amide formation. The mixture was distilled at normal pressure to remove ca. 197 mL (171.5 g) of volatiles (Note: the distillation can also be done under reduced pressure). The batch was adjusted to 40+5 °C, and MeOH (118.6 g) was added. Water (15.0 g) was added and the mixture was stirred at 40+5 °C until crystallization occurred (typically in 30 min), and held for another 1 h. Water (90 g) was charged at 40+5 °C over 1 h, and the batch was cooled to 25+5 °C in 0.5 h, and held for at least 1 h. The solid was filtered, rinsed with a mixture of MeOH (39.5 g), water (100 g), and dried in vacuo (< 200 Torr) at 50+5 °C to give (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3- cyclopentyl- 1 -methyl- lH-indole-6-carboxamido)cyclobutyl)- 1 -methyl- 1H- benzo[if|imidazol-6-yl)acrylate VII (51.82 g, 96.6 % yield) with a HPLC purity of 98.0 A% (240 nm) and 99.0 Wt%.Alternative Process (using compound V from Step 8)To reactor 1 was charged 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (33.6 g), toluene (214 g) and N-methylpyrrolidone (1.37 g). The mixture was heated to 40 °C, then added a solution of thionyl chloride (13 g) in toluene (17 g). The mixture was stirred at 40 °C for at least 0.5 h and cooled to 30 °C. To a second reactor was charged with compound V (the bis-HCl salt toluene solvate from Step 8) (39.4 g), toluene (206 g) and N,N-diisopropylethylamine (70.8 g) at 25 °C. The content of reactor 1 was transferred to reactor 2 at 30 °C and rinsed with toluene (50 g). The mixture was stirred at 30 °C for another 0.5 h, then charged with isopropanol (84 g) and water (108 g) while maintained the temperature at 25 °C. After stirring for 10 min, remove the aqueous phase after phase cutting. To the organic phase was charged isopropanol (43 g), water (54 g) and stirred for 10 min. The aqueous phase was removed after phase cutting. The mixture was distilled under reduced pressure to remove ca.250 mL of volatiles, followed by addition of methyl tert-butyl ether (MTBE, 238 g). The batch was stirred at 65 °C for over 1 hr, then cooled to 20 C over 1 hr and held for another 1 hr at the same temperature. The solid was filtered, rinsed with MTBE (95 g), and dried in vacuo at 80 °C to give (E)-butyl 3-(2-(l-(2- Case No.: 09-0592-PCT(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6-carboxamido)cyclobutyl) methyl- lH-benzo[if|imidazol-6-yl)acrylate VII as a beige solid (50 g, 90 % yield).Step 10. Synthesis of (E)-3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl- lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[</]imidazol-6-yl)acrylic acid (Compound (1))
5) MeOH/H20Notes:The conversion of the acid into acid chloride was achieved using inexpensive thionyl chloride in the presence of catalytic amount of NMP or DMF. An efficient crystallization was developed for the isolation of the desired product in high yield and purity.Procedure (using free base VI):To the suspension of 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (see Step 4) (33.36 g, 90.0 wt %, containing -0.2 equiv of NMP from previous step,75.00 mmol) in THF (133.4 g) was added thionyl chloride (10.71 g). The mixture was stirred at 25+5 °C for at least 1 h. After the conversion was completed as determined by HPLC (as derivative of diethylamine), the mixture was cooled to 10+5 °C and N,N-diisopropylethylamine (378.77 g, 300 mmol) below 25 °C. A solution of (E)-butyl 3-(2-(l-aminocyclobutyl)-l-methyl-lH-benzo[if|imidazol-6-yl)acrylate VI (25.86 g, 97.8 Wt%, 77.25 mmol) dissolved in THF (106.7 g) was added at a rate to maintain the Case No.: 09-0592-PCTtemperature of the content < 25 °C. The mixture was stirred at 25+5 °C for at least 30 min for completion of the amide formation. The mixture was distilled at normal pressure to remove ca. 197 mL (171.5 g) of volatiles (Note: the distillation can also be done under reduced pressure). The batch was adjusted to 40+5 °C, and MeOH (118.6 g) was added. Water (15.0 g) was added and the mixture was stirred at 40+5 °C until crystallization occurred (typically in 30 min), and held for another 1 h. Water (90 g) was charged at 40+5 °C over 1 h, and the batch was cooled to 25+5 °C in 0.5 h, and held for at least 1 h. The solid was filtered, rinsed with a mixture of MeOH (39.5 g), water (100 g), and dried in vacuo (< 200 Torr) at 50+5 °C to give (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3- cyclopentyl- 1 -methyl- lH-indole-6-carboxamido)cyclobutyl)- 1 -methyl- 1H- benzo[if|imidazol-6-yl)acrylate VII (51.82 g, 96.6 % yield) with a HPLC purity of 98.0 A% (240 nm) and 99.0 Wt%.Alternative Process (using compound V from Step 8)To reactor 1 was charged 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (33.6 g), toluene (214 g) and N-methylpyrrolidone (1.37 g). The mixture was heated to 40 °C, then added a solution of thionyl chloride (13 g) in toluene (17 g). The mixture was stirred at 40 °C for at least 0.5 h and cooled to 30 °C. To a second reactor was charged with compound V (the bis-HCl salt toluene solvate from Step 8) (39.4 g), toluene (206 g) and N,N-diisopropylethylamine (70.8 g) at 25 °C. The content of reactor 1 was transferred to reactor 2 at 30 °C and rinsed with toluene (50 g). The mixture was stirred at 30 °C for another 0.5 h, then charged with isopropanol (84 g) and water (108 g) while maintained the temperature at 25 °C. After stirring for 10 min, remove the aqueous phase after phase cutting. To the organic phase was charged isopropanol (43 g), water (54 g) and stirred for 10 min. The aqueous phase was removed after phase cutting. The mixture was distilled under reduced pressure to remove ca.250 mL of volatiles, followed by addition of methyl tert-butyl ether (MTBE, 238 g). The batch was stirred at 65 °C for over 1 hr, then cooled to 20 C over 1 hr and held for another 1 hr at the same temperature. The solid was filtered, rinsed with MTBE (95 g), and dried in vacuo at 80 °C to give (E)-butyl 3-(2-(l-(2- Case No.: 09-0592-PCT(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6-carboxamido)cyclobutyl) methyl- lH-benzo[if|imidazol-6-yl)acrylate VII as a beige solid (50 g, 90 % yield).Step 10. Synthesis of (E)-3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl- lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[</]imidazol-6-yl)acrylic acid (Compound (1)) Notes:In this process, hydrolysis of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl- l-methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate was carried out in mixture of THF/MeOH and aq NaOH. Controlled acidification of the corresponding sodium salt with acetic acid is very critical to obtain easy-filtering crystalline product in high yield and purity.Procedure:To the suspension of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6- yl)acrylate VII (489.0 g, 91.9 Wt%, 633.3 mmol) in THF (1298 g) and MeOH (387 g) was added 50% NaOH (82.7 g, 949.9 mmol), followed by rinse with water (978 g). The mixture was stirred between 65-68 C for about 1 h for complete hydrolysis. The resulting solution was cooled to 35 C, and filtered through an in-line filter (0.5 micron), and rinsed with a pre-mixed solution of water (978 g) and MeOH (387 g). The solution was heated to Case No.: 09-0592-PCT60 +4 C, and acetic acid (41.4 g, 689 mmol) was added over 1 h while the mixture was well agitated. The resulting suspension was stirred at 60 ±4 C for 0.5 h. Another portion of acetic acid (41.4 g, 689 mmol) was charged in 0.5 h, and batch was stirred at 60 ±4 C for additional 0.5 h. The batch was cooled to 26 ±4 C over 1 h and held for 1 h. The batch was filtered, rinsed with a premixed solution of water (1956 g) and MeOH (773.6 g), dried at 50 C under vacuum to give (E)-3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6-yl)acrylic acid (1) (419.0 g, 95 % yield) with > 99.0 A% (240 nm) and 94.1 Wt% by HPLC. Step 11. Formation of Compound (1) Sodium Salt (Type A)
Notes:In this process, hydrolysis of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl- l-methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate was carried out in mixture of THF/MeOH and aq NaOH. Controlled acidification of the corresponding sodium salt with acetic acid is very critical to obtain easy-filtering crystalline product in high yield and purity.Procedure:To the suspension of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6- yl)acrylate VII (489.0 g, 91.9 Wt%, 633.3 mmol) in THF (1298 g) and MeOH (387 g) was added 50% NaOH (82.7 g, 949.9 mmol), followed by rinse with water (978 g). The mixture was stirred between 65-68 C for about 1 h for complete hydrolysis. The resulting solution was cooled to 35 C, and filtered through an in-line filter (0.5 micron), and rinsed with a pre-mixed solution of water (978 g) and MeOH (387 g). The solution was heated to Case No.: 09-0592-PCT60 +4 C, and acetic acid (41.4 g, 689 mmol) was added over 1 h while the mixture was well agitated. The resulting suspension was stirred at 60 ±4 C for 0.5 h. Another portion of acetic acid (41.4 g, 689 mmol) was charged in 0.5 h, and batch was stirred at 60 ±4 C for additional 0.5 h. The batch was cooled to 26 ±4 C over 1 h and held for 1 h. The batch was filtered, rinsed with a premixed solution of water (1956 g) and MeOH (773.6 g), dried at 50 C under vacuum to give (E)-3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6-yl)acrylic acid (1) (419.0 g, 95 % yield) with > 99.0 A% (240 nm) and 94.1 Wt% by HPLC. Step 11. Formation of Compound (1) Sodium Salt (Type A) To a reactor were charged Compound (1) (150 g, mmol), THF (492 mL), H20 (51 mL) and 45% aqueous NaOH solution (20.4 g, mmol). The mixture was stirred for >1 hr at -25 °C to form a clear solution (pH = 9 -11). To the solution was charged a suspension of Charcoal (1.5 g) and H20 (27 mL). The mixture was stirred at -35 °C for >30 min and filtered. The filter was rinsed with THF (108 mL) and H20 (21 mL). The filtrate was heated to 50 °C and charged with methyl ethylketone (MEK) (300 mL). The mixture was seeded with Compound (1) sodium salt MEK solvate (Type A) seeds (0.5 g) and stirred for another 1 hr at 50 °C. To the mixture was charged additional MEK (600 mL). The resultant mixture was stirred for another 1 hr at 50 °C and then cooled to 25 °C. The precipitate was filtered and rinsed with MEK twice (2 x 300 mL). The wet cake was dried in vacuum at 80 °C to give Compound (1) sodium salt (Type A) (145.6 g, 94%). Case No.: 09-0592-PCTThe Compound (1) sodium salt (Type A) MEK solvate seeds used in the above process step can be manufactured by the above process except without using seeds and without drying of the solvate.
To a reactor were charged Compound (1) (150 g, mmol), THF (492 mL), H20 (51 mL) and 45% aqueous NaOH solution (20.4 g, mmol). The mixture was stirred for >1 hr at -25 °C to form a clear solution (pH = 9 -11). To the solution was charged a suspension of Charcoal (1.5 g) and H20 (27 mL). The mixture was stirred at -35 °C for >30 min and filtered. The filter was rinsed with THF (108 mL) and H20 (21 mL). The filtrate was heated to 50 °C and charged with methyl ethylketone (MEK) (300 mL). The mixture was seeded with Compound (1) sodium salt MEK solvate (Type A) seeds (0.5 g) and stirred for another 1 hr at 50 °C. To the mixture was charged additional MEK (600 mL). The resultant mixture was stirred for another 1 hr at 50 °C and then cooled to 25 °C. The precipitate was filtered and rinsed with MEK twice (2 x 300 mL). The wet cake was dried in vacuum at 80 °C to give Compound (1) sodium salt (Type A) (145.6 g, 94%). Case No.: 09-0592-PCTThe Compound (1) sodium salt (Type A) MEK solvate seeds used in the above process step can be manufactured by the above process except without using seeds and without drying of the solvate.- ...............................................................................................................
- 13, FILIBUVIR
- FILIBUVIRPFIZERPF-868554 is an anti-hepatitis C drug candidate which had been in phase II clinical trials at Pfizer; however this research has been discontinued.Li, H.; Tatlock, J.; Linton, A.; et al
Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-(1,2,4)triazolo(1,5-a)pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor
J Med Chem 2009, 52(5): 1255Johnson, S.; Drowns, M.; Tatlock, J.; et al.
Synthetic route optimization of PF-00868554, an HCV polymerase inhibitor in clinical evaluation
Synlett (Stuttgart) 2010, 2010(5): 796WO 2012016995WO 2013101550WO 2011072370WO 2007023381WO 2006018725WO2003095441A1 * 7 mei 2003 20 nov 2003 Melwyn A Abreo Inhibitors of hepatitis c virus rna-dependent rna polymerase, and compositions and treatments using the same WO2006018725A1 * 5 aug 2005 23 feb 2006 Pfizer Inhibitors of hepatitis c virus rna-dependent rna polymerase, and compositions and treatments using the same US20050176701 * 19 nov 2003 11 aug 2005 Agouron Pharmaceuticals, Inc. Inhibitors of hepatitis C virus RNA-dependent RNA polymerase, and compositions and treatments using the sameWO2007023381A1 Example 1 : Preparation of the glycolate salt of (5-amino-1H-1,2,4-triazol-3-yl)methanol glycolate saltGlycolic acid (1 L, 70% in water, 11.51 mol) was added to a 5 L flask. To the solution was slowly added aminoguanidine bicarbonate (783.33 g, 5.755 mol) in portions to control significant bubbling. As solids are added, the solution cools due to endothermic dissolution. The solution was gently heated to maintain an internal temp of 25 °C during addition. Ten minutes after complete addition of aminoguanidine bicarbonate, cone. Nitric acid (6.8 ml_) was carefully added. The solution was heated to an internal temperature of 104-108 0C (mild reflux) for 22 h. The heating was discontinued and the solution allowed to cool, with stirring. At an internal temp of aboutδi °C, solids began to crystallize. After the internal temperature was just below 80 0C, ethanol (absolute, 375 mL) was slowly added to the mixture. After the internal temp had cooled to aboutδδ 0C1the cooling was sped up by the use of an ice/water bath. After cooling below rt, the solution became very thick but remained stirrable at all times. The slurry was stirred for 2h at T<10 0C, then filtered and the solids rinsed with ethanol (900 mL cold, then 250 mL rt). The solids were dried overnight in a vacuum oven (about25 mmHg, 45-50 0C) to provide 815.80 g (75%) of (5-amino-1H-1 ,2,4-triazol-3-yl)methanol as the glycolate salt. 1H (300 MHz, de-DMSO): 3.90 (s, 2), 4.24 (s, 2).Example 2: Preparation of (5,7-dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methanol
glycolate saltGlycolic acid (1 L, 70% in water, 11.51 mol) was added to a 5 L flask. To the solution was slowly added aminoguanidine bicarbonate (783.33 g, 5.755 mol) in portions to control significant bubbling. As solids are added, the solution cools due to endothermic dissolution. The solution was gently heated to maintain an internal temp of 25 °C during addition. Ten minutes after complete addition of aminoguanidine bicarbonate, cone. Nitric acid (6.8 ml_) was carefully added. The solution was heated to an internal temperature of 104-108 0C (mild reflux) for 22 h. The heating was discontinued and the solution allowed to cool, with stirring. At an internal temp of aboutδi °C, solids began to crystallize. After the internal temperature was just below 80 0C, ethanol (absolute, 375 mL) was slowly added to the mixture. After the internal temp had cooled to aboutδδ 0C1the cooling was sped up by the use of an ice/water bath. After cooling below rt, the solution became very thick but remained stirrable at all times. The slurry was stirred for 2h at T<10 0C, then filtered and the solids rinsed with ethanol (900 mL cold, then 250 mL rt). The solids were dried overnight in a vacuum oven (about25 mmHg, 45-50 0C) to provide 815.80 g (75%) of (5-amino-1H-1 ,2,4-triazol-3-yl)methanol as the glycolate salt. 1H (300 MHz, de-DMSO): 3.90 (s, 2), 4.24 (s, 2).Example 2: Preparation of (5,7-dimethyl[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methanol To a 2L, 3-neck flask was charged glycolate salt of (5-amino-1tf-1 ,2,4-triazol-3-yl)methanol (99.93 g, 0.526 mol), 2,4 pentanedione (0.578 mols, 60 mL), acetic acid (6.70 mL), and EtOH (550 mL). The mixture was heated to a slight reflux. One hour after adding the reagents, the resulting solution was cooled to ambient temperature, and CH2CI2 (500 mL) and Celite (25.03 g) were added. After stirring for 1 h, the mixture was filtered through a 4" Buchner funnel packed with celite (20 g) and rinsed with EtOH (100 mL). The solution was distilled to 5 vols then cooled to 0 °C for 1-2 hours. The slurry was filtered and the cake was rinsed with cold EtOH (2x100 mL). The solids were dried to provide 76.67 g (81.7%) of the title compound.1H NMR (300 MHz, d6-DMSO): 2.57 (s, 3), 2.71 (d, 3, J=0.8), 4.63 (uneven d, 2, J=5.7), 5.49(t, 1 , J=6.2), 7.13 (d, 1 , J=0.8).Example 3: Preparation of 5,7-dimethyl[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde
To a 2L, 3-neck flask was charged glycolate salt of (5-amino-1tf-1 ,2,4-triazol-3-yl)methanol (99.93 g, 0.526 mol), 2,4 pentanedione (0.578 mols, 60 mL), acetic acid (6.70 mL), and EtOH (550 mL). The mixture was heated to a slight reflux. One hour after adding the reagents, the resulting solution was cooled to ambient temperature, and CH2CI2 (500 mL) and Celite (25.03 g) were added. After stirring for 1 h, the mixture was filtered through a 4" Buchner funnel packed with celite (20 g) and rinsed with EtOH (100 mL). The solution was distilled to 5 vols then cooled to 0 °C for 1-2 hours. The slurry was filtered and the cake was rinsed with cold EtOH (2x100 mL). The solids were dried to provide 76.67 g (81.7%) of the title compound.1H NMR (300 MHz, d6-DMSO): 2.57 (s, 3), 2.71 (d, 3, J=0.8), 4.63 (uneven d, 2, J=5.7), 5.49(t, 1 , J=6.2), 7.13 (d, 1 , J=0.8).Example 3: Preparation of 5,7-dimethyl[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde To a 10 L reactor was sequentially charged CH2CI2 (5.1 L)1 (5,7- dimethyl[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methanol (680 g, 3.816 mol), and iodobenzene diacetate (1352 g, 4.197 mol). As the iodobenzene diacetate dissolves, there is a significant endotherm (typically down to 15-16 0C). The jacket was set to 23 0C. The mixture was warmed to ambient temperature and Tempo (2,2,6,6-tetramethyl-1-piperidinyloxy, free radical, 43.75 g, 0.28 mol) added in a single charge. The reaction was stirred until 5% of the starting alcohol remained by HPLC. Once the starting material is adjudged to be less than about about5%, the over-oxidized product begins to be observed. Allowing the reaction to run to further completion leads to an overall diminished yield of the desired product. For this reaction, the desired reaction completion was reached in 2.75 h. MTBE (5.1 L) was then slowly charged to the reactor, causing the product to precipitate, and the slurry stirred for an additional 30 mins. The mixture was filtered, washed twice with 1 :1 DCM/MTBE (2 x 1 L), and dried in a vacuum oven overnight at 50 0C to provide 500.3 g (74%) of 5,7- dimethyl[1,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde as an off-white solid. 1H NMR (300 MHz, ds-DMSO): 2.64 (s, 3), 2.78 (d, 3, J=0.8), 7.36 (d, 1 , J=0.9), 10.13 (s, 1). Example 4: Preparation of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one
To a 10 L reactor was sequentially charged CH2CI2 (5.1 L)1 (5,7- dimethyl[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methanol (680 g, 3.816 mol), and iodobenzene diacetate (1352 g, 4.197 mol). As the iodobenzene diacetate dissolves, there is a significant endotherm (typically down to 15-16 0C). The jacket was set to 23 0C. The mixture was warmed to ambient temperature and Tempo (2,2,6,6-tetramethyl-1-piperidinyloxy, free radical, 43.75 g, 0.28 mol) added in a single charge. The reaction was stirred until 5% of the starting alcohol remained by HPLC. Once the starting material is adjudged to be less than about about5%, the over-oxidized product begins to be observed. Allowing the reaction to run to further completion leads to an overall diminished yield of the desired product. For this reaction, the desired reaction completion was reached in 2.75 h. MTBE (5.1 L) was then slowly charged to the reactor, causing the product to precipitate, and the slurry stirred for an additional 30 mins. The mixture was filtered, washed twice with 1 :1 DCM/MTBE (2 x 1 L), and dried in a vacuum oven overnight at 50 0C to provide 500.3 g (74%) of 5,7- dimethyl[1,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde as an off-white solid. 1H NMR (300 MHz, ds-DMSO): 2.64 (s, 3), 2.78 (d, 3, J=0.8), 7.36 (d, 1 , J=0.9), 10.13 (s, 1). Example 4: Preparation of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one DMACL-DBTA NEt3-HOTs + LiBr + NEt3-HBr THF/MTBE
DMACL-DBTA NEt3-HOTs + LiBr + NEt3-HBr THF/MTBE A nitrogen-purged, 5-L, 3-neck flask containing 4-bromo-2,6-diethylpyridine (250.0 g, 0.6472 mol) was sequentially charged with LiBr (112.42 g, 1.2944 mol), 1-cyclopentyl-prop-2- en-1-ol ( 89.84 g, 0.7119 mol), DMAc (625 mL), and H2O (55.0 mL). The mixture was cooled to 5-10 0C and was then purged (subsurface) with N2 for 30 minutes. The flask was charged with Et3N (198.5 mL, 1.4242 mol) and Pd(OaC)2 (3.63 g, 0.0162 mol), followed by a careful purge of the headspace. The reaction was heated until the internal temperature reached 95 0C. After stirring at 95 °C for three hours, an aliquot was removed and analyzed by HPLC, showing >99% conversion to 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one. The reaction was then cooled to 30 0C over 20 min. The flask was charged with H2O (1500 mL), and MTBE (1500 mL). The solution was stirred well for 5 minutes before the mixture was allowed to settle and the aqueous layer was removed. To the organic layer was charged Celite (62.5Og), and Darco G-60 (6.25g). The slurry was stirred for 20 minutes at 20-25 0C. The slurry was then filtered using a Buchner funnel dressed with Celite. The filter cake was rinsed with MTBE (250 mL). The organic layer was extracted with 5% sodium bicarbonate solution (500 mL) and the phases separated. The organic layer was transferred to a 5 L, three-neck flask, and MTBE added to achieve a total reaction volume of 1750 mL. Additional MTBE (1500 mL) was added and atmospherically distilled until an internal volume of 1750 mL was reached. After cooling below 40 0C, a sample was removed for analysis of water content. After cooling to 20-25 0C, MTBE (250 mL) was added to bring the total volume to 2000 mL and the solution was seeded with crystals of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one (130 mg), which were prepared according to this procedure. A solution of dibenzoyl-L-tartaric acid (231.89 g, 0.6472 mol) in THF (900 mL) was added over 25 minutes. The slurry was granulated for 1 hour, the mixture was filtered, and the cake rinsed with MTBE (450 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 366.70 g (92% yield) of the title compound. 1H NMR (300 MHz, d6-DMSO): 1.19 (t, 6, J=7.6), 1.47-1.81 (m, 8), 2.73 (q, 4, J=7.6), 2.73-2.98 (m, 5), 5.86 (s, 2), 7.00 (S1 2), 7.55-7.63 (m, 4), 7.68-7.75 (m, 2), 7.98-8.04 (m, 4).Example 5: Preparation of 3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid
A nitrogen-purged, 5-L, 3-neck flask containing 4-bromo-2,6-diethylpyridine (250.0 g, 0.6472 mol) was sequentially charged with LiBr (112.42 g, 1.2944 mol), 1-cyclopentyl-prop-2- en-1-ol ( 89.84 g, 0.7119 mol), DMAc (625 mL), and H2O (55.0 mL). The mixture was cooled to 5-10 0C and was then purged (subsurface) with N2 for 30 minutes. The flask was charged with Et3N (198.5 mL, 1.4242 mol) and Pd(OaC)2 (3.63 g, 0.0162 mol), followed by a careful purge of the headspace. The reaction was heated until the internal temperature reached 95 0C. After stirring at 95 °C for three hours, an aliquot was removed and analyzed by HPLC, showing >99% conversion to 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one. The reaction was then cooled to 30 0C over 20 min. The flask was charged with H2O (1500 mL), and MTBE (1500 mL). The solution was stirred well for 5 minutes before the mixture was allowed to settle and the aqueous layer was removed. To the organic layer was charged Celite (62.5Og), and Darco G-60 (6.25g). The slurry was stirred for 20 minutes at 20-25 0C. The slurry was then filtered using a Buchner funnel dressed with Celite. The filter cake was rinsed with MTBE (250 mL). The organic layer was extracted with 5% sodium bicarbonate solution (500 mL) and the phases separated. The organic layer was transferred to a 5 L, three-neck flask, and MTBE added to achieve a total reaction volume of 1750 mL. Additional MTBE (1500 mL) was added and atmospherically distilled until an internal volume of 1750 mL was reached. After cooling below 40 0C, a sample was removed for analysis of water content. After cooling to 20-25 0C, MTBE (250 mL) was added to bring the total volume to 2000 mL and the solution was seeded with crystals of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one (130 mg), which were prepared according to this procedure. A solution of dibenzoyl-L-tartaric acid (231.89 g, 0.6472 mol) in THF (900 mL) was added over 25 minutes. The slurry was granulated for 1 hour, the mixture was filtered, and the cake rinsed with MTBE (450 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 366.70 g (92% yield) of the title compound. 1H NMR (300 MHz, d6-DMSO): 1.19 (t, 6, J=7.6), 1.47-1.81 (m, 8), 2.73 (q, 4, J=7.6), 2.73-2.98 (m, 5), 5.86 (s, 2), 7.00 (S1 2), 7.55-7.63 (m, 4), 7.68-7.75 (m, 2), 7.98-8.04 (m, 4).Example 5: Preparation of 3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid A 3-L, 3-neck flask was charged with the dibenzoyl-L-tartaric acid salt of 1- cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one (174.95 g, 0.2832 mol), MTBE (875 mL), water (875 mL), and triethanolamine (113.0 mL, 0.8513 mol). After stirring for 2 h at rt, an aliquot of the aqueous phase was removed and analyzed by HPLC, showing no detectable starting material. The solution was transferred to a separatory funnel and the layers separated. The lower aqueous phase was discarded and the upper org. phase was washed with water (150 mL). The organic layer was added to a flask set up for distillation. The solution was distilled down to approx. 183 mL and an aliquot was removed and analyzed for water content. The dry solution of 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one (th. Wt = 73.47 g) in MTBE was used directly in the next step.A clean 2-L, 3-neck flask was charged with LiHMDS (1.0 M in THF, 355 mL, 0.355 mol) and purged with nitrogen. The flask was cooled to -34 0C. An addition funnel was then charged with EtOAc (35 mL, 0.3583 mol) and this reagent was slowly added to the reaction vessel at such a rate that the low temperature of the vessel could be maintained. After complete EtOAc addition another addition funnel was charged with the 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one solution (crude MTBE soln from prior reaction, theor. 73.47 g, 0.2832 mol) and rinsed over with THF (anhydrous, 5 ml_). The 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one solution was slowly added to the reaction flask at such a rate that the low internal temperature could be maintained. Five minutes after complete addition, a reaction aliquot was removed and analyzed by HPLC, showing less than 1% 1-cyclopentyl-3- (2,6-diethylpyridin-4-yl)propan-1-one. Ten minutes after complete ketone addition, the bath was switched to O 0C. Once the internal temperature had warmed to -10 0C, 1 M NaOH (510 mL) was added. After complete NaOH soln addition, the reaction was heated to 50 0C. After 21 hours the reaction solution was cooled below 30 0C and an aliquot of both layers was removed and analyzed for completion. The mixture was added to a separatory funnel with MTBE (350 mL) and the phases were mixed well and separated. An aliquot of the organic phase was analyzed by HPLC, verifying no significant product, and this layer was discarded. The aqueous phase was added to a flask with CH2CI2(350 mL). Concentrated aqueous HCI (about 100 mL) was slowly added to the aqueous phase until the pH = 5. The mixture was added back to a separatory funnel and mixed well. The phases were separated and the aqueous layer was extracted a second time with CH2CI2 (150 mL). The organic layers were combined and charged to a clean flask set up for distillation. The solution was distilled down to 370 mL then displaced with THF by addition of solvent portions followed by continued distillation down to 370 mL after each addition. When the distillation head temp, held steady at 65 °C for 30 min an aliquot was removed and analyzed by 1H NMR, showing a 12.5:1 ratio of THF:CH2CI2. The solution of 3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid in THF was used directly in the next step.Example 6a: Preparation of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1,3-propanedioI salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid
A 3-L, 3-neck flask was charged with the dibenzoyl-L-tartaric acid salt of 1- cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one (174.95 g, 0.2832 mol), MTBE (875 mL), water (875 mL), and triethanolamine (113.0 mL, 0.8513 mol). After stirring for 2 h at rt, an aliquot of the aqueous phase was removed and analyzed by HPLC, showing no detectable starting material. The solution was transferred to a separatory funnel and the layers separated. The lower aqueous phase was discarded and the upper org. phase was washed with water (150 mL). The organic layer was added to a flask set up for distillation. The solution was distilled down to approx. 183 mL and an aliquot was removed and analyzed for water content. The dry solution of 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one (th. Wt = 73.47 g) in MTBE was used directly in the next step.A clean 2-L, 3-neck flask was charged with LiHMDS (1.0 M in THF, 355 mL, 0.355 mol) and purged with nitrogen. The flask was cooled to -34 0C. An addition funnel was then charged with EtOAc (35 mL, 0.3583 mol) and this reagent was slowly added to the reaction vessel at such a rate that the low temperature of the vessel could be maintained. After complete EtOAc addition another addition funnel was charged with the 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one solution (crude MTBE soln from prior reaction, theor. 73.47 g, 0.2832 mol) and rinsed over with THF (anhydrous, 5 ml_). The 1-cyclopentyl-3-(2,6- diethylpyridin-4-yl)propan-1-one solution was slowly added to the reaction flask at such a rate that the low internal temperature could be maintained. Five minutes after complete addition, a reaction aliquot was removed and analyzed by HPLC, showing less than 1% 1-cyclopentyl-3- (2,6-diethylpyridin-4-yl)propan-1-one. Ten minutes after complete ketone addition, the bath was switched to O 0C. Once the internal temperature had warmed to -10 0C, 1 M NaOH (510 mL) was added. After complete NaOH soln addition, the reaction was heated to 50 0C. After 21 hours the reaction solution was cooled below 30 0C and an aliquot of both layers was removed and analyzed for completion. The mixture was added to a separatory funnel with MTBE (350 mL) and the phases were mixed well and separated. An aliquot of the organic phase was analyzed by HPLC, verifying no significant product, and this layer was discarded. The aqueous phase was added to a flask with CH2CI2(350 mL). Concentrated aqueous HCI (about 100 mL) was slowly added to the aqueous phase until the pH = 5. The mixture was added back to a separatory funnel and mixed well. The phases were separated and the aqueous layer was extracted a second time with CH2CI2 (150 mL). The organic layers were combined and charged to a clean flask set up for distillation. The solution was distilled down to 370 mL then displaced with THF by addition of solvent portions followed by continued distillation down to 370 mL after each addition. When the distillation head temp, held steady at 65 °C for 30 min an aliquot was removed and analyzed by 1H NMR, showing a 12.5:1 ratio of THF:CH2CI2. The solution of 3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid in THF was used directly in the next step.Example 6a: Preparation of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1,3-propanedioI salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid A 2-L, 3-neck flask was sequentially charged with a solution of 3-cyclopentyl-5-(2,6- diethylpyridin-4-yl)-3-hydroxypentanoic acid (crude from last step, theoretical 95.28 g, 0.1792 mol, in about300 mL), (1 R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol (38.03 g, 0.1792 moles) and THF (415 mL). A seed crystal of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid, prepared according to this procedure, was added and the mixture was stirred and heated to 65 0C, then held at this temperature for 16 h. The slurry was cooled slowly to rt and stirred for at least 1 h. The slurry was filtered and the cake rinsed with THF (100 mL). The filtrate (solution of (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid in THF) was used directly in the next procedure. The solids were dried to provide 67.09 g (42 %) of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6- diethylpyridin-4-yl)-3-hydroxypentanoic acid as an off-white crystalline solid. Chiral HPLC analysis of the product showed a 92.1:7.9 ratio of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)- 1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid to (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid. HPLC conditions: The solid was dissolved in methanol. HPLC conditions: Chirobiotic TAG column, 4.6 x 250 mm, 40 0C column chamber, flow rate = 0.5 mL/min, mobile phase = 100% MeOH (0.05% TEA, 0.05% HOAc). Gradient: Initial flow rate = 0.5 mL/min; 10 min flow rate = 0.5 mL/min; 10.10 min flow rate = 2.00 mL/min; 35 min flow rate = 2.00 mL/min; 36 min flow rate = 0.5 mLΛnin. Percentages reported are at 265 nm. Retention times: (1 R,2R)-(-)-2- amino-1-(4-nitrophenyl)-1 ,3-propanediol = >30 min; (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4- yl)-3-hydroxypentanoic acid = 5.8 min; ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid = 7.2 min. 1H NMR (300 MHz, d6-DMSO): 1.19 (t, 6, J=7.6), 1.38-1.62 (m, 8), 1.65-1.75 (m, 2), 1.93-2.07 (m, 1), 2.23 (d, 1 , J=14.4), 2.31 (d, 1 , J=14.4), 2.56 (m, 2), 2.64 (q, 4, J=7.6), 2.91-2.99 (m, 1), 3.22 (dd, 1 , J=5.8, 11.1), 3.42 (dd, 1 , J=4.8, 11.1), 4.77 (d, 1 , J=6.2), 6.0 (br s, 6), 6.84 (s, 2), 7.62 (d, 2, J=8.7), 8.20 (d, 2, J=8.8). Example 6b: Recrystallization of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acidA 2-L, 3-neck flask was charged with the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (66.20 g, 0.1245 moles) and 2B EtOH (970 mL absolute EtOH + 5 mL toluene). The slurry was stirred and heated to reflux. After holding at reflux for 40 min, all the solids had dissolved and the solution was cooled to an internal temp of about 65 0C over 30 min, and the solution was then seeded with crystals of the title compound. The solution was allowed to cool to 50 0C and held for an additional 2h. The solution was then cooled slowly to room temperature over about 2 hours. The cooled solution was stirred at rt for an additional 10 h. The mixture was then filtered and the solids rinsed with 2B EtOH (75 mL). The solids were dried to provide 52.72 g (80%) of product as an off-white crystalline solid that was then dried under vacuum (30 mm Hg) with a nitrogen bleed at 50 0C for 12 h. Chiral HPLC analysis showed product with 96% ee. For determination of e.e., the solid was dissolved in MeOH. HPLC conditions: Chirobiotic TAG column, 4.6 x 250 mm, 40 0C column chamber, flow rate = 0.5 ml_/min, 100% MeOH (0.05% TEA, 0.05% HOAc). Gradient: Initial flow rate = 0.5 mL/min; 10 min flow rate = 0.5 mL/min; 10.10 min flow rate = 2.00 mL/min; 35 min flow rate = 2.00 mL/min; 36 min flow rate = 0.5 mL/min. Percentages reported are at 265 nm. Retention times: (1 R,2R)-(-)-2- amino-1-(4-nitrophenyl)-1 ,3-propanediol = >30 min, (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4- yl)-3-hydroxypentanoic acid = 5.8 min, ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid = 7.2 min.Example 7: Preparation of 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one from (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid
A 2-L, 3-neck flask was sequentially charged with a solution of 3-cyclopentyl-5-(2,6- diethylpyridin-4-yl)-3-hydroxypentanoic acid (crude from last step, theoretical 95.28 g, 0.1792 mol, in about300 mL), (1 R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol (38.03 g, 0.1792 moles) and THF (415 mL). A seed crystal of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid, prepared according to this procedure, was added and the mixture was stirred and heated to 65 0C, then held at this temperature for 16 h. The slurry was cooled slowly to rt and stirred for at least 1 h. The slurry was filtered and the cake rinsed with THF (100 mL). The filtrate (solution of (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid in THF) was used directly in the next procedure. The solids were dried to provide 67.09 g (42 %) of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6- diethylpyridin-4-yl)-3-hydroxypentanoic acid as an off-white crystalline solid. Chiral HPLC analysis of the product showed a 92.1:7.9 ratio of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)- 1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid to (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid. HPLC conditions: The solid was dissolved in methanol. HPLC conditions: Chirobiotic TAG column, 4.6 x 250 mm, 40 0C column chamber, flow rate = 0.5 mL/min, mobile phase = 100% MeOH (0.05% TEA, 0.05% HOAc). Gradient: Initial flow rate = 0.5 mL/min; 10 min flow rate = 0.5 mL/min; 10.10 min flow rate = 2.00 mL/min; 35 min flow rate = 2.00 mL/min; 36 min flow rate = 0.5 mLΛnin. Percentages reported are at 265 nm. Retention times: (1 R,2R)-(-)-2- amino-1-(4-nitrophenyl)-1 ,3-propanediol = >30 min; (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4- yl)-3-hydroxypentanoic acid = 5.8 min; ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid = 7.2 min. 1H NMR (300 MHz, d6-DMSO): 1.19 (t, 6, J=7.6), 1.38-1.62 (m, 8), 1.65-1.75 (m, 2), 1.93-2.07 (m, 1), 2.23 (d, 1 , J=14.4), 2.31 (d, 1 , J=14.4), 2.56 (m, 2), 2.64 (q, 4, J=7.6), 2.91-2.99 (m, 1), 3.22 (dd, 1 , J=5.8, 11.1), 3.42 (dd, 1 , J=4.8, 11.1), 4.77 (d, 1 , J=6.2), 6.0 (br s, 6), 6.84 (s, 2), 7.62 (d, 2, J=8.7), 8.20 (d, 2, J=8.8). Example 6b: Recrystallization of the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acidA 2-L, 3-neck flask was charged with the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (66.20 g, 0.1245 moles) and 2B EtOH (970 mL absolute EtOH + 5 mL toluene). The slurry was stirred and heated to reflux. After holding at reflux for 40 min, all the solids had dissolved and the solution was cooled to an internal temp of about 65 0C over 30 min, and the solution was then seeded with crystals of the title compound. The solution was allowed to cool to 50 0C and held for an additional 2h. The solution was then cooled slowly to room temperature over about 2 hours. The cooled solution was stirred at rt for an additional 10 h. The mixture was then filtered and the solids rinsed with 2B EtOH (75 mL). The solids were dried to provide 52.72 g (80%) of product as an off-white crystalline solid that was then dried under vacuum (30 mm Hg) with a nitrogen bleed at 50 0C for 12 h. Chiral HPLC analysis showed product with 96% ee. For determination of e.e., the solid was dissolved in MeOH. HPLC conditions: Chirobiotic TAG column, 4.6 x 250 mm, 40 0C column chamber, flow rate = 0.5 ml_/min, 100% MeOH (0.05% TEA, 0.05% HOAc). Gradient: Initial flow rate = 0.5 mL/min; 10 min flow rate = 0.5 mL/min; 10.10 min flow rate = 2.00 mL/min; 35 min flow rate = 2.00 mL/min; 36 min flow rate = 0.5 mL/min. Percentages reported are at 265 nm. Retention times: (1 R,2R)-(-)-2- amino-1-(4-nitrophenyl)-1 ,3-propanediol = >30 min, (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4- yl)-3-hydroxypentanoic acid = 5.8 min, ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid = 7.2 min.Example 7: Preparation of 1-cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one from (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid A flask was charged with a solution of (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid (crude from last step, theoretical 15 g, 0.0470 mol, in about 200 mL THF) and ethanol (100 ml_, 1.7126 mol). To the solution, H2SO4 (5.0 ml_, 0.0938 mol) was added slowly. The solution was heated at reflux for 18 h. When the reaction was judged to be complete by HPLC, the solution was cooled and added to a separatory funnel with 0.5M NaOH (400 mL) and then extracted with MTBE (200 mL). The phases were separated and the organic layer was washed with aqueous acetic acid H2O (100 mL H2O + 3.0 mL HOAc). The phases were separated and the organic layer was washed with 0.5 M NaOH (100 mL). The phases were separated and the organic layer was washed with saturated aqueous NaCI solution (25 mL). The organic layer was distilled at atmospheric pressure down to an internal volume of 150 mL. The solvent was displaced by toluene via atmospheric distillation by adding toluene (100 mL), distilling down to 200 mL internal volume, and repeating this procedure two more times. The final solution was distilled down to an internal volume of 130 mL. An aliquot was removed and analyzed by KF titration. The solution was cooled to rt and a solution of KotBu (1.0M in THF, 4.7 mL, 0.0047 mol) was added in one portion. After 5 min, an aliquot was removed and analyzed by HPLC. The solution was added to a separatory funnel with 1M HCI (60 mL). The phases were mixed well and separated, transferring the product to the aqueous phase. The organic phase was extracted once with water (10 mL) and the aqueous phases combined. The organic phase was discarded. To the aqueous phase was added MTBE (60 mL) and 1 M NaOH (70 mL) and the phases mixed well. The phases were separated and the organic phase extracted with saturated aqueous NaCI solution (25 mL). MTBE was added to bring the volume up to 125 mL. The solution was cooled to rt and seeded with crystals of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6-diethylpyridin- 4-yl)propan-1-one (prepared according to Example 4). In a separate vessel, L-DBTA (16.89 g, 0.0471 mol) was dissolved in THF (65 ml_). The solution of L-DBTA was added to the 1- cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one solution over 45 min, and the slurry granulated for 1 h. The slurry was filtered and the cake washed with MTBE (50 mL). The solids were dried to provide 19.54 g of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3- (2,6-diethylpyridin-4-yl)propan-1-one (67 %) as an off-white solid. Example 8a: Preparation of the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one
A flask was charged with a solution of (S)-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3- hydroxypentanoic acid (crude from last step, theoretical 15 g, 0.0470 mol, in about 200 mL THF) and ethanol (100 ml_, 1.7126 mol). To the solution, H2SO4 (5.0 ml_, 0.0938 mol) was added slowly. The solution was heated at reflux for 18 h. When the reaction was judged to be complete by HPLC, the solution was cooled and added to a separatory funnel with 0.5M NaOH (400 mL) and then extracted with MTBE (200 mL). The phases were separated and the organic layer was washed with aqueous acetic acid H2O (100 mL H2O + 3.0 mL HOAc). The phases were separated and the organic layer was washed with 0.5 M NaOH (100 mL). The phases were separated and the organic layer was washed with saturated aqueous NaCI solution (25 mL). The organic layer was distilled at atmospheric pressure down to an internal volume of 150 mL. The solvent was displaced by toluene via atmospheric distillation by adding toluene (100 mL), distilling down to 200 mL internal volume, and repeating this procedure two more times. The final solution was distilled down to an internal volume of 130 mL. An aliquot was removed and analyzed by KF titration. The solution was cooled to rt and a solution of KotBu (1.0M in THF, 4.7 mL, 0.0047 mol) was added in one portion. After 5 min, an aliquot was removed and analyzed by HPLC. The solution was added to a separatory funnel with 1M HCI (60 mL). The phases were mixed well and separated, transferring the product to the aqueous phase. The organic phase was extracted once with water (10 mL) and the aqueous phases combined. The organic phase was discarded. To the aqueous phase was added MTBE (60 mL) and 1 M NaOH (70 mL) and the phases mixed well. The phases were separated and the organic phase extracted with saturated aqueous NaCI solution (25 mL). MTBE was added to bring the volume up to 125 mL. The solution was cooled to rt and seeded with crystals of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3-(2,6-diethylpyridin- 4-yl)propan-1-one (prepared according to Example 4). In a separate vessel, L-DBTA (16.89 g, 0.0471 mol) was dissolved in THF (65 ml_). The solution of L-DBTA was added to the 1- cyclopentyl-3-(2,6-diethylpyridin-4-yl)propan-1-one solution over 45 min, and the slurry granulated for 1 h. The slurry was filtered and the cake washed with MTBE (50 mL). The solids were dried to provide 19.54 g of the dibenzoyl-L-tartaric acid salt of 1-cyclopentyl-3- (2,6-diethylpyridin-4-yl)propan-1-one (67 %) as an off-white solid. Example 8a: Preparation of the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one i. CDI, DWIAP O OIi. KO-^^OEt MgCI2
i. CDI, DWIAP O OIi. KO-^^OEt MgCI2 A nitrogen-purged flask containing the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (20.00 g, 0.0376 mol) was charged with CH2CI2 (200 mL) and H2O (100 mL). The pH of the mixture was adjusted to pH 4.75 with 40% aqueous citric acid (10 mL) and was stirred for 60 minutes. The layers were allowed to settle for 30 minutes and separated. The upper (aqueous) layer was charged CH2CI2 (50 mL), stirred 15 minutes, and was then allowed to settle. The organic layer was combined with the first organic layer and dried with sodium sulfate. The dried organic was concentrated under reduced pressure. The ®-3-cyclopentyl-5-(2,6-diethylpyridin- 4-yl)-3-hydroxypentanoic acid residue was dissolved in THF (47 mL) and this solution added to a slurry of carbonyl diimidazole (9.00 g, 0.0555 mol) and 4-N,N-dimethylaminopyridine (DMAP, 0.45 g, 0.0037 mol) in THF (106 mL) over 5 minutes. Upon complete acyl-imidazole formation, the solution was added to a slurry of potassium ethyl malonate (12.57 g, 0.0738 mol) and magnesium chloride (7.38 g, 0.0775 mol) in 106 mL THF over 5 minutes. The slurry was allowed to stir at 20-25 0C for 30 hours. An aliquot was removed and analyzed by HPLC, showing 96% conversion to ©-ethyl 5-cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3- oxoheptanoate. The flask was charged with H2O (64 mL), and MTBE (118 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. To the organic layer was charged brine (52 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. The organic layer was then displaced via atmospheric distillation with methanol (2 x 210 mL) until a total volume of 140 mL was achieved. MTBE (105 mL) was added followed by powdered potassium carbonate (7.65 g, 0.0554 mol), and the slurry heated to reflux for 12 hours. After cooling to 40 °C, MTBE (140 mL) and water (140 mL) were added. The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was isolated. The organic layer was extracted with water (30 mL) and the aqueous layers were combined. CH2CI2 (140 mL) was added to the aqueous layer and the pH adjusted to 6.4 with 40% aqueous citric acid (29 mL). The aqueous layer was extracted a second time with CH2CI2 (25 mL). The combined organic layers were then displaced fully into MTBE (140 mL final volume) via atmospheric distillation, cooled, and added slowly to a solution of dibenzoyl-D-tartaric acid (9.92 g, 0.0277 mol) in MTBE (100 mL). The slurry was heated to reflux for 1 hour, then allowed to cool to 20-25 0C. The mixture was filtered, and the cake rinsed with MTBE (50 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 16.40 g (62%) of the title compound.Example 8b: Preparation of the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-oneA nitrogen-purged flask containing the (1 R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (50.00 g, 0.0940 mol) was charged with CH2CI2 (500 mL) and H2O (250 mL). The pH of the resulting suspension was adjusted to pH 4.6 to 4.8 (a measured pH of 4.75 is preferred) with 40% aqueous citric acid (21 mL) and was stirred for 30 minutes. The layers were allowed to settle for 30 minutes and separated. The upper (aqueous) layer was charged with CH2CI2 (100 mL), stirred 15 minutes, and allowed to settle. The organic layer was combined with the first organic layer. The upper (aqueous) layer was again charged with CH2CI2 (100 mL), stirred 15 minutes, and allowed to settle. This organic layer was also combined with the first organic layer. A sample of each of the combined organic layers and the aqueous layer was taken for HPLC analysis. The combined organic layers were atmospherically distilled until a total volume of 120 mL was reached. THF (100 mL) was charged and atmospheric distillation continued until a total volume of 120 mL was reached. The THF charge and displacement was repeated 3 times. A sample was removed and analyzed by NMR and KF. The resulting solution was added to a slurry of CDI (22.86 g, 0.1410 mol) and DMAP (1.15 g, 0.0094 mol) in THF (250 mL) over 15 minutes. The addition funnel was then rinsed with 10 mL THF which was then added to the CDI slurry. After stirring 15 minutes, a sample was removed and analyzed by HPLC. Upon complete acyl-imidazole formation, the solution was added to a slurry of potassium ethyl malonate (32.00 g, 0.1880 mol) and magnesium chloride (18.80 g, 0.1974 mol) in 250 mL THF at 20-25 0C over 25 minutes. The slurry was allowed to stir at 20-25 0C for 21 hours. An aliquot was removed and analyzed by HPLC, showing 96% conversion to ®- ethyl 5-cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3-oxoheptanoate. The flask was charged with H2O (162 mL), and MTBE (300 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the yellow aqueous (lower) layer was removed. To the organic layer was charged brine (100 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. The organic layer was then atmospherically distilled down to 350 mL total volume. MTBE (250 mL) was charged and the solution distilled to 350 mL total volume. Additional MTBE (250 mL) was charged and the solution distilled at a temperature of at least 55 0C to 350 mL total volume. A sample was removed for KF titration. Methanol (250 mL) was charged and the solution was then atmospherically distilled until a total volume of 350 mL was achieved. Methanol (250 mL) was charged and then the solution was atmospherically distilled until a total volume of 350 mL was achieved and a temperature of ~66 0C was achieved. Powdered potassium carbonate (19.49 g, 0.1410 mol) was added and the slurry heated to reflux for 4 hours. A sample was removed for HPLC analysis showing >99% completion. After cooling to 22 0C, MTBE (350 mL) and water (350 mL) were added. The mixture was stirred well for 5 minutes before it was allowed to settle and the product rich aqueous (lower) layer was isolated. The organic layer was extracted with water (100 mL) and the aqueous layers were combined. To the combined aqueous layers was charged MTBE (100 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the product rich aqueous (lower) layer was isolated. CH2CI2 (350 mL) was added to the aqueous layer and the pH adjusted to 6.0-6.4 with 40% aqueous citric acid (75 mL). The aqueous layer was extracted a second time with CH2CI2 (100 mL). The combined organic layers were then atmospherically distilled to 250 mL total volume. MTBE (400 mL) was charged and the solution was atmospherically distilled at a temperature of at least 55 0C until 250 mL final volume was reached. After cooling the solution to 20-25 0C, a prepared solution of dibenzoyl-D-tartaric acid (23.58 g, 0.0658 mol) in MTBE (125 mL) was added over 10 minutes. The resulting slurry was heated to reflux for 4 hours, then allowed to cool to 20-25 0C and stirred an additional 4 hours. The slurry was filtered, and the cake rinsed with MTBE (125 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 38.19 g (58%) of the title compound. HPLC conditions: aliquots were withdrawn and dissolved in CH3CN/H2O (40:60). HPLC conditions: Kromasil C4 column, 5 μm, 4.6x150mm, 40 0C column chamber, flow rate= 1.0 mL/min, 40% CHsCN/60% aqueous (1.OmL 70% HcIO4 in 1 L H2O) isocratic. Percentages reported are at 254 nm. Approximate retention times: ®-3- cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid = 3.4 min; ©-ethyl 5- cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3-oxoheptanoate = 7.3 min; ®-6-cyclopentyl- 3-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one = 3.9 min; D-DBTA = 5.5 min. Example 9a: Preparation of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7- dimethyl-ri^.^triazoloII.S-alpyrimidini-Z-yOmethylH-hydroxy-S.e-clihyclropyran^-one
A nitrogen-purged flask containing the (1R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3- propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (20.00 g, 0.0376 mol) was charged with CH2CI2 (200 mL) and H2O (100 mL). The pH of the mixture was adjusted to pH 4.75 with 40% aqueous citric acid (10 mL) and was stirred for 60 minutes. The layers were allowed to settle for 30 minutes and separated. The upper (aqueous) layer was charged CH2CI2 (50 mL), stirred 15 minutes, and was then allowed to settle. The organic layer was combined with the first organic layer and dried with sodium sulfate. The dried organic was concentrated under reduced pressure. The ®-3-cyclopentyl-5-(2,6-diethylpyridin- 4-yl)-3-hydroxypentanoic acid residue was dissolved in THF (47 mL) and this solution added to a slurry of carbonyl diimidazole (9.00 g, 0.0555 mol) and 4-N,N-dimethylaminopyridine (DMAP, 0.45 g, 0.0037 mol) in THF (106 mL) over 5 minutes. Upon complete acyl-imidazole formation, the solution was added to a slurry of potassium ethyl malonate (12.57 g, 0.0738 mol) and magnesium chloride (7.38 g, 0.0775 mol) in 106 mL THF over 5 minutes. The slurry was allowed to stir at 20-25 0C for 30 hours. An aliquot was removed and analyzed by HPLC, showing 96% conversion to ©-ethyl 5-cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3- oxoheptanoate. The flask was charged with H2O (64 mL), and MTBE (118 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. To the organic layer was charged brine (52 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. The organic layer was then displaced via atmospheric distillation with methanol (2 x 210 mL) until a total volume of 140 mL was achieved. MTBE (105 mL) was added followed by powdered potassium carbonate (7.65 g, 0.0554 mol), and the slurry heated to reflux for 12 hours. After cooling to 40 °C, MTBE (140 mL) and water (140 mL) were added. The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was isolated. The organic layer was extracted with water (30 mL) and the aqueous layers were combined. CH2CI2 (140 mL) was added to the aqueous layer and the pH adjusted to 6.4 with 40% aqueous citric acid (29 mL). The aqueous layer was extracted a second time with CH2CI2 (25 mL). The combined organic layers were then displaced fully into MTBE (140 mL final volume) via atmospheric distillation, cooled, and added slowly to a solution of dibenzoyl-D-tartaric acid (9.92 g, 0.0277 mol) in MTBE (100 mL). The slurry was heated to reflux for 1 hour, then allowed to cool to 20-25 0C. The mixture was filtered, and the cake rinsed with MTBE (50 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 16.40 g (62%) of the title compound.Example 8b: Preparation of the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-oneA nitrogen-purged flask containing the (1 R,2R)-(-)-2-amino-1-(4-nitrophenyl)-1 ,3-propanediol salt of ®-3-cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid (50.00 g, 0.0940 mol) was charged with CH2CI2 (500 mL) and H2O (250 mL). The pH of the resulting suspension was adjusted to pH 4.6 to 4.8 (a measured pH of 4.75 is preferred) with 40% aqueous citric acid (21 mL) and was stirred for 30 minutes. The layers were allowed to settle for 30 minutes and separated. The upper (aqueous) layer was charged with CH2CI2 (100 mL), stirred 15 minutes, and allowed to settle. The organic layer was combined with the first organic layer. The upper (aqueous) layer was again charged with CH2CI2 (100 mL), stirred 15 minutes, and allowed to settle. This organic layer was also combined with the first organic layer. A sample of each of the combined organic layers and the aqueous layer was taken for HPLC analysis. The combined organic layers were atmospherically distilled until a total volume of 120 mL was reached. THF (100 mL) was charged and atmospheric distillation continued until a total volume of 120 mL was reached. The THF charge and displacement was repeated 3 times. A sample was removed and analyzed by NMR and KF. The resulting solution was added to a slurry of CDI (22.86 g, 0.1410 mol) and DMAP (1.15 g, 0.0094 mol) in THF (250 mL) over 15 minutes. The addition funnel was then rinsed with 10 mL THF which was then added to the CDI slurry. After stirring 15 minutes, a sample was removed and analyzed by HPLC. Upon complete acyl-imidazole formation, the solution was added to a slurry of potassium ethyl malonate (32.00 g, 0.1880 mol) and magnesium chloride (18.80 g, 0.1974 mol) in 250 mL THF at 20-25 0C over 25 minutes. The slurry was allowed to stir at 20-25 0C for 21 hours. An aliquot was removed and analyzed by HPLC, showing 96% conversion to ®- ethyl 5-cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3-oxoheptanoate. The flask was charged with H2O (162 mL), and MTBE (300 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the yellow aqueous (lower) layer was removed. To the organic layer was charged brine (100 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the aqueous (lower) layer was removed. The organic layer was then atmospherically distilled down to 350 mL total volume. MTBE (250 mL) was charged and the solution distilled to 350 mL total volume. Additional MTBE (250 mL) was charged and the solution distilled at a temperature of at least 55 0C to 350 mL total volume. A sample was removed for KF titration. Methanol (250 mL) was charged and the solution was then atmospherically distilled until a total volume of 350 mL was achieved. Methanol (250 mL) was charged and then the solution was atmospherically distilled until a total volume of 350 mL was achieved and a temperature of ~66 0C was achieved. Powdered potassium carbonate (19.49 g, 0.1410 mol) was added and the slurry heated to reflux for 4 hours. A sample was removed for HPLC analysis showing >99% completion. After cooling to 22 0C, MTBE (350 mL) and water (350 mL) were added. The mixture was stirred well for 5 minutes before it was allowed to settle and the product rich aqueous (lower) layer was isolated. The organic layer was extracted with water (100 mL) and the aqueous layers were combined. To the combined aqueous layers was charged MTBE (100 mL). The mixture was stirred well for 5 minutes before it was allowed to settle and the product rich aqueous (lower) layer was isolated. CH2CI2 (350 mL) was added to the aqueous layer and the pH adjusted to 6.0-6.4 with 40% aqueous citric acid (75 mL). The aqueous layer was extracted a second time with CH2CI2 (100 mL). The combined organic layers were then atmospherically distilled to 250 mL total volume. MTBE (400 mL) was charged and the solution was atmospherically distilled at a temperature of at least 55 0C until 250 mL final volume was reached. After cooling the solution to 20-25 0C, a prepared solution of dibenzoyl-D-tartaric acid (23.58 g, 0.0658 mol) in MTBE (125 mL) was added over 10 minutes. The resulting slurry was heated to reflux for 4 hours, then allowed to cool to 20-25 0C and stirred an additional 4 hours. The slurry was filtered, and the cake rinsed with MTBE (125 mL). The solids were dried in a vacuum oven at 50 0C for 12 h to provide 38.19 g (58%) of the title compound. HPLC conditions: aliquots were withdrawn and dissolved in CH3CN/H2O (40:60). HPLC conditions: Kromasil C4 column, 5 μm, 4.6x150mm, 40 0C column chamber, flow rate= 1.0 mL/min, 40% CHsCN/60% aqueous (1.OmL 70% HcIO4 in 1 L H2O) isocratic. Percentages reported are at 254 nm. Approximate retention times: ®-3- cyclopentyl-5-(2,6-diethylpyridin-4-yl)-3-hydroxypentanoic acid = 3.4 min; ©-ethyl 5- cyclopentyl-7-(2,6-diethylpyridin-4-yl)-5-hydroxy-3-oxoheptanoate = 7.3 min; ®-6-cyclopentyl- 3-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one = 3.9 min; D-DBTA = 5.5 min. Example 9a: Preparation of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7- dimethyl-ri^.^triazoloII.S-alpyrimidini-Z-yOmethylH-hydroxy-S.e-clihyclropyran^-one BHe-pyridine
BHe-pyridine A flask was charged with the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one (this material contained 1.5 eq DBTA counterion, 4.00 g, theor. 0.00454 mol), 2-MeTHF (40 ttiL), MTBE (40 mL), and water (20 mL). A solution of 5% aq NaHCO3 (about 20 mL) was added until the pH was 7.4. The solution pH was back-adjusted to pH = 7.2 with a small amount of 40% citric acid solution. The phases were separated and the aqueous layer was extracted with 2-MeTHF (25 mL). The combined organic layers were dried with Na2SO4 and concentrated to an oil. The oil was used directly in the subsequent condensation. To the crude ®-6-cyclopentyl-6-(2-(2,6- diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one was added methanol (32 mL) and the solution cooled to -40 0C. To the cold solution was added pyridine-borane complex (1.30 mL, 0.01287 mol) and 5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde (1.41 g, 0.00800 mol). The solution was warmed to 0 0C over 45 min then stirred for an additional 2 h. The reaction was quenched by the addition of water (10 mL) and the mixture stirred at rt overnight. To the mixture was added 1M HCI (10 mL), and the solution was stirred for 3 h. lsopropyl acetate (57 mL) was added and the pH adjusted to 7 by the addition of 1 M NaOH. The phases were separated and the organic layer extracted with water (25 mL x 2). The aqueous phases were extracted further with CH2CI2 (100 ml, 2 x 25 mL). The combined IPAc and CH2CI2 layers were dried (Na2SO4), filtered, and concentrated to yield 3.41 g of crude ®-6- cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2- yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one. To the residue was added isopropyl acetate (46 mL) and EtOH (2.5 mL) and the mixture heated to reflux until homogeneous. The solution was allowed to cool slowly to rt and stirred overnight. The slurry was filtered, the solids rinsed with IPAc (13 mL), and dried to provide 1.74 g (76 %) of ®-6-cyclopentyl-6-(2-(2,6- diethylpyridin-4-yl)ethyl)-3-((5J-dirnethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one as an off-white solid.Example 9b: Preparation of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7- dimethyl-[1,2,4]triazolot1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-oneA 500 mL flask was charged with the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one (15.00 g, 0.02137 moles), THF (75 mL), MeOH (75 mL), pyridine-borane (4.25 mL, 0.034 moles), and 5,7- dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde (5.65 g, 0.03207 moles) was added last. The resulting mixture was stirred at rt and an aliquot was removed after 1.25 h and analyzed by HPLC showing 13.5% ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4- hydroxy-5,6-dihydropyran-2-one. Stirring was continued for an additional 2 h, and HPLC analysis of an aliquot then showed 4.8% of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one remaining. The reaction solution was charged with CH2CI2(150 mL) and water (150 mL), and the phases were stirred overnight. The lower organic layer was removed and to the upper aqueous layer was charged CH2CI2 (25 mL), the phases were mixed well and separated and the aqueous layer was discarded. The organic layers were combined and charged to a flask containing water (150 mL) and triethanolamine (7.1 mL,0.0535 mol), mixed well then separated. The lower organic layer was removed and to the upper aqueous layer was charged CH2CI2 (25 mL), the phases were mixed well, separated, and the aqueous layer was discarded. To the combined organic layers was charged water(100 mL) and 1M NaOH (25 mL), the phases were mixed well, separated, and the lower organic layer was discarded. To the upper aqueous layer was charged CH2CI2 (75 mL) and1N HCI was added until the pH=6.91 (~25 mL added), the phases were mixed well, separated, and the aqueous layer was discarded. The combined organic layers were extracted with water (3.2 volumes). The layers were separated and the organic layer was transferred to a;lean flask marked with a 75 mL volume line. The organic layer was distilled atmospherically0 75 mL. To the flask was charged isopropyl acetate (75 mL x 2) followed by distillation down0 75 mL total volume after each addition. The flask was seeded and cooled to rt and stirred)vemight. The reaction was filtered and the cake was washed with isopropyl acetate (25 ml)."he solids were dried to provide 7.20 g (67%) of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-'l)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6- lihydropyran-2-one as an off-white powder, which was dried in a vacuum oven (~25 inHg at0 0C) for 12 h. For HPLC monitoring, aliquots were withdrawn and dissolved in CH3CN/H2O1-0:60). HPLC conditions: Kromasil C4 column, 5 μm, 4.6x150 mm, 40 0C column chamber, ow rate= 1.0 mL/min, 40% CH3CN/60% aqueous (1.0 mL 70% HcIO4 in 1L H2O) isocratic.'ercentages reported are at 254 nm. Retention times: ®-6-cyclopentyl-6-(2-(2,6- iethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one = 3.85 min; ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4- hydroxy-5,6-dihydropyran-2-one = 3.56 min; DBTA= 5.14 min; BH3 «pyr=3.36 min.Example 10: Recrystallization of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2- oneA 200 mL flask was charged with ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3- ((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (10.05 g, 0.01995 mol) and THF (70 mL). The mixture was stirred and heated to 30 to 35 0C to provide a homogeneous solution. The solution was filtered through a 0.45 μm Teflon filter, and rinsed with THF (10 mL). The filtrate was added to a flask set up for atmospheric distillation and isopropyl acetate (IPAC, 50 mL) was added. The solution was concentrated by distillation to an internal volume of 100 mL. Isopropyl acetate (50 mL) was added and distillation continued at atmospheric pressure until the internal volume reached 100 mL. The solution was seeded with ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl- [1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one and additional IPAC (30 mL) was added. The solution was again distilled to an internal volume of 100 mL and was cooled over about 1 h to 50 0C. The solution was held at 50 0C for an additional 1.5 h, cooled over about 2 h to rt, and stirred overnight. The resulting slurry was filtered and rinsed with IPAC (30 mL). The resulting solids were dried to provide 9.41 g (94%) of the title compound as an off-white powder that was vacuum dried (~25 in Hg, 50 0C) for 12 h.
A flask was charged with the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one (this material contained 1.5 eq DBTA counterion, 4.00 g, theor. 0.00454 mol), 2-MeTHF (40 ttiL), MTBE (40 mL), and water (20 mL). A solution of 5% aq NaHCO3 (about 20 mL) was added until the pH was 7.4. The solution pH was back-adjusted to pH = 7.2 with a small amount of 40% citric acid solution. The phases were separated and the aqueous layer was extracted with 2-MeTHF (25 mL). The combined organic layers were dried with Na2SO4 and concentrated to an oil. The oil was used directly in the subsequent condensation. To the crude ®-6-cyclopentyl-6-(2-(2,6- diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one was added methanol (32 mL) and the solution cooled to -40 0C. To the cold solution was added pyridine-borane complex (1.30 mL, 0.01287 mol) and 5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde (1.41 g, 0.00800 mol). The solution was warmed to 0 0C over 45 min then stirred for an additional 2 h. The reaction was quenched by the addition of water (10 mL) and the mixture stirred at rt overnight. To the mixture was added 1M HCI (10 mL), and the solution was stirred for 3 h. lsopropyl acetate (57 mL) was added and the pH adjusted to 7 by the addition of 1 M NaOH. The phases were separated and the organic layer extracted with water (25 mL x 2). The aqueous phases were extracted further with CH2CI2 (100 ml, 2 x 25 mL). The combined IPAc and CH2CI2 layers were dried (Na2SO4), filtered, and concentrated to yield 3.41 g of crude ®-6- cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2- yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one. To the residue was added isopropyl acetate (46 mL) and EtOH (2.5 mL) and the mixture heated to reflux until homogeneous. The solution was allowed to cool slowly to rt and stirred overnight. The slurry was filtered, the solids rinsed with IPAc (13 mL), and dried to provide 1.74 g (76 %) of ®-6-cyclopentyl-6-(2-(2,6- diethylpyridin-4-yl)ethyl)-3-((5J-dirnethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one as an off-white solid.Example 9b: Preparation of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7- dimethyl-[1,2,4]triazolot1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-oneA 500 mL flask was charged with the dibenzoyl-L-tartaric acid salt of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one (15.00 g, 0.02137 moles), THF (75 mL), MeOH (75 mL), pyridine-borane (4.25 mL, 0.034 moles), and 5,7- dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidine-2-carbaldehyde (5.65 g, 0.03207 moles) was added last. The resulting mixture was stirred at rt and an aliquot was removed after 1.25 h and analyzed by HPLC showing 13.5% ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4- hydroxy-5,6-dihydropyran-2-one. Stirring was continued for an additional 2 h, and HPLC analysis of an aliquot then showed 4.8% of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one remaining. The reaction solution was charged with CH2CI2(150 mL) and water (150 mL), and the phases were stirred overnight. The lower organic layer was removed and to the upper aqueous layer was charged CH2CI2 (25 mL), the phases were mixed well and separated and the aqueous layer was discarded. The organic layers were combined and charged to a flask containing water (150 mL) and triethanolamine (7.1 mL,0.0535 mol), mixed well then separated. The lower organic layer was removed and to the upper aqueous layer was charged CH2CI2 (25 mL), the phases were mixed well, separated, and the aqueous layer was discarded. To the combined organic layers was charged water(100 mL) and 1M NaOH (25 mL), the phases were mixed well, separated, and the lower organic layer was discarded. To the upper aqueous layer was charged CH2CI2 (75 mL) and1N HCI was added until the pH=6.91 (~25 mL added), the phases were mixed well, separated, and the aqueous layer was discarded. The combined organic layers were extracted with water (3.2 volumes). The layers were separated and the organic layer was transferred to a;lean flask marked with a 75 mL volume line. The organic layer was distilled atmospherically0 75 mL. To the flask was charged isopropyl acetate (75 mL x 2) followed by distillation down0 75 mL total volume after each addition. The flask was seeded and cooled to rt and stirred)vemight. The reaction was filtered and the cake was washed with isopropyl acetate (25 ml)."he solids were dried to provide 7.20 g (67%) of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-'l)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6- lihydropyran-2-one as an off-white powder, which was dried in a vacuum oven (~25 inHg at0 0C) for 12 h. For HPLC monitoring, aliquots were withdrawn and dissolved in CH3CN/H2O1-0:60). HPLC conditions: Kromasil C4 column, 5 μm, 4.6x150 mm, 40 0C column chamber, ow rate= 1.0 mL/min, 40% CH3CN/60% aqueous (1.0 mL 70% HcIO4 in 1L H2O) isocratic.'ercentages reported are at 254 nm. Retention times: ®-6-cyclopentyl-6-(2-(2,6- iethylpyridin-4-yl)ethyl)-4-hydroxy-5,6-dihydropyran-2-one = 3.85 min; ®-6-cyclopentyl-6-(2- (2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4- hydroxy-5,6-dihydropyran-2-one = 3.56 min; DBTA= 5.14 min; BH3 «pyr=3.36 min.Example 10: Recrystallization of ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2- oneA 200 mL flask was charged with ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3- ((5,7-dimethyl-[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (10.05 g, 0.01995 mol) and THF (70 mL). The mixture was stirred and heated to 30 to 35 0C to provide a homogeneous solution. The solution was filtered through a 0.45 μm Teflon filter, and rinsed with THF (10 mL). The filtrate was added to a flask set up for atmospheric distillation and isopropyl acetate (IPAC, 50 mL) was added. The solution was concentrated by distillation to an internal volume of 100 mL. Isopropyl acetate (50 mL) was added and distillation continued at atmospheric pressure until the internal volume reached 100 mL. The solution was seeded with ®-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl- [1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one and additional IPAC (30 mL) was added. The solution was again distilled to an internal volume of 100 mL and was cooled over about 1 h to 50 0C. The solution was held at 50 0C for an additional 1.5 h, cooled over about 2 h to rt, and stirred overnight. The resulting slurry was filtered and rinsed with IPAC (30 mL). The resulting solids were dried to provide 9.41 g (94%) of the title compound as an off-white powder that was vacuum dried (~25 in Hg, 50 0C) for 12 h.CAS 877130-28-4 FILIBUVIR (R)-6-Cyclopentyl-6-[2-(2,6-diethylpyridin-4-yl)ethyl]-3-[(5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl]-4-hydroxy-5,6-dihydro-2H-pyran-2-one Filibuvir;Pf-00868554;Unii-198J479Y2l;(6R)-6-Cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl(1,2,4)triazolo(1,5-A)pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydro-2H-pyran-2-one;(R)-6-Cyclopentyl-6-[2-(2,6-diethylpyridin-4-yl)ethyl]-3-[(5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl]-4-hydroxy-5,6-dihydro-2H-pyran-2-one;2H-Pyran-2-one, 6-cyclopentyl-6-(2-(2,6-diethyl-4-pyridinyl)ethyl)-3-((5,7-dimethyl(1,2,4)triazolo(1,5-A)pyrimidin-2-yl)methyl)-5,6-dihydro-4-hydroxy-, (6R)- MF C29H37N5O3 MW 503.64 - .....................................................
- 14 FAVIPIRAVIR
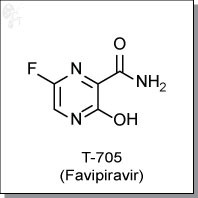 FAVIPIRAVIRToyama (Originator)RNA-Directed RNA Polymerase (NS5B) Inhibitors
FAVIPIRAVIRToyama (Originator)RNA-Directed RNA Polymerase (NS5B) InhibitorsChemical Formula: C5H4FN3O2 CAS #: 259793-96-9 Molecular Weight: 157.1 Anti-influenza compound
clinical trials http://clinicaltrials.gov/search/intervention=Favipiravir Chemical Name: 6-fluoro-3-hydroxy-2-pyrazinecarboxamide Synonyms: T-705, T705, Favipiravir T-705 is an RNA-directed RNA polymerase (NS5B) inhibitor which has been filed for approval in Japan for the oral treatment of influenza A (including avian and H1N1 infections) and for the treatment of influenza B infection.The compound is a unique viral RNA polymerase inhibitor, acting on viral genetic copying to prevent its reproduction, discovered by Toyama Chemical. In 2005, Utah State University carried out various studies under its contract with the National Institute of Allergy and Infectious Diseases (NIAID) and demonstrated that T-705 has exceptionally potent activity in mouse infection models of H5N1 avian influenza.T-705 (Favipiravir) is an antiviral pyrazinecarboxamide-based, inhibitor of of the influenza virus with an EC90 of 1.3 to 7.7 uM (influenza A, H5N1). EC90 ranges for other influenza A subtypes are 0.19-1.3 uM, 0.063-1.9 uM, and 0.5-3.1 uM for H1N1, H2N2, and H3N2, respectively. T-705 also exhibits activity against type B and C viruses, with EC90s of 0.25-0.57 uM and 0.19-0.36 uM, respectively. (1) Additionally, T-705 has broad activity against arenavirus, bunyavirus, foot-and-mouth disease virus, and West Nile virus with EC50s ranging from 5 to 300 uM.Studies show that T-705 ribofuranosyl triphosphate is the active form of T-705 and acts like purines or purine nucleosides in cells and does not inhibit DNA synthesisIn 2012, MediVector was awarded a contract from the U.S. Department of Defense's (DOD) Joint Project Manager Transformational Medical Technologies (JPM-TMT) to further develop T-705 (favipiravir), a broad-spectrum therapeutic against multiple influenza viruses.Several novel anti-influenza compounds are in various phases of clinical development. One of these, T-705 (favipiravir), has a mechanism of action that is not fully understood but is suggested to target influenza virus RNA-dependent RNA polymerase. We investigated the mechanism of T-705 activity against influenza A (H1N1) viruses by applying selective drug pressure over multiple sequential passages in MDCK cells. We found that T-705 treatment did not select specific mutations in potential target proteins, including PB1, PB2, PA, and NP. Phenotypic assays based on cell viability confirmed that no T-705-resistant variants were selected. In the presence of T-705, titers of infectious virus decreased significantly (P < 0.0001) during serial passage in MDCK cells inoculated with seasonal influenza A (H1N1) viruses at a low multiplicity of infection (MOI; 0.0001 PFU/cell) or with 2009 pandemic H1N1 viruses at a high MOI (10 PFU/cell). There was no corresponding decrease in the number of viral RNA copies; therefore, specific virus infectivity (the ratio of infectious virus yield to viral RNA copy number) was reduced. Sequence analysis showed enrichment of G→A and C→T transversion mutations, increased mutation frequency, and a shift of the nucleotide profiles of individual NP gene clones under drug selection pressure. Our results demonstrate that T-705 induces a high rate of mutation that generates a nonviable viral phenotype and that lethal mutagenesis is a key antiviral mechanism of T-705. Our findings also explain the broad spectrum of activity of T-705 against viruses of multiple families.favipiravirFavipiravir also known as T-705 is an experimental anti-viral drug with activity against many RNA viruses. It, like some other experimental antiviraldrugs—T-1105 and T-1106, is apyrazinecarboxamide derivative. Favipiravir is active against influenza viruses, West Nile virus, yellow fever virus, foot-and-mouth disease virus as well as other flaviviruses, arenaviruses, bunyavirusesand alphaviruses.[1]The mechanism of its actions is thought to be related to the selective inhibition of viral RNA-dependent RNA polymerase. Favipiravir does not inhibit RNA of DNA synthesis in mammalian cells and is not toxic to them.[1]- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D. F.; Barnard, D. L.; Gowen, B. B.; Julander, J. G.; Morrey, J. D. (2009). "T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections". Antiviral Research 82 (3): 95–102. doi:10.1016/j.antiviral.2009.02.198. PMID 19428599. edit
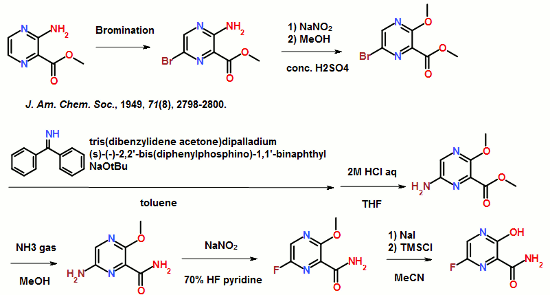
- WO 2000010569
- WO 2008099874
- WO 201009504
- WO 2010104170
- WO 2012063931
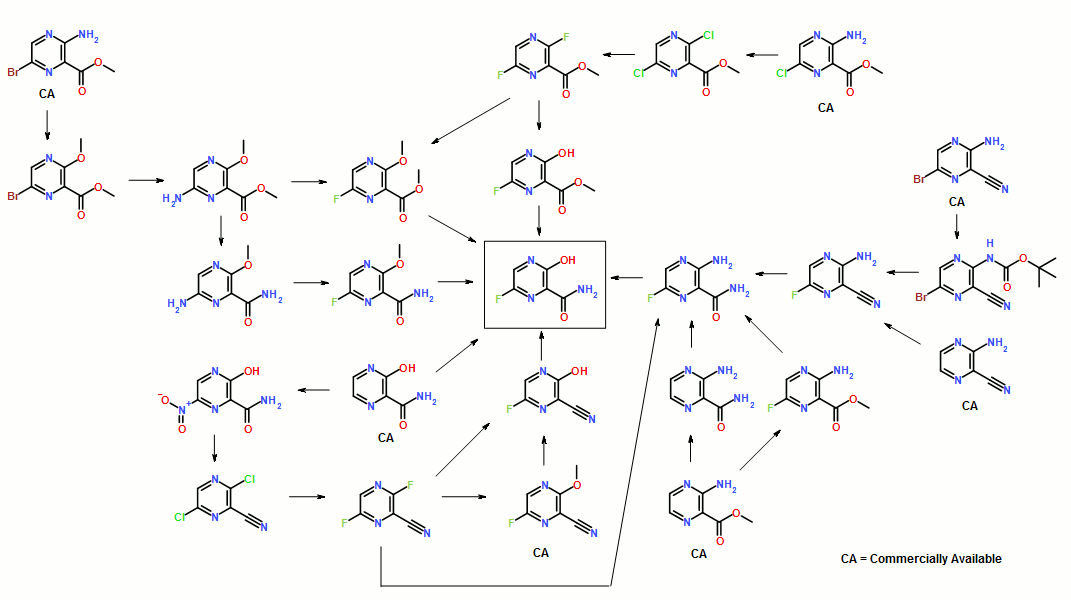
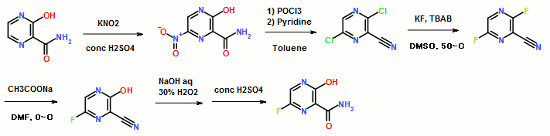
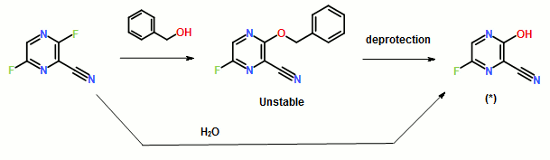
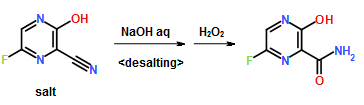
................................................................................................
Influenza virus is a central virus of the cold syndrome, which has attacked human being periodically to cause many deaths amounting to tens millions. Although the number of deaths shows a tendency of decrease in the recent years owing to the improvement in hygienic and nutritive conditions, the prevalence of influenza is repeated every year, and it is apprehended that a new virus may appear to cause a wider prevalence.
For prevention of influenza virus, vaccine is used widely, in addition to which low molecular weight substances such as Amantadine and Ribavirin are also used
.....................................

Synthesis of Favipiravir
ZHANG Tao1, KONG Lingjin1, LI Zongtao1,YUAN Hongyu1, XU Wenfang2*
(1. Shandong Qidu PharmaceuticalCo., Ltd., Linzi 255400; 2. School of Pharmacy, Shandong University, Jinan250012)
ABSTRACT: Favipiravir was synthesized from3-amino-2-pyrazinecarboxylic acid by esterification, bromination with NBS,diazotization and amination to give 6-bromo-3-hydroxypyrazine-2-carboxamide,which was subjected to chlorination with POCl3, fluorination with KF, andhydrolysis with an overall yield of about 22%.
......................................
US6787544

subs G1 G2 G3 G4 R2
.....................compd 32 N CH C—CF3 N H
EP2192117

Example 1-1

To a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile, a 3.8 ml water solution of 7.83 g of potassium acetate was added dropwise at 25 to 35° C., and the solution was stirred at the same temperature for 2 hours. 0.38 ml of ammonia water was added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of this solution was adjusted to 9.4 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added. Then 7.71 g of dicyclohexylamine was added dropwise and the solution was stirred at 20 to 30° C. for 45 minutes. Then 15 ml of water was added dropwise, the solution was cooled to 10° C., and the precipitate was filtered and collected to give 9.44 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyradinecarbonitrile as a lightly yellowish white solid product.
1H-NMR (DMSO-d6) δ values: 1.00-1.36 (10H, m), 1.56-1.67 (2H, m), 1.67-1.81 (4H, m), 1.91-2.07 (4H, m), 3.01-3.18 (2H, m), 8.03-8.06 (1H, m), 8.18-8.89 (1H, broad)
Example 1-2
4.11 ml of acetic acid was added at 5 to 15° C. to a 17.5 ml N,N-dimethylformamide solution of 5.0 g of 3,6-difluoro-2-pyrazinecarbonitrile. Then 7.27 g of triethylamine was added dropwise and the solution was stirred for 2 hours. 3.8 ml of water and 0.38 ml of ammonia water were added to the reaction mixture, and then 15 ml of water and 0.38 g of active carbon were added. The insolubles were filtered off and the filter cake was washed with 11 ml of water. The filtrate and the washing were joined, the pH of the joined solution was adjusted to 9.2 with ammonia water, and 15 ml of acetone and 7.5 ml of toluene were added to the solution, followed by dropwise addition of 7.71 g of dicyclohexylamine. Then 15 ml of water was added dropwise, the solution was cooled to 5° C., and the precipitate was filtered and collected to give 9.68 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile as a slightly yellowish white solid product.
Examples 2 to 5
The compounds shown in Table 1 were obtained in the same way as in Example 1-1.
TABLE 1 
Example No. Organic amine Example No. Organic amine 2 Dipropylamine 4 Dibenzylamine 3 Dibutylamine 5 N-benzylmethylamine
Dipropylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.39 (6H, t, J=7.5 Hz), 1.10 (4H, sex, J=7.5 Hz), 2.30-2.38 (4H, m), 7.54 (1H, d, J=8.3 Hz)
Dibutylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) 6 values: 0.36 (6H, t, J=7.3 Hz), 0.81 (4H, sex, J=7.3 Hz), 0.99-1.10 (4H, m), 2.32-2.41 (4H, m), 7.53 (1H, d, J=8.3 Hz)
Dibenzylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 4.17 (4H, s), 7.34-7.56 (10H, m), 8.07 (1H, d, J=8.3 Hz)
N-benzylmethylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile
1H-NMR (DMSO-d6) δ values: 2.57 (3H, s), 4.14 (2H, s), 7.37-7.53 (5H, m), 8.02-8.08 (1H, m)
Preparation Example 1

300 ml of toluene was added to a 600 ml water solution of 37.5 g of sodium hydroxide. Then 150 g of dicyclohexylamine salt of 6-fluoro-3-hydroxy-2-pyrazinecarbonitrile was added at 15 to 25° C. and the solution was stirred at the same temperature for 30 minutes. The water layer was separated and washed with toluene, and then 150 ml of water was added, followed by dropwise addition of 106 g of a 30% hydrogen peroxide solution at 15 to 30° C. and one-hour stirring at 20 to 30° C. Then 39 ml of hydrochloric acid was added, the seed crystals were added at 40 to 50° C., and 39 ml of hydrochloric acid was further added dropwise at the same temperature. The solution was cooled to 10° C. the precipitate was filtered and collected to give 65.6 g of 6-fluoro-3-hydroxy-2-pyrazinecarboxamide as a slightly yellowish white solid.
1H-NMR (DMSO-d6) δ values: 8.50 (1H, s), 8.51 (1H, d, J=7.8 Hz), 8.75 (1H, s), 13.41 (1H, s)
......................
jan 2014Investigational flu treatment drug has broad-spectrum potential to fight multiple viruses
First patient enrolled in the North American Phase 3 clinical trials for investigational flu treatment drugBioDefense Therapeutics (BD Tx)—a Joint Product Management office within the U.S. Department of Defense (DoD)—announced the first patient enrolled in the North American Phase 3 clinical trials for favipiravir (T-705a). The drug is an investigational flu treatment candidate with broad-spectrum potential being developed by BD Tx through a contract with Boston-based MediVector, Inc.Favipiravir is a novel, antiviral compound that works differently than anti-flu drugs currently on the market. The novelty lies in the drug's selective disruption of the viralRNA replication and transcription process within the infected cell to stop the infection cycle."Favipiravir has proven safe and well tolerated in previous studies," said LTC Eric G. Midboe, Joint Product Manager for BD Tx. "This first patient signifies the start of an important phase in favipiravir's path to U.S. Food and Drug Administration (FDA) approval for flu and lays the groundwork for future testing against other viruses of interest to the DoD."In providing therapeutic solutions to counter traditional, emerging, and engineered biological threats, BD Tx chose favipiravir not only because of its potential effectiveness against flu viruses, but also because of its demonstrated broad-spectrum potential against multiple viruses. In addition to testing favipiravir in the ongoing influenzaprogram, BD Tx is testing the drug's efficacy against the Ebola virus and other viruses considered threats to service members. In laboratory testing, favipiravir was found to be effective against a wide variety of RNA viruses in infected cells and animals."FDA-approved, broad-spectrum therapeutics offer the fastest way to respond to dangerous and potentially lethal viruses," said Dr. Tyler Bennett, Assistant Product Manager for BD Tx.MediVector is overseeing the clinical trials required by the FDA to obtain drug licensure. The process requires safety data from at least 1,500 patients treated for flu at the dose and duration proposed for marketing of the drug. Currently, 150 trial sites are planned throughout the U.S.SOURCE BioDefense TherapeuticsMalpani Y, Achary R, Kim SY, Jeong HC, Kim P, Han SB, Kim M, Lee CK, Kim JN, Jung YS.
Eur J Med Chem. 2013 Apr;62:534-44. doi: 10.1016/j.ejmech.2013.01.015. Epub 2013 Jan 29.
US3631036 * Nov 4, 1969 Dec 28, 1971 American Home Prod 5-amino-2 6-substituted-7h-pyrrolo(2 3-d) pyrimidines and related compounds US3745161 * Apr 20, 1970 Jul 10, 1973 Merck & Co Inc Phenyl-hydroxy-pyrazine carboxylic acids and derivatives US4404203 * May 14, 1981 Sep 13, 1983 Warner-Lambert Company Substituted 6-phenyl-3(2H)-pyridazinones useful as cardiotonic agents US4545810 * Mar 25, 1983 Oct 8, 1985 Sds Biotech Corporation Herbicidal and plant growth regulant diphenylpyridazinones US4565814 * Jan 18, 1984 Jan 21, 1986 Sanofi Pyridazine derivatives having a psychotropic action and compositions US4661145 * Sep 20, 1984 Apr 28, 1987 Rohm And Haas Company Plant growth regulating 1-aryl-1,4-dihydro-4-oxo(thio)-pyridazines US5420130 May 16, 1994 May 30, 1995 Synthelabo 2-aminopyrazine-5-carboxamide derivatives, their preparation and their application in therapeutics US5459142 * Aug 23, 1993 Oct 17, 1995 Otsuka Pharmaceutical Co., Ltd. Pyrazinyl and piperazinyl substituted pyrazine compounds US5597823 Jun 5, 1995 Jan 28, 1997 Abbott Laboratories Tricyclic substituted hexahydrobenz [e]isoindole alpha-1 adrenergic antagonists US6159980 * Sep 15, 1997 Dec 12, 2000 Dupont Pharmaceuticals Company Pyrazinones and triazinones and their derivatives thereof EP0023358A1 * Jul 28, 1980 Feb 4, 1981 Rohm And Haas Company Process for the preparation of pyridazine derivatives GB1198688A Title not available HU9401512A Title not available JPH09216883A * Title not available JPS5620576A Title not available - ...............................................
- 15 ASUNAPREVIR
 ASUNAPREVIR630420-16-5 CASTHERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES1. Cyclopropanecarboxamide, N-[(1,1-dimethylethoxy)carbonyl]-3-methyl-L-valyl-(4R)-4-[(7-chloro-4-methoxy-1-isoquinolinyl)oxy]-L-prolyl-1-amino-N-(cyclopropylsulfonyl)-2-ethenyl-, (1R,2S)-2. 1,1-dimethylethyl [(1S)-1-{[(2S,4R)-4-(7-chloro-4methoxyisoquinolin-1-yloxy)-2-({(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)pyrrolidin-1-yl]carbonyl}-2,2-dimethylpropyl]carbamateMOLECULAR FORMULA C35H46ClN5O9SMOLECULAR WEIGHT 748.3SPONSOR Bristol-Myers SquibbCODE DESIGNATION ...........BMS-650032CAS REGISTRY NUMBER 630420-16-5Asunaprevir (formerly BMS-650032) is an experimental drug candidate for the treatment of hepatitis C. It is undergoing development by Bristol-Myers Squibb and is currently inPhase III clinical trials.[1]In 2013, the company Bristol-Myers Squibb received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and BMS-791325.Asunaprevir is being tested in combination with pegylated interferon and ribavirin, as well as in interferon-free regimens with other direct-acting antiviral agents includingdaclatasvir[3][4][5]Asunaprevir is an antiviral agent originated by Bristol-Myers Squibb undergoing the registration in Japan for the treatment of chronic hepatitis C virus infection in combination with daclatasvir in patients who are non-responsive to interferon plus ribavirin and interferon based therapy ineligible naive/intolerant
ASUNAPREVIR630420-16-5 CASTHERAPEUTIC CLAIM Treatment of hepatitis CCHEMICAL NAMES1. Cyclopropanecarboxamide, N-[(1,1-dimethylethoxy)carbonyl]-3-methyl-L-valyl-(4R)-4-[(7-chloro-4-methoxy-1-isoquinolinyl)oxy]-L-prolyl-1-amino-N-(cyclopropylsulfonyl)-2-ethenyl-, (1R,2S)-2. 1,1-dimethylethyl [(1S)-1-{[(2S,4R)-4-(7-chloro-4methoxyisoquinolin-1-yloxy)-2-({(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)pyrrolidin-1-yl]carbonyl}-2,2-dimethylpropyl]carbamateMOLECULAR FORMULA C35H46ClN5O9SMOLECULAR WEIGHT 748.3SPONSOR Bristol-Myers SquibbCODE DESIGNATION ...........BMS-650032CAS REGISTRY NUMBER 630420-16-5Asunaprevir (formerly BMS-650032) is an experimental drug candidate for the treatment of hepatitis C. It is undergoing development by Bristol-Myers Squibb and is currently inPhase III clinical trials.[1]In 2013, the company Bristol-Myers Squibb received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and BMS-791325.Asunaprevir is being tested in combination with pegylated interferon and ribavirin, as well as in interferon-free regimens with other direct-acting antiviral agents includingdaclatasvir[3][4][5]Asunaprevir is an antiviral agent originated by Bristol-Myers Squibb undergoing the registration in Japan for the treatment of chronic hepatitis C virus infection in combination with daclatasvir in patients who are non-responsive to interferon plus ribavirin and interferon based therapy ineligible naive/intolerant- "A Phase 3 Study in Combination With BMS-790052 and BMS-650032 in Japanese Hepatitis C Virus (HCV) Patients". ClinicalTrials.gov.
- C. Reviriego (2012). Drugs of the Future 37 (4): 247–254.doi:10.1358/dof.2012.37.4.1789350.
- Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- "Bristol-Myers' Daclatasvir, Asunaprevir Cured 77%: Study". Bloomberg. Apr 19, 2012.
- AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Bioorganic and Medicinal Chemistry Letters, 2011 , vol. 21, 7 pg. 2048 - 2054
WO 2003099274, WO 2003099274, WO 2009085659US8202996 6-20-2012 Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide US8163921 4-25-2012 Hepatitis C Virus Inhibitors US7915291 3-30-2011 HEPATITIS C VIRUS INHIBITORS US7449479 11-12-2008 Hepatitis C virus inhibitors US6995174 2-8-2006 Hepatitis C virus inhibitors ..........Hepatitis C virus (HCV) is a major human pathogen, infecting an estimated 170 million persons worldwide—roughly five times the number infected by human immunodeficiency virus type 1. A substantial fraction of these HCV infected individuals develop serious progressive liver disease, including cirrhosis and hepatocellular carcinoma.Presently, the most effective HCV therapy employs a combination of alpha-interferon and ribavirin, leading to sustained efficacy in 40 percent of patients. Recent clinical results demonstrate that pegylated alpha-interferon is superior to unmodified alpha-interferon as monotherapy. However, even with experimental therapeutic regimens involving combinations of pegylated alpha-interferon and ribavirin, a substantial fraction of patients do not have a sustained reduction in viral load. Thus, there is a clear and unmet need to develop effective therapeutics for treatment of HCV infection.
 ........................Compound 277Compound 277 was prepared by following Scheme 2 of Example 269 except that 3- (4-chloro-phenyl)-3-methoxy-acrylic acid was used in place of 2- trifluormethoxycinnamic acid in step 1.Step 1:Modifications: 4.24 g 3-(4-chloro-phenyl)-3-methoxy-acrylic acid used, 130 mg product obtained (3% yield) Product:
........................Compound 277Compound 277 was prepared by following Scheme 2 of Example 269 except that 3- (4-chloro-phenyl)-3-methoxy-acrylic acid was used in place of 2- trifluormethoxycinnamic acid in step 1.Step 1:Modifications: 4.24 g 3-(4-chloro-phenyl)-3-methoxy-acrylic acid used, 130 mg product obtained (3% yield) Product: Data: 1H NMR(400 MHz, CD3OD) δ ppm 3.96 (s, 3 H), 7.19 (dd, 7=8.80, 2.45 Hz, 1 H), 7.28 (d, 7=2.45 Hz, 1 H), 7.34 (s, 1 H), 8.25 (d, 7=9.05 Hz, 1 H); MS: (M+H)+ 210.Step 2:Modifications: 105 mg 7-chloro-4-methoxy-2H-isoquinolin-l-one used, 60 mg product obtained (71% yield). Product:
Data: 1H NMR(400 MHz, CD3OD) δ ppm 3.96 (s, 3 H), 7.19 (dd, 7=8.80, 2.45 Hz, 1 H), 7.28 (d, 7=2.45 Hz, 1 H), 7.34 (s, 1 H), 8.25 (d, 7=9.05 Hz, 1 H); MS: (M+H)+ 210.Step 2:Modifications: 105 mg 7-chloro-4-methoxy-2H-isoquinolin-l-one used, 60 mg product obtained (71% yield). Product: Data: Η NMR (400 Hz, CDC13) δ ppm 4.05 (s, 3 H), 7.67 (dd, 7=8.80, 1.96 Hz, 1 H), 7.80 (s, 1 H), 8.16 (d, 7=9.05 Hz, 1 H), 8.24 (d, 7=1.96 Hz, 1 H); MS: (M+H)+ 229.Step 3:Modifications: 46 mg l,7-dichloro-4-methoxy-isoquinoline and 113 mg { l-[2-(l- cyclopropanesulfonylaminocarbonyl-2-vinyl-cyclopropylcarbamoyl)-4-hydroxy- pyrrolidine-1 -carbon yl]-2,2-dimethyl-propyl} -carbamic acid tert-butyl ester used, 50 mg product obtained (31% yield). Product:
Data: Η NMR (400 Hz, CDC13) δ ppm 4.05 (s, 3 H), 7.67 (dd, 7=8.80, 1.96 Hz, 1 H), 7.80 (s, 1 H), 8.16 (d, 7=9.05 Hz, 1 H), 8.24 (d, 7=1.96 Hz, 1 H); MS: (M+H)+ 229.Step 3:Modifications: 46 mg l,7-dichloro-4-methoxy-isoquinoline and 113 mg { l-[2-(l- cyclopropanesulfonylaminocarbonyl-2-vinyl-cyclopropylcarbamoyl)-4-hydroxy- pyrrolidine-1 -carbon yl]-2,2-dimethyl-propyl} -carbamic acid tert-butyl ester used, 50 mg product obtained (31% yield). Product: Compound 277Data: 1H NMR (400 Hz, CD3OD) δ ppm 1.06 (m, 11 H), 1.16 (s, 9 H), 1.24 (m, 2 H), 1.44 (dd, 7=9.54, 5.38 Hz, 1 H), 1.88 (dd, 7=8.07, 5.62 Hz, 1 H), 2.28 (m, 2 H), 2.59 (dd, 7=13.69, 6.85 Hz, 1 H), 2.94 (m, 1 H), 4.00 (s, 3 H), 4.05 (d, 7=11.74 Hz, 1 H), 4.19 (s, 1 H), 4.43 (d, 7=11.49 Hz, 1 H), 4.56 (dd, 7=10.03, 6.85 Hz, 1 H), 5.12 (d, 7=11.49 Hz, 1 H), 5.30 (d, 7=17.12 Hz, 1 H), 5.76 (m, 2 H), 7.57 (s, 1 H), 7.67 (d, 7=8.56 Hz, 1 H), 8.04 (s, 1 H), 8.08 (d, 7=8.80 Hz, 1 H); MS: (M+H)+ 749...............
Compound 277Data: 1H NMR (400 Hz, CD3OD) δ ppm 1.06 (m, 11 H), 1.16 (s, 9 H), 1.24 (m, 2 H), 1.44 (dd, 7=9.54, 5.38 Hz, 1 H), 1.88 (dd, 7=8.07, 5.62 Hz, 1 H), 2.28 (m, 2 H), 2.59 (dd, 7=13.69, 6.85 Hz, 1 H), 2.94 (m, 1 H), 4.00 (s, 3 H), 4.05 (d, 7=11.74 Hz, 1 H), 4.19 (s, 1 H), 4.43 (d, 7=11.49 Hz, 1 H), 4.56 (dd, 7=10.03, 6.85 Hz, 1 H), 5.12 (d, 7=11.49 Hz, 1 H), 5.30 (d, 7=17.12 Hz, 1 H), 5.76 (m, 2 H), 7.57 (s, 1 H), 7.67 (d, 7=8.56 Hz, 1 H), 8.04 (s, 1 H), 8.08 (d, 7=8.80 Hz, 1 H); MS: (M+H)+ 749...............
 ..................WO 2003099274
..................WO 2003099274 .........................
.........................
 Preparation of Compound CDMSO (264 ml) was added to a mixture of Compound A (6 g, 26.31 mmol, 1.0 eq, 96.5% potency), Compound B (6.696 g, 28.96 mmol, 1.1 eq) and KOtBu (8.856 g, 78.92 mmol, 3 eq) under nitrogen and stirred at 36° C. for 1 h. After cooling the dark solution to 16° C., it was treated with water (66 ml) and EtOAc (132 ml). The resulting biphasic mixture was acidified to pH 4.82 with 1N HCl (54 ml) at 11.2-14.6° C. The phases were separated. The aqueous phase was extracted once with EtOAc (132 ml). The organic phases were combined and washed with 25% brine (2×132 ml). Rich organic phase (228 ml) was distilled at 30-40° C./50 mbar to 37.2 ml. A fresh EtOAc (37.2 ml) was added and distilled out to 37.2 ml at 30-35° C./50 nm bar. After heating the final EtOAc solution (37.2 ml) to 50° C., heptane ((37.2 ml) was added at 46-51° C. and cooled to 22.5° C. over 2 h. It was seeded with 49 mg of Compound C and held at 23° C. for 15 min to develop a thin slurry. It was cooled to 0.5° C. in 30 min and kept at 0.2-0.5° C. for 3 h. After the filtration, the cake was washed with heptane (16.7 ml) and dried at 47° C./80 mm/15.5 h to give Compound C as beige colored solids (6.3717 g, 58.9% corrected yield, 99.2% potency, 97.4 AP).Preparation of Compound EDIPEA (2.15 ml, 12.3 mmol, 1.3 eq followed by EDAC (2 g, 10.4 mmol, 1.1 eq) were added to a mixture of Compound C (4 g, 9.46 mmol, 97.4% potency, 98.5 AP), Compound D (4.568 g, 11.35 mmol, 1.20 eq), HOBT-H2O (0.86 g, 4.18 mmol, 0.44 eq) in CH2Cl2 (40 ml) at 23-25° C. under nitrogen. The reaction was complete after 3 h at 23-25° C. It was then washed with 1N HCl (12 ml), water (12 ml) and 25% brine (12 ml). MeOH (80 ml) was added to the rich organic solution at 25° C., which was distilled at atmospheric pressure to ˜60 ml to initiate the crystallization of the product. The crystal slurry was then cooled from 64° C. to 60° C. in 5 min and stirred at 60° C. for 1 h. It was further cooled to 24° C. over 1.5 h and held at 24° C. for 2 h. After the filtration, the cake was washed with MeOH (12 ml) and dried at 51° C./20-40 nm i/18 h to give Compound E (5.33 g, 89% yield, 97.7% potency, 99.1 AP).Preparation of Compound F5-6N HCl in IPA (10.08 ml, 50.5 mmol, Normality: 5N) was added in four portions in 1 h to a solution of Compound E (8 g, 12.6 mmol, 97.7% potency, 99.1 AP) in IPA (120 ml) at 75° C. After stirring for 1 h at 75° C., the resulting slurry was cooled to 21° C. in 2 h and stirred at 21° C. for 2 h. It was filtered and the cake was washed with IPA (2×24 ml). The wet cake was dried at 45° C./House vacuum/16 h to give Compound F as an off-white solid (6.03 g, 84.5% yield, 98.5% potency, 100 AP).Preparation of Compound (I)DIPEA (9.824 ml) followed by HATU (7.99 g) were added to a stirred mixture of Compound F (10 g, 99.2% potency, 99.6 AP) and Compound G (4.41 g) in CH2Cl2 (100 ml) at 2.7-5° C. under nitrogen. The resulting light brown solution was stirred at 0.2-3° C. for 1.5 h, at 3-20° C. in 0.5 h and at 20-23° C. for 15.5 h for a reaction completion. It was quenched with 2N HCl (50 ml) at 23° C. and stirred for 20 min at 23-24° C. The biphasic mixture was polish filtered through diatomaceous earth (Celite®) (10 g) to remove insoluble solids of HOAT and HATU. The filter cake was washed with 20 ml of CH2Cl2. After separating the organic phase from the filtrates, it was washed with 2N HCl (5×50 ml) and water (2×50 ml). The organic phase (115 ml) was concentrated to ˜50 ml, which was diluted with absolute EtOH (200 proof, 100 ml) and concentrated again to ˜50 ml. Absolute EtOH (50 ml) was added to bring the final volume to 100 ml. It was then warmed to 50° C. to form a clear solution and held at 50° C. for 35 min. The ethanolic solution was cooled from 50 to 23° C. over 15 min to form the crystal slurry. The slurry was stirred at 23 CC for 18 h, cooled to 0.3° C. over 30 min and kept at 0.2-0.3° C. for 2 h. After the filtration, the cake was washed with cold EtOH (2.7° C., 2×6 ml) and dried at 53° C./72 mm/67 h to give Compound (I) in Form T1F-1/2 as an off white solid (10.49 g, 80.7% yield, 99.6 AP).https://www.google.co.in/patents/US20090202476?dq=WO+2009085659&ei=dzy5UpL_LMXXrQewxYG4Dw&cl=en.........extra infoHepatitis C virus (HCV) infection is the principal cause of chronic liver disease that can lead to cirrhosis, carcinoma and liver failure.1 More than 200 million people worldwide are chronically infected by this virus. Currently, the most effective treatment for HCV infection is based on a combination therapy of injectable pegylated interferon-α (PEG IFN-α) and antiviral drug ribavirin. This treatment, indirectly targeting the virus, is associated with significant side effects often leading to treatment discontinuation in certain patient populations.2 In addition, this treatment regimen cures only less than 50% of patients infected with genotype-1 which is the predominant genotype (while genotype 1a is most abundant in the US, the majority of sequences in Europe and Japan are from genotype 1b).3 Limited efficacy and adverse side effects of current treatment, and high prevalence of infection worldwide highlight an urgent need for more effective, convenient, and well-tolerated treatments.4HCV NS3 serine protease plays a critical role in the HCV replication by cleaving downstream sites (with the assistance of the cofactor NS4A) along the HCV viral polyprotein to produce functional proteins. Recently, NS3/4A protease inhibitors have emerged as a promising treatment for HCV infection.5 There are two distinct classes of NS3 protease inhibitors in clinical development. The first class is comprised of serine-trap inhibitors, exemplified by VX-950 (telaprevir)6 and SCH-503034 (boceprevir).7 The second class is represented by reversible noncovalent inhibitors such as macrocyclic inhibitors BILN-2061 (ciluprevir),8 ITMN-191 (danoprevir),9 TMC-43535010 and MK-7009 (vaniprevir).11 Due to concern over cardiac issues in animals treated with macrocyclic BILN-2061,12 newer acyclic inhibitors have recently been developed exemplified by BI-20133513 and BMS-650032.14 However, a rapid development of viral resistance has been observed for patients treated with HCV NS3 protease inhibitors.15 Therefore, the discovery of new NS3 protease inhibitors with novel binding paradigm and thus potentially differentiated resistance profile is highly desirable.
Preparation of Compound CDMSO (264 ml) was added to a mixture of Compound A (6 g, 26.31 mmol, 1.0 eq, 96.5% potency), Compound B (6.696 g, 28.96 mmol, 1.1 eq) and KOtBu (8.856 g, 78.92 mmol, 3 eq) under nitrogen and stirred at 36° C. for 1 h. After cooling the dark solution to 16° C., it was treated with water (66 ml) and EtOAc (132 ml). The resulting biphasic mixture was acidified to pH 4.82 with 1N HCl (54 ml) at 11.2-14.6° C. The phases were separated. The aqueous phase was extracted once with EtOAc (132 ml). The organic phases were combined and washed with 25% brine (2×132 ml). Rich organic phase (228 ml) was distilled at 30-40° C./50 mbar to 37.2 ml. A fresh EtOAc (37.2 ml) was added and distilled out to 37.2 ml at 30-35° C./50 nm bar. After heating the final EtOAc solution (37.2 ml) to 50° C., heptane ((37.2 ml) was added at 46-51° C. and cooled to 22.5° C. over 2 h. It was seeded with 49 mg of Compound C and held at 23° C. for 15 min to develop a thin slurry. It was cooled to 0.5° C. in 30 min and kept at 0.2-0.5° C. for 3 h. After the filtration, the cake was washed with heptane (16.7 ml) and dried at 47° C./80 mm/15.5 h to give Compound C as beige colored solids (6.3717 g, 58.9% corrected yield, 99.2% potency, 97.4 AP).Preparation of Compound EDIPEA (2.15 ml, 12.3 mmol, 1.3 eq followed by EDAC (2 g, 10.4 mmol, 1.1 eq) were added to a mixture of Compound C (4 g, 9.46 mmol, 97.4% potency, 98.5 AP), Compound D (4.568 g, 11.35 mmol, 1.20 eq), HOBT-H2O (0.86 g, 4.18 mmol, 0.44 eq) in CH2Cl2 (40 ml) at 23-25° C. under nitrogen. The reaction was complete after 3 h at 23-25° C. It was then washed with 1N HCl (12 ml), water (12 ml) and 25% brine (12 ml). MeOH (80 ml) was added to the rich organic solution at 25° C., which was distilled at atmospheric pressure to ˜60 ml to initiate the crystallization of the product. The crystal slurry was then cooled from 64° C. to 60° C. in 5 min and stirred at 60° C. for 1 h. It was further cooled to 24° C. over 1.5 h and held at 24° C. for 2 h. After the filtration, the cake was washed with MeOH (12 ml) and dried at 51° C./20-40 nm i/18 h to give Compound E (5.33 g, 89% yield, 97.7% potency, 99.1 AP).Preparation of Compound F5-6N HCl in IPA (10.08 ml, 50.5 mmol, Normality: 5N) was added in four portions in 1 h to a solution of Compound E (8 g, 12.6 mmol, 97.7% potency, 99.1 AP) in IPA (120 ml) at 75° C. After stirring for 1 h at 75° C., the resulting slurry was cooled to 21° C. in 2 h and stirred at 21° C. for 2 h. It was filtered and the cake was washed with IPA (2×24 ml). The wet cake was dried at 45° C./House vacuum/16 h to give Compound F as an off-white solid (6.03 g, 84.5% yield, 98.5% potency, 100 AP).Preparation of Compound (I)DIPEA (9.824 ml) followed by HATU (7.99 g) were added to a stirred mixture of Compound F (10 g, 99.2% potency, 99.6 AP) and Compound G (4.41 g) in CH2Cl2 (100 ml) at 2.7-5° C. under nitrogen. The resulting light brown solution was stirred at 0.2-3° C. for 1.5 h, at 3-20° C. in 0.5 h and at 20-23° C. for 15.5 h for a reaction completion. It was quenched with 2N HCl (50 ml) at 23° C. and stirred for 20 min at 23-24° C. The biphasic mixture was polish filtered through diatomaceous earth (Celite®) (10 g) to remove insoluble solids of HOAT and HATU. The filter cake was washed with 20 ml of CH2Cl2. After separating the organic phase from the filtrates, it was washed with 2N HCl (5×50 ml) and water (2×50 ml). The organic phase (115 ml) was concentrated to ˜50 ml, which was diluted with absolute EtOH (200 proof, 100 ml) and concentrated again to ˜50 ml. Absolute EtOH (50 ml) was added to bring the final volume to 100 ml. It was then warmed to 50° C. to form a clear solution and held at 50° C. for 35 min. The ethanolic solution was cooled from 50 to 23° C. over 15 min to form the crystal slurry. The slurry was stirred at 23 CC for 18 h, cooled to 0.3° C. over 30 min and kept at 0.2-0.3° C. for 2 h. After the filtration, the cake was washed with cold EtOH (2.7° C., 2×6 ml) and dried at 53° C./72 mm/67 h to give Compound (I) in Form T1F-1/2 as an off white solid (10.49 g, 80.7% yield, 99.6 AP).https://www.google.co.in/patents/US20090202476?dq=WO+2009085659&ei=dzy5UpL_LMXXrQewxYG4Dw&cl=en.........extra infoHepatitis C virus (HCV) infection is the principal cause of chronic liver disease that can lead to cirrhosis, carcinoma and liver failure.1 More than 200 million people worldwide are chronically infected by this virus. Currently, the most effective treatment for HCV infection is based on a combination therapy of injectable pegylated interferon-α (PEG IFN-α) and antiviral drug ribavirin. This treatment, indirectly targeting the virus, is associated with significant side effects often leading to treatment discontinuation in certain patient populations.2 In addition, this treatment regimen cures only less than 50% of patients infected with genotype-1 which is the predominant genotype (while genotype 1a is most abundant in the US, the majority of sequences in Europe and Japan are from genotype 1b).3 Limited efficacy and adverse side effects of current treatment, and high prevalence of infection worldwide highlight an urgent need for more effective, convenient, and well-tolerated treatments.4HCV NS3 serine protease plays a critical role in the HCV replication by cleaving downstream sites (with the assistance of the cofactor NS4A) along the HCV viral polyprotein to produce functional proteins. Recently, NS3/4A protease inhibitors have emerged as a promising treatment for HCV infection.5 There are two distinct classes of NS3 protease inhibitors in clinical development. The first class is comprised of serine-trap inhibitors, exemplified by VX-950 (telaprevir)6 and SCH-503034 (boceprevir).7 The second class is represented by reversible noncovalent inhibitors such as macrocyclic inhibitors BILN-2061 (ciluprevir),8 ITMN-191 (danoprevir),9 TMC-43535010 and MK-7009 (vaniprevir).11 Due to concern over cardiac issues in animals treated with macrocyclic BILN-2061,12 newer acyclic inhibitors have recently been developed exemplified by BI-20133513 and BMS-650032.14 However, a rapid development of viral resistance has been observed for patients treated with HCV NS3 protease inhibitors.15 Therefore, the discovery of new NS3 protease inhibitors with novel binding paradigm and thus potentially differentiated resistance profile is highly desirable.References and notes
- Rev. Med. Virol., 13 (2003), p. 57
- Hepatology, 36 (2002), p. S237
- Am. J. Med., 117 (2004), p. 344
- For a recent review on HCV anti-viral agents, see: Expert Opin. Invest. Drugs, 19 (2010), p. 63
- Curr. Opin. Pharmacol., 8 (2008), p. 522
- For a recent review on HCV NS3/4A protease inhibitors, see: Curr. Opin. Invest. Drugs, 10 (2009), p. 821
- Expert Rev. Anti. Infect. Ther., 7 (2009), p. 537
- Infect. Disord. Drug Targets, 6 (2006), p. 3
- Acc. Chem. Res., 41 (2008), p. 50
- J. Med. Chem., 47 (2004), p. 1605
- Antimicrob. Agents Chemother., 52 (2008), p. 4432
- Bioorg. Med. Chem. Lett., 18 (2008), p. 4853
- J. Med. Chem., 53 (2010), p. 2443
- Gastroenterology, 127 (2004), p. 1347
- J. Med. Chem., 53 (2010), p. 6466
- (a)Chemical and Engineering News (April 12, 2010 issue), 88, pp 30–33.
- (b)Perrone, R.K.; Wang, C.; Ying, W.; Song, A.I. WO 2009085659
- Sci. Transl. Med., 2 (2010), p. 30ra32
- ....................................................
- CONTD ON
- http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
- ....................................................................
- THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.DGLENMARK SCIENTIST , NAVIMUMBAI, INDIAdid you feel happy, a head to toe paralysed man’s soul in action for you round the clockneed help, email or call meMOBILE-+91 9323115463web linkI was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
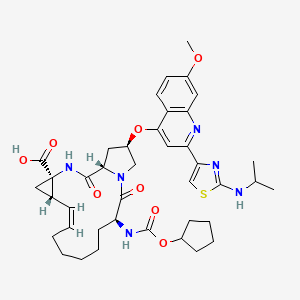
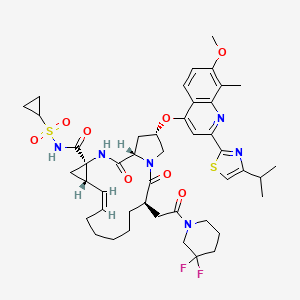
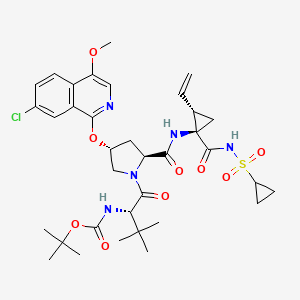
Our trained chemists work on custom projects designed specifically for the needs of each client. N-hexylpyridinium bis((trifluoromethyl)sulfonyl)imide
ReplyDeleteMulțumim pentru sugestiile minunate, acesta este într-adevăr un blog minunat și aceste sfaturi sunt foarte utile pentru pacienții cu cancer
ReplyDeletemyhep lvir,myhep lvir comprimat,myhep lvir pret in India
My life is beautiful thanks to you, Mein Helfer. Lord Jesus in my life as a candle light in the darkness. You showed me the meaning of faith with your words. I know that even when I cried all day thinking about how to recover, you were not sleeping, you were dear to me. I contacted the herbal center Dr Itua, who lived in West Africa. A friend of mine here in Hamburg is also from Africa. She told me about African herbs but I was nervous. I am very afraid when it comes to Africa because I heard many terrible things about them because of my Christianity. god for direction, take a bold step and get in touch with him in the email and then move to WhatsApp, he asked me if I can come for treatment or I want a delivery, I told him I wanted to know him I buy ticket in 2 ways to Africa To meet Dr. Itua, I went there and I was speechless from the people I saw there. Patent, sick people. Itua is a god sent to the world, I told my pastor about what I am doing, Pastor Bill Scheer. We have a real battle beautifully with Spirit and Flesh. Adoration that same night. He prayed for me and asked me to lead. I spent 2 weeks and 2 days in Africa at Dr Itua Herbal Home. After the treatment, he asked me to meet his nurse for the HIV test when I did it. It was negative, I asked my friend to take me to another nearby hospital when I arrived, it was negative. I was overwhite with the result, but happy inside of me. We went with Dr. Itua, I thank him but I explain that I do not have enough to show him my appreciation, that he understands my situation, but I promise that he will testify about his good work. Thank God for my dear friend, Emma, I know I could be reading this now, I want to thank you. And many thanks to Dr. Itua Herbal Center. He gave me his calendar that I put on my wall in my house. Dr. Itua can also cure the following diseases, HIV, Herpes, Hepatitis B, Inflammatory Liver, Diabetes,Bladder cancer,Brain cancer,Esophageal cancer,Gallbladder cancer,Gestational trophoblastic disease,Head and neck cancer,Hodgkin lymphoma
ReplyDeleteIntestinal cancer,Kidney cancer,Leukemia,Liver cancer,Lung cancer,Melanoma,Mesothelioma,Multiple myeloma,Neuroendocrine tumors
Non-Hodgkin lymphoma,Oral cancer,Ovarian cancer,Sinus cancer,Skin cancer,Soft tissue sarcoma,Spinal cancer,Stomach cancer
Testicular cancer,Throat cancer,Thyroid Cancer,Uterine cancer,Vaginal cancer,Vulvar cancerBipolar Disorder, Bladder Cancer,Colorectal Cancer,HPV,Breast Cancer,Anal cancer.Appendix cancer.Kidney Cancer,Prostate Cancer,Glaucoma., Cataracts,Macular degeneration,Adrenal cancer.Bile duct cancer,Bone cancer.Cardiovascular disease,Lung disease.Enlarged prostate,OsteoporosisAlzheimer's disease,Brain cancer,Bunyavirus,Dementia.Weak Erection,Love Spell,Leukemia,Fibroid,Infertility,Parkinson's disease,Inflammatory bowel disease ,Fibromyalgia, recover your ex. You can contact him by email or drituaherbalcenter@gmail.com, ..WhatsApp phone number+ 2348149277967 .. He is a good doctor, talk to him kindly. I'm sure he will also listen to you.
Nice job, it’s a great post. The info is good to know! https://techealthinfo.com/omni-health-30-day-weight-management-system/ Excellent read, I just passed this into a colleague who was doing a little research on that. And he actually bought me lunch because I found it for him smile So let me rephrase that.
ReplyDelete