
TELAPREVIR
MF C36H53N7O6
MolWeight: 679.85
CAS No.: 402957-28-2
NMR..........http://www.abmole.com/download/vx-950-hnmr.pdf
AND
http://file.selleckchem.com/downloads/nmr/S153802-Telaprevir-NMR-Selleck.pdf
1H NMR

13C NMR
.png)



Chem. Commun., 2010,46, 7918-7920
DOI: 10.1039/C0CC02823A
http://pubs.rsc.org/en/Content/ArticleLanding/2010/CC/c0cc02823a#!divAbstract'
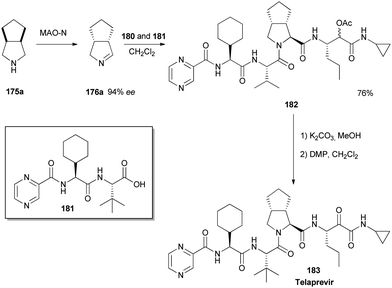
A very short and efficient synthesis of the important drug candidate telaprevir, featuring a biocatalytic desymmetrization and two multicomponent reactions as the key steps, is presented. The classical issue of lack of stereoselectivity in Ugi- and Passerini-type reactions is circumvented. The atom economic and convergent nature of the synthetic strategy require only very limited use of protective groups .

Telaprevir (1). 250 l of saturated K2CO3 was added to a solution of 14 (0.514 g, 0.75 mmol) in MeOH (20 ml) at room temperature. The reaction mixture was stirred for 30 minutes at room temperature resulting in a pale yellow suspension. After full conversion (as judged by TLC analysis), the reaction mixture was washed with 20 ml brine, the aqueous layer was washed again with 10 ml CH2Cl2 (2x). The organic layers were collected, dried with MgSO4 and concentrated in vacuo, to yield a pale yellow solid. The yellow solid was dissolved in CH2Cl2 (10 ml) and Dess-Martin periodinane (0.650 g, 1.532 mmol) was added at room temperature. The reaction mixture was stirred overnight before adding saturated NaHCO3 solution (10 ml) and saturated Na2S2O3 solution (10 ml). This mixture was stirred for 10 minutes, separated and the aqueous layers were washed with EtOAc (2 x 10 ml). The organic layers were collected, dried with MgSO4 and concentrated in vacuo to give the crude product as an 83:13:4 mixture of diastereomers. After silica gel flash chromatography (1% MeOH in CH2Cl2), 1 (0.412 mg, 0.61 mmol, 80%) was obtained as a white solid.
1H NMR (500.23 MHz, DMSO-d6): δ = 9.19 (d, J = 1.4 Hz, 1H), 8.91 (d, J = 24.5 Hz, 1H), 8.76 (dd, J = 1.5, 2.5 Hz, 1H), 8.71 (d, J = 5.3 Hz, 1H), 8.49 (d, J = 9.2 Hz, 1H), 8.25 (d, J = 6.8 Hz, 1H), 8.21 (d, J = 8.9 Hz, 1H), 4.94 (m, 1H), 4.68 (dd, J = 6.5, 9.0 Hz, 1H), 4.53 (d, J = 9.0 Hz, 1H), 4.27 (d, J = 3.5 Hz, 1H), 3.74 (dd, J = 8.0, 10 Hz, 1H), 2.74 (m, 1H), 3.64 (d, J = 3.5 Hz, 1H), 0.92 (s, 9H), 0.87 (t, 3H), 0.84-1.40 (m, 23H), 0.65 (m, 2H), 0.56 (m, 2H);
13C NMR (125.78 MHz, CDCl3): δ = 197.0 (C), 171.8 (C), 170.4 (C), 169.0 (C), 162.1 (C), 161.9 (C), 147.9 (CH), 144.0 (C), 143.4 (CH), 56.4 (CH), 56.3 (CH), 54.2 (CH), 53.4 (CH), 42.3 (CH), 41.3 (CH), 32.1 (CH), 31.8 (CH), 31.6 (CH), 29.1 (CH), 28.0 (CH), 26.4 (CH3);
max (cm -1 ): 3302 (m), 2928 (m), 2858 (w), 1658 (s), 1620 (s), 1561 (s), 1442 (m);
HRMS (ESI, 4500 V): m/z calcd. for C36H53N7O6Na + ([M + Na] + ) 702.3950, found 702.3941.
SYN
Reference:

WO2011/103932 A1, ; Page/Page column 50; 51 ;

WO2011/103932 A1, ;
 AND
AND 
WO2013/135870 A1, ;
 WO2013/135870 A1, ;
WO2013/135870 A1, ; WO2011/103932 A1, ;
WO2011/103932 A1, ;
...................
t mpound XVIII,

(XVIII).
This compound, which also known as Telaprevir, could be prepared in higher yields and with higher efficiency than any previously disclosed processes. Furthermore, the chiral information used for the preparation was derived from readily available simple building blocks, making the process a highly effective approach to such prolyl dipeptides and similar peptidomimetics.
EXAMPLE 22

(5)-Methyl 2-cyclohexyl-2-(pyrazine-2-carboxamido)acetate (9).
Pyrazinecarboxylic acid (2.72 g, 21.9 mmol) was added to a solution of L- cyclohexylglycine methyl ester (4.13 g, 19.9 mmol) in CH2CI2 (100 ml) at room temperature under N2, forming a white suspension. Triethylamine (6.33 ml, 4.62 g, 45.8 mmol) was added, followed by benzotriazol-l-yloxy-tris-(dimethylamino)- phosphonium hexafluorophosphate (BOP; 9.69 g, 21.9 mmol), which turned the reaction mixture from purple to an orange solution. After two days of stirring at room temperature the reaction mixture was washed two times with 50 ml saturated Na2CC>3, followed by the washing of the aqueous layers with CH2CI2 (2 χ 50 ml). The organic layers were collected and dried with MgSC , followed by concentration in vacuo. Purification by silica gel flash chromatography (c-Hex:EtOAc = 2: 1 with 0.5% triethylamine) afforded 9 (5.28 g, 19.03 mmol, 96%) as a yellow oil that solidified upon standing to give a white solid.
[a f° = +42.5 (c= 1.13, CHC13); *H NMR (250.13 MHz, CDCI3) δ = 9.39 (d, J= 1.25 Hz, 1H), 8.76 (d, J = 2.5 Hz, 1H), 8.57 (t, J = 1.5 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 4.74 (dd, J= 5.5, 9.3 Hz, 1H), 3.78 (s, 3H), 1.96 (m, 1H), 1.77 (m, 5H), 1.24 (m, 5H); 13C NMR (62.90 MHz, CDCI3): δ= 172.0 (C), 162.8 (C), 147.4 (CH), 144.5 (CH), 144.1 (C), 142.7(CH), 57.0 (CH), 52.3 (CH3), 41.2 (CH), 29.7 (CH2), 28.4 (CH2), 26.0 (CH2); IR (neat): v^cm ) = 3374 (m), 2920 (s), 2845 (w), 1740 (s), 1665 (s); HRMS (ESI, 4500 V): m/z calcd. for Ci4Hi9N303Na+ ([M + Na]+) 300.1319, found 300.1319.
Example 23 :

(5)-2-cyclohexyl-2-(pyrazine-2-carboxamido)acetic acid (10). A solution of 1 M NaOH (12 ml, 12 mmol) was added to a solution of 9 (2.77 g, 10 mmol) in THF (25 ml) at 0°C. MeOH was added to the formed suspension, to give a clear, colorless solution. The reaction mixture was stirred overnight at room temperature, followed by concentration in vacuo. The pH of the aqueous layer was set on 3.5 with a 1 M KHSO4 solution and was extracted with EtOAc (2 χ 25 ml). The mixture was dried with Na2S04, filtered, and concentrated in vacuo, to give 10 (2.49 g, 9.45 mmol, 95%) as a white solid.
[a £° = +50.9 (c= 1.06, CHC13); H NMR (250.13 MHz, CDCI3): δ = 9.38 (d, J = 1.5
Hz, 1H), 8.78 (d, J= 2.5 Hz, 1H), 8.58 (dd, J= 1.5, 2.5 Hz, 1H), 8.27 (d, J= 9.0, 1H), 4.77 (dd, J = 4.3, 5.0 Hz, 1H), 2.00 (m, 1H), 1.76 (m, 5H), 1.37 (m, 5H); 13C NMR (62.90 MHz, CDCI3): δ = 175.7 (C), 163.0 (C), 147.2 (CH), 144.3 (CH), 144.2 (C), 142.0 (CH), 56.9 (CH), 40.9 (CH), 29.7 (CH2), 28.1 (CH2), 25.9 (CH2); IR (neat): vmax (cm4) = 3383 (m), 2928 (s), 2852 (w), 1713 (m), 1676 (s), 1518 (s); HRMS (ESI, 4500 V): m/z calcd. For Ci3H17N303Na+ ([M + Na]+) 286.1162, found 286.1158.
Example 23 :

(S)-methyl 2-((S)-2-cyclohexyl-2-(pyrazine-2-carboxamido)-acetamido)-3,3- dimethylbutanoate (11). 10 (0.653 g, 4.5 mmol) was added to a solution of H-Tle- OMe (0.653 g, 4.5 mmol) in DMF (40 ml). l-Ethyl-3-(3-dimethylaminopropyl)- carbodiimide-HCl (EDOHC1; 0.919 g, 6.75 mmol) was added to this colorless solution followed by 1 -hydroxy-7-azabenzotriazole (HOAt; 1.035 g, 5.4 mmol) giving a bright yellow solution. The reaction mixture was stirred for 3 days and afterwards concentrated in vacuo. The formed yellow solid was dissolved in EtOAc, washed with 40 ml saturated aqueous ammonium chloride solution and 40 ml of saturated aqueous NaHCC>3 solution. The organic layers were collected, dried with MgSC^ and concentrated in vacuo to give 11 (1.48 g, 3.78 mmol, 84%) as a white solid.
[a f° = -2.0 (c= 1.0, CHC13); ¾ NMR (250.13 MHz, CDC13): δ = 9.39 (d, J = 1.5 Hz, 1H), 8.76 (d, J = 2.3 Hz, 1H), 8.55 (dd, J = 2.4, 1.8 Hz, 1H), 8.29 (d, J = 8.1, 1H), 6.40 (d, J= 9.3 Hz, 1H), 4.46 (m, 2H), 3.74 (s, 3H), 1.81 (m, 1H), 1.76 (m, 4H), 1.24 (m, 6H), 0.96 (s, 12H); 13C NMR (62.90 MHz, CDC13): δ = 171.7 (C) , 170.4 (C), 163.0 (C), 147.5 (CH), 144.5 (CH), 144.2 (C), 142.7 (CH), 60.2 (CH3), 58.4 (CH), 51.9 (CH), 40.5 (CH), 31.7 (C), 29.7 (CH2), 28.7 (CH2), 26.6 (CH3), 25.9 (CH2); IR (neat): v„(cmJ) = 3350 (m), 2928 (m), 2853 (w), 1738 (s), 1686 (s), 1640 (s), 1520 (s); HRMS (ESI, 4500 V): m/z calcd. for C2oH3oN404Na+ ([M + Na]+) 413.2159, found 413.2169.
Example 24:

(S)-2-((S)-2-cyclohexyl-2-(pyrazine-2-carboxamido)acetamido)-3,3- dimethylbutanoic acid (2). A solution of 1 M NaOH (0.94 ml, 0.94 mmol) was added to a solution of 11 (0.31 g, 0.78 mmol) in THF (3 ml) at 0°C. MeOH was added to the formed suspension, to give a clear and colourless solution. The reaction mixture was stirred overnight at room temperature, followed by concentration in vacuo. The pH of this aqueous layer was set to 3.5 with 1 M KHSO4 and subsequently extracted with EtOAc (2 χ 10ml). The mixture was dried with Na2S04, filtered, and concentrated in vacuo, to give 2 (0.28 g, 0.75 mmol, 95%) as a white solid.
[a f° = +21.7 (c= 1.015, CHC13); *H NMR (250.13 MHz, CDC13): δ = 9.39 (d, J= 1.3
Hz, 1H), 8.77 (d, J = 2.5 Hz, 1H), 8.57 (dd, J = 1.5, 2.5 Hz, 1H), 8.35 (d, J = 9 Hz, 1H), 6.70 (d, J = 9.0 Hz, 1H), 4.45 (t, J = 8.8 Hz, 1H), 4.42 (d, J = 9.2 Hz, 1H), 1.94 (m, 1H), 1.71 (m, 5H), 1.20 (m, 5H), 1.01 (s, 9H); 13C NMR (62.90 MHz, CDCI3): δ = 173.4 (C), 170.5 (C), 163.3 (C), 147.4 (CH), 144.4 (CH), 144.2 (C), 142.8 (CH), 58.4 (CH), 51.9 (CH), 40.4 (CH), 34.7 (C), 29.8 (CH2), 28.6 (CH2), 26.6 (CH3), 25.8 (CH2); IR (neat): v„(cm ) = 3335 (w), 2930 (m), 1726 (m), 1663 (s), 1514 (s); HRMS (ESI, 4500 V): m/z calc. for Ci9H29N404Na+ ([M + Na]+) 399.2003, found 399.2013.
Example 25:

(5)-2-formamido-l-pentanol (12). (5)-2-amino-l-pentanol (1.00 g, 9.7 mmol) was dissolved in ethylformate (7.84 ml, 7.19 g, 97 mmol). This reaction mixture was refluxed at 80 °C for 4 hours, followed by stirring overnight at room temperature. The colourless solution was concentrated in vacuo and stirred for 1 hour in a 10 mol% K2C03 in MeOH (25 ml). Afterwards, the pH was set to 7 with DOWEX 50wx8, followed by filtration and concentration in vacuo to give 12 (1.26 g, 9.61 mmol, 99%). [a f° = -29.6 (c = 1.15, CHCI3); H NMR (250.13 MHz, CDC13): δ = 8.20 (s, 1H), 5.81 (bs, 1H), 4.04 (m, 1H), 2.11 (b, 1H), 1.47 (m, 4H), 0.94 (t, J = 7.0 Hz, 3H); 13C NMR (62.90 MHz, CDCI3): 161.8 (C), 65.1 (CH2), 50.6 (CH), 33.2 (CH2), 19.2 (CH2), 13.9 (CH3); IR (neat): vmax (cm ) = 3248 (s), 2957 (m), 1651 (s), 1528 (m), 1381 (m); HRMS (ESI, 4500 V): m/z calcd. for C6Hi3N02Na+ ([M + Na]+) 154.0838, found 154.0835.
Example 26:

(5)-2-formamidopentanal. (7). Dess-Martin periodinane (5.514 g, 13 mmol) was added to a solution of (5)-2-formamido-l-pentanol (12, 1.31 g, 10 mmol) in CH2C12 (100 ml) at room temperature. The white suspension was stirred for 2 days and subsequently 35 ml MeOH was added and stirred for 30 minutes. The resulting suspension was filtrated and the filtrate was concentrated in vacuo. The crude product was purified by silica gel flash chromatography (cHex:EtOAc = 1 :4) to give 7 (1.08 g,
8.29 mmol, 83%) as a white solid. NMR analysis indicates that 7 is in equilibrium with its cyclic dimer.
[a f° = +37.6 ( c= 0.745, CHC13); lU NMR assigned to the monomer (250.13 MHz, CDCI3): δ = 8.22 (s, 1H), 7.84 (s, 1H), 7.10 (m, 1H), 5.31 (m, 1H), 1.52 (m, 4H), 0.95 (m, 3H); 13C NMR assigned to the monomer (100.61 MHz, CDC13): 198.8 (CH), 161.7 (CH), 57.4 (CH), 30.8 (CH2), 18.4 (CH2), 13.7 (CH3); lH NMR assigned to the dimer (400.13 MHz, CDC13) 8.22 (s, 2H), 5.26 (m, 2H), 3.72 (m, 2H) 1.52 (m, 8H), 0.95 (m, 6Η;) 13C NMR (100.61 MHz, CDCI3) assigned to the dimer: 161.7 (CH), 89.8 (CH), 63.1 (CH), 30.8 (CH2), 18.4 (CH2), 13.7 (CH3); IR (neat): Vmax (cm ): 3325 (s), 2959 (s), 1649 (s), 1530 (s), 1381 (m), 1123 (w); HRMS (ESI, 4500 V): m/z calc. for C6Hi2N02 + ([M + H]+) 130.0863, found 130.0858.
It was noted that the dimer exists as a mixture of diastereomers.
Example 27:

(35)-2-acetoxy- V-cyclopropyl-3-formamidohexanoyl amide (13). From 7: Aldehyde 7 (0.892 g, 6.91 mmol) was added to a solution of cyclopropyl isocyanide (0.410 g, 6.12 mmol) in CH2C12 (110 ml) and stirred for 5 minutes at room temperature. Acetic acid (0.711 ml, 0.747 g, 12.44 mmol) was added and the yellow reaction mixture was stirred for 3 days at room temperature. The reaction mixture was washed twice with 100 ml saturated Na2C03, followed by drying with Na2S04 and concentration in vacuo. The crude was purified by silica gel flash chromatography (5% MeOH in CH2C12, 1% triethylamine). (3S)-2-acetoxy-N-cyclopropyl-3- formamidohexanoyl amide (0.99 g, 3.87 mmol, 56%) was obtained as a white solid as a 78:22 mixture of diastereomers.
From 12: Dess Martin periodinane (5.66 g, 12.3 mmol) was added to a solution of (S)-N-(l hydroxypentan-2-yl)formamide (1.15 g, 8.8 mmol) in CH2C12 (12 ml) at room temperature. The white suspension was stirred for 60 minutes and subsequently cyclopropyl isocyanide (0.74 g, 10.0 mmol) was added and stirred for 48 hours. The resulting suspension was filtrated and washed twice with 10 ml saturated Na2C03, followed by drying with Na2SC>4 and concentration in vacuo. The crude product was purified by silica gel flash chromatography (5% MeOH in CH2CI2, 1% triethylamine) to give 13 (1.34 g, 5.22 mmol, 60%) as a pale yellow solid as a 78:22 mixture of diastereomers.
*H NMR (130 °C, 400.13 MHz, DMSO-i¾: δ = 8.03 (s, 1H), 7.52 (m, 1H), 7.30 (m, 1H), 4.89 (d, J= 4.4, 1H), 4.28 (m, 1H), 2.65 (m, 1H), 2.17(s, 3H), 1.27-1.47 (m, 4H), 0.89 (t, J= 7.2, 3H), 0.63 (m, 2H), 0.48 (m, 2H); 13C NMR (125.78 MHz, DMSO-i 6): δ = 169.8 (C), 168.5 (C), 160.6 (CH), 74.4 (CH), 47.5 (CH), 22.2 (CH), 18.4 (CH3), 13.6 (CH3), 5.7 (CH2); IR (neat): vmax (cm ) 3283 (s), 2961 (w), 1744 (m), 1661 (s), 1530 (s), 1238 (s); HRMS (ESI, 4500 V): m/z calcd. for Ci2H2oN204Na+ ([M + Na]+) 279.1315, found 279.1325.
Example 28:

(35)-2-acetoxy-7V-cyclopropyl-3-isocyano-hexanoyl amide (4). N- methylmorpholine (0.57 ml, 0.562 g, 5.56 mmol) was added to a solution of (5)-l- (cyclopropylamino)-3-formamido-l-oxohexan-2-yl acetate (0.713 g, 2.78 mmol) in CH2CI2 (40 ml) at room temperature. The reaction mixture was cooled to -78 °C and triphosgene (0.289 g, 0.97 mmol) was quickly added and stirred for 5 minutes at this temperature. The resulting yellow solution was warmed up to -30 °C and was stirred for another 3 h. Subsequently, the reaction was quenched with water and extracted twice with CH2CI2 (40 ml). The organic layers were collected, dried with Na2SC>4 and concentrated in vacuo. The crude product was purified by silica gel flash chromatography (2% MeOH in CH2C12) to give 4 (0.578 g, 2.42 mmol, 87%) as a white solid. lU NMR (250.13 MHz, CDC13): δ = 6.28 (s, 1H), 5.25 (d, J = 2.5 Hz, 1H), 4.2 (m, 1H), 2.74 (m, 1H), 2.24 (s, 3H), 1.55 (m, 4H), 0.96 (m, 3H), 0.84 (m, 2H), 0.60 (m, 2H); 13C NMR (62.90 MHz, CDCI3): δ= 169.7 (C), 168.3 (C), 74.4 (CH), 47.5 (CH), 22.0 (CH), 20.6 (CH3), 18.5 (CH2), 13.5 (CH3), 5.5 (CH2); IR (neat): Vmas(cm ): 3267 (s), 2959 (m), 1745 (m), 1643 (s), 1512 (m), 1221 (s); HRMS (ESI, 4500 V): m/z calcd. for Ci2H18N203Na+ ([M + Na]+) 261.1210, found 261.1214.
Example 29:

Compound 14. Isocyanide 4 (0.549 g, 2.3 mmol) was dropwise added to a solution of imine 3 (0.252 g, 2.3 mmol) and carboxylic acid 2 (0.602 g, 1.60 mmol) in CH2C12 (5 ml) at room temperature. This yellow solution was stirred for 72 hours and afterwards diluted with 5 ml CH2C12. The reaction mixture was washed twice with saturated Na2C03 solution (10 ml) and twice with saturated NH4CI. The organic layers were collected, dried with MgS04 and concentrated in vacuo. The crude product was purified by silica gel flash chromatography (5% MeOH in CH2C12) to give 14 (0.876 g, 1.21 mmol, 76%) as a mixture of diastereomers.
lU NMR (500.23 MHz, CDC13): δ = 9.50 (s, 1H), 8.75 (d, J = 2.5, 1H), 8.59 (s, 1H), 8.35 (d, J = 9.0, 1H), 6.84 (d, J= 9.0, 1H), 6.44 (s, 1H), 5.20 (d, J= 3.0, 1H), 4.74 (d, J= 9.5, 1H), 4.58 (t, J = 7.5, 1H), 4.38 (m, 1H), 3.37 (d, J= 6.0, 1H), 2.82 (m, 1H), 2.69 (m, 1H), 2.11 (s, 3H), 1.26 (s, 2H), 0.97 (s, 9H), 0.86 (m, 3H), 0.84-2.00 (m, 21H), 0.76 (m, 2H), 0.51 (m, 2H);13C NMR (125.78 MHz, CDC13): δ = 170.5 (C), 169.3 (C), 162.9 (C), 147.4 (CH), 144.6 (CH), 144.2 (C), 142.8 (CH), 74.4 (CH), 66.6 (CH), 58.3 (CH), 56.6 (CH), 54.5 (CH2), 44.9 (CH), 43.0 (CH), 41.3 (CH), 35.5 (C), 26.4 (CH3), 20.8 (CH3), 19.1 (CH2), 13.8 (CH3), 6.6 (CH2); vmax (cm4): 3306 (m), 2928 (m), 2931 (m), 1743 (w), 1655 (s), 1520 (m), 1219 (m); HRMS (ESI, 4500 V): m/z calcd. for C38H57N707Na+ ([M + Na]+) 746.4212, found 746.4107.
Example 30:

Telaprevir (1). 250 μΐ of saturated K2C03 was added to a solution of 14 (0.514 g, 0.75 mmol) in MeOH (20 ml) at room temperature. The reaction mixture was stirred for 30 minutes at room temperature resulting in a pale yellow suspension. After full conversion (as judged by TLC analysis), the reaction mixture was washed with 20 ml brine, the aqueous layer was washed again with 10 ml CH2C12 (2x). The organic layers were collected, dried with MgS04 and concentrated in vacuo, to yield a pale yellow solid. The yellow solid was dissolved in CH2CI2 (10 ml) and Dess-Martin periodinane (0.650 g, 1.532 mmol) was added at room temperature. The reaction mixture was stirred overnight before adding saturated NaHC03 solution (10 ml) and saturated Na2S203 solution (10 ml). This mixture was stirred for 10 minutes, separated and the aqueous layers were washed with EtOAc (2 x 10 ml). The organic layers were collected, dried with MgSC^ and concentrated in vacuo to give the crude product as an 83: 13:4 mixture of diastereomers. After silica gel flash chromatography (1% MeOH in CH2C12), 1 (0.412 mg, 0.61 mmol, 80%) was obtained as a white solid. lU NMR (500.23 MHz, DMSO-i¾: 5 = 9.19 (d, J= 1.4 Hz, 1H), 8.91 (d, J= 24.5 Hz, 1H), 8.76 (dd, J = 1.5, 2.5 Hz, 1H), 8.71 (d, J= 5.3 Hz, 1H), 8.49 (d, J= 9.2 Hz, 1H), 8.25 (d, J = 6.8 Hz, 1H), 8.21 (d, J = 8.9 Hz, 1H), 4.94 (m, 1H), 4.68 (dd, J= 6.5, 9.0 Hz, 1H), 4.53 (d, J = 9.0 Hz, 1H), 4.27 (d, J = 3.5 Hz, 1H), 3.74 (dd, J = 8.0, 10 Hz, 1H), 2.74 (m, 1H), 3.64 (d, J = 3.5 Hz, 1H), 0.92 (s, 9H), 0.87 (t, 3H), 0.84-1.40 (m, 23H), 0.65 (m, 2H), 0.56 (m, 2H);
13C NMR (125.78 MHz, CDC13): δ = 197.0 (C), 171.8 (C), 170.4 (C), 169.0 (C), 162.1 (C), 161.9 (C), 147.9 (CH), 144.0 (C), 143.4 (CH), 56.4 (CH), 56.3 (CH), 54.2 (CH), 53.4 (CH), 42.3 (CH), 41.3 (CH), 32.1 (CH), 31.8 (CH), 31.6 (CH), 29.1 (CH), 28.0 (CH), 26.4 (CH3); (cm4): 3302 (m), 2928 (m), 2858 (w), 1658 (s), 1620 (s), 1561 (s), 1442 (m);
HRMS (ESI, 4500 V): m/z calcd. for C36H53N706Na+ ([M + Na]+) 702.3950, found 702.3941.
..................
telaprevir according to Formula 1

Telaprevir is a protease inhibitor that can be used as antiviral drug. By way of example, telaprevir inhibits the hepatitis C virus NS3-4A serine protease.
Although some processes for the synthesis of telaprevir or its pharmaceutical acceptable salts are available, it is an object of the present invention to provide an alternative process, in particular an enhanced process that overcomes at least one of the problems of the prior art processes.
Y. Yip et al. Bioorg. Med. Chem. Lett., 2004, 14, 5007 discloses the preparation of a 1 :1 mixture of isomers defined by Formula 5a (see Scheme 1 ) which isomers appear to have a stereochemical configuration other than that of telaprevir. WO 2007/022459 A2 discloses a process for preparing telaprevir, wherein in a first coupling step, a bicyclic pyrrolidine derivative is reacted with a protected amino acid, followed by a stepwise extension of the chain of the amino acid to provide a tripeptide as shown in Formula 2. Subsequently, a β-amino acid is added to the carbon chain-end opposite to said previously built chain. Finally, telaprevir is obtained in an oxidation step. Turner et al. (Chemical Communications 2010, 46(42), 7918) discloses a process for the preparation of teiaprevir by applying an Ugi reaction type process which reacts a compound of Formula 2

a chiral imine, namely (3aS,6aR)-1 ,3a,4,5,6,6a-hexahydrocyclopenta[c]pyrrole, which is obtained by enzyme technology and is thus difficult to prepare and is instabile and a relatively unstable isonitrile derivative of formula 4.
Summary of the invention The known processes for the preparation of teiaprevir are based on long linear sequences or require the use of labile, highly reactive agents and specific enzymes. The process described herein may for example allow to avoid the use of said labile, highly reactive reactants and specific enzymes. It was surprisingly found within the context of the present invention that teiaprevir may be prepared in a smaller number of process steps in a convergent manner by using stabile precursors (see an example process in Figure 1 ). The present invention may also contribute to preserving the desired stereochemical configuration during the process of preparing teiaprevir. In particular, it has been found that the desired stereochemical configuration may be preserved during the process of peptide bond formation in the compound according to Formula 5 when using the coupling agents described herein, in particular when using 2, 4,6-tripropyl-1 ,3,5,2,4,6- trioxatriphosphorinane-2,4,6-trioxide (T3P) or related compounds in dichloromethane. It is also possible to use a combination of a diimide coupling reagent, including but not being limited to dicyclohexylcarbodiimide (DCC), diispropylcarbodiimide (DIC) and 1-ethyl-3-(3- dimethy!aminopropyl)carbodiimide hydrochloride (EDC), with 1-hydroxy-benzotriazole (HOBt) or 1-hydroxy-7-aza-benzotriazole (HOAt) or related reagents for preparing teiaprevir. It has been found that the coupling agents are particularly effective when used in the presence of a lewis acid such as a copper salt. It was also unexpectedly found that the choice of solvent for carrying out the coupling reaction may further enhance the preservation of the stereochemical configuration during peptide bond formation in the compound according to Formula 5.
Furthermore, the expensive compound according to Formula 3

3
is used at a later stage of the process compared to the process of WO 2007/022459 A2, namely for coupling to the compound according to Formula 2 which already represents a dipeptide. Considering the yields of the single process steps, a smaller amount of the compound according to Formula 3 is required according to the invention, and, thus, the process may be less costly. Compared to the above method of Turner et al., it is not required to use a toxic and instable isonitrile compound. It has also been found that the process for preparing telaprevir may provide an advantage since fewer impurities such as epimeric forms and other byproducts may be formed.
Example 1b - (1S,3aR,6aS)-tert-butyl 2-((S)-2-((S)-2-cyclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3 dimethylbutanovDoctahydrocvclopentafclpyrrole-l- carboxylate (5b)

A round bottom flask is charged with 41 mg of 2 (0.11 mmol, 1 eq.) and 1 ml. of DCM is added. Then, 29 mg of 3b (0.16 mmol, 1.5 eq.) are added. After stirring for 5 min 190μΙ of T3P (50% in EtOAc, 0.32 mmol, 3 eq.) are added and the reaction mixture is stirred for 21 h at room temperature.
The reaction is then diluted with DCM and quenched with water. The aqueous layer is separated and re-extracted with DCM. The combined organic layers are washed with brine, dried over Na2S04, filtered and concentrated in vacuo. Purification by flash chromatography yielded 5b (26 mg, 43% yield), (d.r. = 7.5:1 ).
Example 1c - (1S,3aR.6aS)-tert-butyl 2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3 dimethylbutanoyl)octahvdrocvclopentarc1pyrrole-1- carboxylate (5b)

A round bottom flask is charged with 1g of 2 (2.66 mmol, 1 eq.) and 20 ml_ of DCM is added. Then, 0.73g of 3b (3.98 mmol, 1.5 eq.) are added. After stirring for 5 min 4.75mL of T3P (50% in EtOAc, 7.98 mmol, 3 eq.) are added and the reaction mixture is stirred for 21 h at room temperature. The reaction is then quenched with water. The aqueous layer is separated and re-extracted with DCM. The combined organic layers are washed with sat. NaHC03-solution, brine and then dried over Na2S04, filtered and concentrated in vacuo. Purification by flash chromatography yielded 5b (0.70g, 49% yield), (d.r. = 5.6:1 ).
Example 1d - (1S,3aR,6aS)-tert-butyl 2-gS)-2-((S)-2-cvclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3.3 dimethylbutanoyl)octahvdrocvclopentaMpyrrole-1- carboxylate (5b)

A round bottom flask is charged with 1.0g of 2 (2.66 mmol, 1 eq.) and 0.58g of 3b (3.19 mmol, 1.2 eq), then 8 mL of DMF is added and the mixture cooled to 0°C using an ice-bath. In a second flask, 0.36g CuCI2 (2.66 mmol, 1 eq.) are dispersed in 5mL DMF, cooled to 0°C and the previously prepared solution is added to it. Now, 0.36g HOBt (2.66 mmol, 1 eq.) and 2.0g EDC HCI (10.43 mmol, 4 eq.) are added and the mixture is then stirred at r.t. for 16h.
The reaction is then quenched with 10ml_ 10% NH3-solution and then extracted 4 times with a total of 60mL of EtOAc. The combined organic layers are then washed 3 times with dilute hydrochloric acid, once with sat. NaHC03-solution and brine, then dried over Na2S04, filtered and concentrated in vacuo. Purification by flash chromatography yielded 5b (0.78g, 54% yield), (d.r. = 53 : 1 ).
Example 1e - (1S.3aR.6aS)-tert-butyl 2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3 dimethylbutanoyl)octahvdrocvclopentarclpyrrole-1- carboxylate (5b)

A round bottom flask is charged with 1.25g PS-supported HOBt (1.07 mmol/mg) and 0.30g of 3b (1.65 mmol, 1.2 eq) and 0.36g CuCI2 (2.66 mmol, 1 eq.). Then 15 ml_ of DMF are added and the mixture cooled to 0°C using an ice-bath, while mixing with a mechanical stirrer. In a second flask, 0.5g of 2 (1.3 mmol, 1 eq.) and 1.0g EDC HCI (5.21 mmol, 4 eq.) are dispersed in 12mL DMF, cooled to 0°C and added to the previously prepared solution. The mixture is then stirred at r.t. for 22h.
The reaction is then filtered and the filter washed with 15mL DMF. 50ml_ EtOAc are added to the filtrate, followed by 35ml_ 5% NH3-solution. The aqueous layer is then separated and extracted 3 times with a total of 45ml_ of EtOAc. The combined organic layers are then washed once with 10% NH3-solution, dilute hydrochloric acid, sat. NaHC03-solution and brine, then dried over Na2S04, filtered and concentrated in vacuo. Purification by flash chromatography yielded 5b (0.43g, 61 % yield), (d.r. = 18 : 1 ).
Example 1f - (1S.3aR,6aS)-tert-butyl 2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3 dimethylbutanovDoctahydrocvclopentafclpyrrole-l- carboxylate (5b)

A round bottom flask is charged with 2.0g of 2 (5.3 mmol, 1 eq.) and 1.17g of 3b (6.4 mmol, 1.2 eq), then 10 mL of DMF is added and the mixture cooled to 0°C using an ice-bath, then 0.72g CuCI2 (5.3 mmol, 1 eq.) are added. In a second flask 0.72g HOBt (5.3 mmol, 1 eq.) and 1.34g DIC (10.6 mmol, 2 eq.) are dissolved in 5mL DMF, cooled to 0°C and added to the previously prepared solution. The mixture is then stirred at r.t. for 5h.
The reaction is then quenched with 30mL 2% NH3-solution and then extracted 3 times with a total of 60ml_ of EtOAc. The combined organic layers are then washed 3 times with a total of 60mL of dilute hydrochloric acid, once with 20ml_ sat. NaHC03-solution and 20ml_ of brine, then dried over Na2S04, filtered and concentrated in vacuo. Purification by flash chromatography yielded 5b (2.59g, 90% yield), (d.r. = 340 : 1 )
Example 1g - Use of HOAT as anti-isomerisation reagent
To 4.4ml of a 0.6M HOAT solution in DMF (1.1eq, 2.63mmol) were added 0.6ml DMF.
Afterwards 1g of 2 (90% content, 1eq, 2.39mmol), 567mg of 3b (1.3eq, 3.11 mmol) and 391 mg DIC (1.3eq, 3.11 mmol) were added at room temperature. The reaction was stirred at room temperature for 23h. After 19h 86% conversion to 5b, with a d.r. 3.9/1 was observed. After 23h, with 87% conversion to 5b, and a d.r. 4.1/1 the conversion had stalled and the product was not isolated. Example 1 h - Use of CuCI? with in situ generation of AOC-Et from its HCI salt
353mg water free CuCI2 (1.1 eq, 2.63mmol) was dissolved in 5ml DMF. To the solution 1 g 2 (90% content, 1 eq, 2.39mmol), 683mg 3b. HCI (1 .3eq, 3.1 1 mmol), 315mg NMM (1.3eq,
3.1 1 mmol) and 391 mg DIC (1.3eq, 3.1 1 mmol) were added at room temperature. The reaction mixture was stirred at room temperature. After two hours 2.6area% 2 was detected and yield was 96.5% (calculated via internal standard). After 5h less than 0.5area% 2 was detected and yield was 98.1 %. d.r. at both IPCs was 108/1.
Example 1 i - HOAT without CuCI? in DMF
To a solution of 0.5g 2 (90%, 1 eq, 1 .19mmol) and 179mg HOAT (1.1eq, 1.32mmol) in 2.5ml DMF 284mg 3b (1.3eq, 1 .55mmol) was added. Afterwards
241 μΙ DIC (1 .3eq, 1.55mmol) was added. Reaction was stirred at room temperature. After2.5h 91 % conversion and DR of 4.3/1 was observed. After 5h complete conversion
Was observed and DR of 4.1/1. No work was performed.
Example 1j - HOAT without CuCI, in THF/MED
To a suspension of 0.5g 2 (90, 1 eq. 1.19mmol) and 179mg HOAT (1 .1 eq, 1 ,32mmol) in 2.5ml of a 1/1 mixture of THF/MED (methylene chloride) 284mg 3b (1.3eq, 1.55mmol) was added. Afterwards, 241 μΙ DIC (1.3eq, 1 .55mmol) was added. Reaction was stirred at room
temperature. After2.5h 7.5% conversion and DR of 6.7/1 was observed. After 5h 80 conversion was observed and DR of 6.2/1 . After 19h 86% conversion and DR of 6.0/1 was found. No work was performed. Example 1 k - HOAT with CuCI? in DMF
177mg CuCI2 (1 .1 eq, 1 .31 mmol) was dissolved in 2.5ml DMF. To the solution 0.5g 2 (90%, 1 eq, 1.19mmol), 179mg HOAT (1.1 eq, 1 .32mmol), 284mg 3b (1.3eq, 1.55mmol) and 241 μΙ DIC (1.3eq, 1.55mmol) was added. The reaction was stirred at room temperature for 13h. 95% conversion and DR of 48/1 was observed. No work up was performed.
Example 11 - Substochiometric amounts of CuCI? in DMF without HOAT
177mg CuCI2 (0.55eq, 1.31 mmol) was dissolved in 5ml DMF. To the solution 1.0g 2 (90%, 1 eq, 2.39mmol), 683mg 3b.HCI (1.3eq, 3.11 mmol), 340μΙ NMM (1.3eq, 3.11 mmol) and 481 μΙ DIC (1.3eq, 3.11 mmol) was added. The reaction was stirred at room temperature for 4.5h, complete conversion and DR of 76/1 was observed. No separated work up was performed. Example 2a - Synthesis of the compound according to Formula 7
(1S.3aR.6aS)-2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2-carboxamido)acetamido)-3,3- dimethylbutanoyl)octahvdrocvclopentarclpyrrole-1 -carboxylic acid (7)

A round-bottom flask was charged with 6g of 5b (11.08 mmol, 1 eq.) and 85mL of THF and 26mL of H20 was added. Then 2.20g LiOH H20 (52.43 mmol, 4.7 eq.) were added and the mixture was stirred at r.t. for 18h.
Then 50ml_ EtOAc and 50mL H20 were added, and the aqueous layer separated. The organic layer was washed once more with 40ml_ H20. To the combined aqueous layers 50ml_ of EtOAc were added, and by slow addition of 2M HCI the pH was adjusted to 1.89. After separation of the aqueous layer, it was extracted once more with 50ml_ EtOAc, and the combined organic layers washed with brine, then dried over Na2S04, filtered and concentrated in vacuo.
Purification by flash chromatography yielded 7 (4.83g, 85% yield).
Example 2b - Synthesis of the compound according to Formula 7
(1S,3aR,6aS)-2-((S)-2-((S)-2-cyclohexyl-2-(pyrazine-2-carboxamido)acetamido)-3,3- dimethylbutanoyl)octahvdrocvclopenta[c1pyrrole-1 -carboxylic acid (7)

A round bottom flask is charged with 2.0g of 2 (5.3 mmol, 1 eq.) and 1.17g of 3b (6.4 mmol, 1.2 eq), then 10 mL of DMF is added and the mixture cooled to 0°C using an ice-bath, then 0.72g CuCI2 (5.3 mmol, 1 eq.) are added. In a second flask 0.72g HOBt (5.3 mmol, 1 eq.) and 1 .34g DIC (10.6 mmol, 2 eq.) are dissolved in 3ml_ DMF, cooled to 0°C and added to the previously prepared solution. The mixture is then stirred at r.t. for 5h.
The reaction is then quenched with 30mL 2% NH3-solution and then extracted 3 times with a total of 70ml_ of EtOAc. The combined organic layers are then washed with 15mL 2% NH3- solution, 1 time with 20ml_ 1 M HCI, 3 times with a total of 60ml_ of dilute hydrochloric acid, once with 20ml_ sat. NaHC03-solution and 20ml_ of brine, then dried over Na2S04, filtered and concentrated in vacuo. The residue (compound 5b - 2.59g, 4.78 mmol, 1 eq.) was dissolved in 27ml_ of a 1 :1 mixture THF/H20. Then 0.48g NaOH (1 1.95 mmol, 2.5 eq.) were added and the mixture was stirred at r.t. for 18h.
Then 20ml_ EtOAc and 10ml_ H20 were added, and the aqueous layer separated. The organic layer was washed once more with 20ml_ H20. To the combined aqueous layers 20ml_ of EtOAc were added, and by slow addition of 2M HCI the pH was adjusted to 1.27. After separation of the aqueous layer, it was extracted once more with 20ml_ EtOAc, and the combined organic layers washed with brine, then dried over Na2S04, filtered and concentrated in vacuo to give 7 (2.82g, 93% yield). Trace metal analysis using ICP-OES showed residual copper < 1 ppm, wherein the following method was used:
Digestion: about 250mg of sample material was digested under pressure with a mixture of HNO3+HCI in a closed quartz container which can be heated by microwave radiation.
Determination of Cu:
Measurement was performed with ICP-OES at 324,754nm, Axialplasm, simultaneous background correction; calibration with external standards, certified elemental standard of Merck, Device: Fabr. Thermo Electron, Type: IRIS Intrepid XSP II, Duo. Example 2c - Synthesis of the compound according to Formula 7
(1 S,3aR,6aSV2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2-carboxamido)acetamido)-3.3- dimethylbutanoyl)octahvdrocyclopenta[clpyrrole-1 -carboxylic acid (7)

7.07g water free CuCI2 (1.1eq, 52.6mmol) was dissolved in 100ml DMF. To the solution 20g of 2 (90% content, 1eq, 47.82mmol), 10.51g of 3b (d.r. = 9:1) (1.2eq, 57.38mmol), 6.3ml NMM (1.2eq, 57.38mmol) and 9.6ml DIC (1.3eq, 62.16mmol) were added at 0°C. the reaction mixture was warmed to 40°C in 1.5h and stirred at that temperature until complete conversion was observed. To the reaction mixture isopropyl acetate was added followed by the addition of 10% HCI. The organic phase was separated and washed with 5% ammonia and 2% NaCI. The organic solvent was removed to dryness and 26.95g was isolated as a diasteromeric mixture of 9:1 detected via NMR.
6g of this material was dissolved in 15ml ethanol and 15ml water. To the mixture 1.15g sodium hydroxide was added. The reaction mixture was stirred at room temperature until no further conversion was observed. Ethanol was removed via distillation and water was added. The basic aqueous phase was washed with isopropyl acetate, the organic phase was re-extracted with water. To the combined aqueous phase Isopropyl acetate was added an pH was adjusted to 1.5 via addition of 10% HCI. The organic phase was separated and solvent was removed to dryness to yield 5.29g of compound 7 as a single diastereomer according to NMR analysis.
Example 2d - Use of CuCI? as anti isomerisation reagent (without any triazol reagent) 353mg water free CuCI2 (1.1eq, 2.63mmol) was dissolved in 5ml DMF. To the solution 1g 2 (90% content, 1eq, 2.39mmol), 567mg 3b (1.3eq, 3.11 mmol) and 391 mg DIC (1.3eq,
3.1 1 mmol) were added at room temperature. The reaction mixture was stirred at room temperature for 19h full conversion to 5b with d.r. 116/1 was observed. To the reaction mixture 50ml of ethyl acetate was added and the occurring precipitation was removed via filtration. The organic phase was washed with 5% ammonia and the aqueous phase was reextracted with 50ml ethyl acetate. The combined organic phase was washed with 40ml 2M HCI and 40ml brine. After drying with sodium sulfate and filtration the organic solvent was removed via evaporation. The solid residue was dissolved in 20ml methylene chloride and again the solvent was removed to dryness. After drying (rt, 40mbar), 1.373g of a slightly yellow amorphous solid (NMR content 81.6%, yield 86.4%).
Example 3 - Synthesis of the compound according to Formula 6
(1S,3aR,6aSV2-((S)-2- SV2-cvclohexyl-2-(pyrazine-2-carboxamido)acetamidoV3,3- dimethylbutanoyl)-N-((S)-1-(cvclopropylamino)-2-hvdroxy-1-oxohexan-3- yl)octahvdrocvclopentarclpyrrole-1-carboxamide (6)

A round-bottom flask was charged with 11.87g of 7 (23.11 mmol, 1 eq.), 5.32g of EDC*HCI (27.73mmol, 1.2 eq.), 3,74g of HOBt (27.73 mmol, 1.2 eq.) and 80 mL DCM were added. The mixture was cooled with an ice-bath and a suspension of 5.66g of 4 (25.42mmol, 1.1 eq.) in 50 mL of DCM containing 2.75g NEt3 (27.73 mmol, 3.88mL, 1.2 eq.) was added. This mixture was then stirred at r.t. for 6h when conversion was complete.
The reaction was quenched by addition of 50ml_ H20, followed by dropwise addition of 6M HCI to adjust the pH to 1.45. The aqueous layer was separated and extracted once with 50ml_ DCM. The combined organic layers were washed with 50ml_ sat. NaHC03 solution and 50mL brine, dried over Na2S04, filtered and concentrated in vacuo. Purification by flash
chromatography yielded 6 (14.89g, 94% yield).
Example 4 - Synthesis of the compound according to Formula 1
(1S,3aR,6aS)-2-((S)-2-((S)-2-cvclohexyl-2-(pyrazine-2-carboxamido)acetamido)-3.3- dimethylbutanoyl)-N-((S)-1-(cvclopropylamino)-1 ,2-dioxohexan-3- yl)octahvdrocvclopentaFclpyrrole-1-carboxamide (Telaprevir) (1)

A round-bottom flask was charged with 2.00g of 6 (2.93 mmol, 1 eq.) and 20mL of DCM and then cooled with an ice-bath. 200μΙ of 15% KBr-solution and 800μΙ of sat. NaHC03-solution were added, followed by 1 1 mg of TEMPO (0.07mmol, 0.025 eq.) and 600μΙ 10% NaOCI- solution. After stirring at r.t. for 18h, another 1.2ml_ of 10% NaOCI-solution were added - after another 2h the reaction was complete.
The reaction mixture was then diluted with 10mL of H20. After separation of the aqueous layer it was extracted with 10ml_ of DCM. The combined organic layers were washed with 10ml_ of 1 % Na2S03 and 10ml_ of H20, dried over Na2S04, filtered and concentrated in vacuo.
The residue was then stirred in 40ml_ Et20, filtered, washed with 10ml_ of cold Et20 and then dried in vacuo to give crystalline 1 (1 .41g, 71 %).
Cited literature
WO 2007/022459 A2; Turner et al. (Chemical Communications 2010, 46(42), 7918); WO2010/126881 ; Y. Yip et al. Bioorg. Med. Chem. Lett., 2004, 14, 5007; Harbeson, S. L. et al. J. Med. Chem. 1994, 37, 2918-2929.
| WO2007022459A2 | 18 Aug 2006 | 22 Feb 2007 | Vertex Pharma | Processes and intermediates |
| WO2010126881A1 | 27 Apr 2010 | 4 Nov 2010 | Vertex Pharmaceuticals Incorporated | Processes and intermediates |
..................
PATENT
http://www.google.im/patents/WO2010126881A1?cl=en







5 6 7 8 (rac)
9 10 1
Scheme I
Scheme II
a cyclopropylamide of Formula 18 is prepared using the Passeπni reaction (see, e.g., A. Doemling et al., Angew. Chem., 2000, 1 12, 3300-3344). Scheme IV
Scheme V
34
The invention further relates to a process for piepaπng a compound of Formula 4

[0060] In some embodiments, the process for preparing compounds of Formula 4 includes the steps of i) providing an N-alkoxycarbonyl-S-azabicycloP 3 0]octane, ii) forming a 2-anion of the N-alkoxycarbonyl-3-azabicyclo[3 3 0]octane in the presence of a chelating agent, iii) treating the anion of step ii) with carbon dioxide to produce a cis /trans mixture of N-alkoxycarbonyl-octahydrocyclopenta[c]pyrrole- l-carboxyhc acids, iv) treating the mixture of step iii) with a strong base to produce an essentially pure trans- N-alkoxycarbonyl-octahydrocyclopenta[c]ρyrrole-l -carboxyhc acid, v) forming a salt of the carboxylic acid with an optically active amine, vi) crystallizing the salt, vii) esteπfying the salt provided in step vi), viii) removing the N-alkoxycarbonyl gioup to produce (lS,3aR,6aS)-f-butyl- octahydiocyclopenta[c)pyiτole-l -carboxylate, /-butyl ester, ix) reacting the bicyclic of step viii) with a protected amino acid of Formula 26,

26 wherein Z is an amine protecting group, in the piesence of a coupling reagent, to pioduce an amide-ester of Formula 27,

27 x) removing the protecting group Z from the amide-ester of step ix) to produce the amino compound of Formula 28,

28 xi) reacting the amino compound of Formula 28 with a protected amino acid of Formula 29
Z-HN^/CO2H
29 in the presence of a coupling reagent to produce a tripeptide of Formula 30;

30 xii) removing the protecting group Z in the tripeptide of Formula 30 to produce a free amino-tripeptide of Formula 31;

31 xiii) reacting the amino-tripeptide of Formula 31 with pyrazine-2-carboxylic acid in the presence of a coupling reagent to produce an amide-tripeptide ester of Formula 33;

33 xiv) hydrolyzing the ester of the amide-tripeptide ester of Formula 33 to produce an amide-tripeptide acid of Formula 34;

34
XV) reacting the amide-tπpeptide acid of Formula 34 with an aminohydroxy-amide of Formula 18


35 xvi) oxidizing the hydroxy gioup of Formula 35 to produce the compound of Formula 4

Example 13: (lS,3aR,6aS)-2-((S)-2-((S)-2-cyclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3-dimethylbutanoyl)-N-((S)-l-(cyclopropylamino)-l,2- dioxohexan-3-yl)octahydrocyclopenta[c]pyrrole-l -carboxamide (4)

35 4
Method 1
[00256] A 500 inL 3-neck round bottomed flask equipped with an overhead stirrer, condenser, thermocouple, and nitrogen outlet was purged with nitrogen for several minutes A methylene chloride solution of the hydroxyamide peptide amide 35 (128 64 g, 16-17wt%, 20 6 g and 30 mmol of 35) in methylene chloride was added to the reaction flask, followed by the addition of 15% w/w aq NaBr (13 mL) and 7 5% w/w aq NaHCO3 (52 mL) The solution was cooled to 5±3 0C in an ice bath TEMPO (0 7 g) dissolved in methylene chloride (3 mL) was added to the reaction mixture In a separate Erlenmeyer flask, 10- 13% NaOCI solution (23 25 mL, titer = 108 mg/mL, 2 51 g, 33 7 mmol, 1 12 molar eq ) was diluted with water (70 mL) The NaOCI solution was charged to the reaction mixture via addition funnel at a rate that maintained the temperature below 8 0C The reaction mixture was allowed to stir at 5±3 0C for lhour The layers were separated and the organic layer was quenched with 10% (w/w) aq Na2SOs (100 mL) and washed with water (100 mL) The organic phase was reduced to dryness at reduced pressure and the solid triturated with ethyl acetate (100 mL) and filtered on a Buchner funnel to give the title compound Method 2
[00257] TEMPO (1 09 g, 6 95 mmol) was added to the methylene chloπde solution of 35 from Example 12, Method 2, followed by a solution of sodium bicarbonate (21 89 g, 260 5 mmol) in water (400 mL) and the mixture cooled to 0-5 0C A solution of sodium hypochlorite (122 17 g, 11 64 wt%, 191 04 mmol) was added over 2 hours while maintaining a temperature of 0-5 0C The mixture was stirred for 1 hour at 0-5 0C, then the phases separated The organic phase was washed with water (500 mL), 1 wt% aqueous sodium bisulfite (500 mL) and water (500 mL), then polish filtered The mixture was distilled at 38- 42 0C, 710 mm Hg, to a volume of about 320 mL Ethyl acetate (44 mL) was added followed immediately by 1 5 g of seed ciystals of 4 and the mixture was stirred for 15 minutes at 38-42 0C Ethyl acetate (800 mL) was added over 3 hours while maintaining a temperature of 38-42 0C The mixture was then distilled at 38-42 0C, 200-250 mm Hg, to a volume of about 400 mL Additional ethyl acetate (200 mL) was added over 0 5 hour The resultant slurry was cooled over 1 hour to 20-25 0C and stirred an additional hour at the same temperature The mixture was filtered and the filter cake washed with ethyl acetate (twice, 300 mL each) and dned under vacuum with a nitrogen bleed at 45-55 0C to give the title compound 4 as a white solid. Method 3
[00258] TEMPO (0 06 eq) was added to the CH2CI2 solution of 35 from Example 12, method 3, and the solution was stirred at 20-25 0C until all TEMPO dissolved To this solution was added a solution of NaHCOi (1 5 eq ) in water (4 vol ) The resulting biphasic mixture was cooled to 0-5 0C While maintaining the reaction temperature at 0-5 0C, a 10-13 wt% NaOCl solution (1 10 eq ) was added over 2-3 hours and the mixture stirred for additional one hour The layers were separated and the organic layer was washed at 0-5 0C with H2O (5 vol ), 1 wt% Na2SO3 (5 vol ), and H2O (5 vol ) Glacial acetic acid (0 12 eq ) was added to the solution of compound 4 in CH2Cl2 to stabilize compound 4
Example 14: Recrystallization of Compound of Formula 4.
[00259] The solution of Compound 4 from Example 13, Method 3, was filtered through Cehte, and the filtrate solution was reduced to 3 1 -3 3 volumes by vacuum distillation at lower than 20 0C After distillation, the solution was brought to 38-420C before EtOAc (0 80 vol ) was added, followed by the addition of Compound 4 seed (1 5 wt% relative to 34, Example 12) The resulting mixture was stirred for 15 minutes at 38-42 0C EtOAc (8 vol ) was added over 3 hours to this mixture while maintaining a temperature of 38-42 0C The total volume of the slurry was then reduced to 3 9-4 1 volumes by vacuum distillation at 38- 42 0C To this mixture was added EtOAc (2 vol ) over 30 minutes while maintaining the batch temperature at 38-42 0C The resulting slurry was then cooled to 20-25 0C over 1 hour and stirred at 20-25 0C for additional 1 hour. The slurry was filtered. The filter cake was washed with EtOAc (twice, 3 vol. each) and dried under vacuum with a nitrogen bleed at 45- 55 0C for 6 hours.
[00260] To the dried filter cake was added 2.2-2.4 volumes of CH2Ch to a total volume of 3.1-3.3 volumes. The mixture was broughl to 38-42 0C to give a homogeneous solution. EtOAc (0.80 vol) was added, followed by the addition of Compound 4 seed ( 1.5 wt% relative to 34, Example 12). The resulting mixture was stirred for 15 minutes at 38-42 0C. EtOAc (8 vol.) was added over 3 hours to this mixture while maintaining a temperature of 38-42 0C. The total volume of the slurry was then reduced to 3.9-4.1 volumes by vacuum distillation at 38-420C. EtOAc (2 vol.) was added over 30 minutes to this mixture while maintaining the batch temperature at 38-42°C. The resulting slurry was then cooled to 20-25 0C over 1 hour and stirred at 20-25 0C for additional one hour. The slurry was filtered and the filter cake was washed with EtOAc (twice, 3 vol. each) and dried under vacuum with a nitrogen bleed at 45-55 0C for 12 hour to give purified Compound 4. 1H NMR (500 MHz, CDCI3) 0.78 (m, 2H), 0.87 (m, 2H), 0.91 (s, 9H), 0.91 (t Lobscured], 3H), 0.98 (m, 4H), 1.08 (m, IH), 1.20 (m, 4H), 1.29 (m, IH), 1.40 (m, IH), 1..42 (m, 2H), 1.46 (m, IH), 1.48 (m, IH), 1.60 (m, IH), 1.70 (m, IH), 1.79 (m, IH), 1.83 (m, 2H), 1.88 (m, IH), 1.94 (m, IH), 2.67 (m, IH), 2.89 (bs, IH), 2.96 (bs, IH), 3.63 (d, IH), 3.99 (d, IH), 4.70 (s, IH), 4.82 (d, IH), 4.89 (t, IH), 5.65 (bs, IH), 7.74 (bs, IH), 8.00 (bs, I H), 8.06 (bs, IH), 8.29 (bs, IH), 8.60 (s, IH), 8.77 (s, I H), 9.42 (s, IH).
Example 15: (lS,3a#,6aS)-2-((,S>2-((S)-2-cyclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3-dimethylbutanoyl)-Λ'-((S)-l-(cyclopropylamino)-l,2-dioxo- hexan-3-yl)octahydrocyclopenta[c]pyrrole-l-carboxamide (4)

...........................
PATENT
Example 1: N-/-butyloxycarbonyI-3-azabicycIo[3.3.0]octane (6)

6
Method 1
[00177] To a 2 L 3-necked round-bottom flask under nitrogen fitted with a mechanical stirrer, a 500 niL addition funnel, and a thermometer was charged 3-azabicyclo[3.3.0]nonane hydrochloride (100 g, 0.677 mol), potassium carbonate (187 g, 1.35 mol), /-butyl methyl ether (220 mL) and water (160 mL), with stirring. The mixture was cooled to 14-16 °C. In a separate 500 mL erylenmeyer flask was charged Boc2O (di-t-butyl dicarbonate) (145 g, 0.644 mol) and t-butyl methyl ether (190 mL). The mixture was stirred until complete dissolution was obtained. The solution was poured into the addition funnel and added to the above reaction mixture, keeping the reaction temperature below 25 °C. Water (290 mL) was added to dissolve solids, and the mixture was stirred for 10-15 minutes. After separating the lower aqueous phase, the organic phase was washed with 5% aq. NaHSO4 (twice, 145 mL each), then water (145 mL). The organic phase was concentrated and methyl t-butyl ether was added (1.3 L) to give a solution of the title compound in t-butyl methyl ether. See, e.g., R.Griot, HeIv. CMm. Acta., 42, 67 (1959).
Method 2
[00178] A solution of potassium carbonate (187 g, 1.35 mol) in water (160 mL) was added to a mixture of 3-azabicyclo[3.3.0]octane hydrochloride (100 g, 0.677 mol) and t-butyl methyl ether (220 mL), and the resultant mixture was cooled to 14-16 °C. A solution of Boc2O (145 g, 0.644 mol) in t-butyl methyl ether (190 mL) was added while maintaining a temperature below 35 0C. After the addition, the mixture was stirred for 1 hour then filtered. The solids were washed with MTBE (50 mL). The phases were separated and the organic phase washed with 5% aq. NaHSO4 (twice, 145 mL each) and water (145 mL) and concentrated to 300 mL under vacuum. MTBE (300 mL) was added and the mixture concentrated to remove water to less than 550 ppm. The concentrate was diluted with MTBE (400 mL) to provide a solution of the title compound in MTBE.
Example 2: mc-2-(/-butoxycarbonyl)octahydrocyclopenta[c]pyrrole-l-carboxylic acid (7)

6 7
Method 1
[00179] The solution from Example 1, Method 1, was charged to a 5 L 4-necked flask fitted with a mechanical stirrer, an addition funnel, a ReactIR probe, and a thermometer. 3,7- Dipropyl-3,7-diazabicyclo[3.3.1]nonane (183 g, 0.88 mol) was charged to the flask. Data collection was started on the ReactIR instrument, and the solution was cooled to -72 to -75 0C. sec-Butyllithium (600 mL, 1.6 M in cyclohexane) was slowly added to the reaction mixture, keeping the reaction temperature below -69 0C. The addition was monitored with the ReactIR instrument, and the addition was stopped after the absorbance at 1698 cm"1 had disappeared and the absorbance at 1654 cm"1 ceased to increase for three consectutive scans (2-minute intervals). The solution was agitated for 3 hours at -75 to -72 °C. A 10% mixture of CO2 in nitrogen was carefully sparged into the reaction mixture, keeping the reaction temperature below -70 °C. The sparge was stopped after the absorbance for CO2 appeared in the ReactIR spectrum (2350 cm"1). The mixture was warmed to 0-5 0C, and a solution of 30 wt% NaHSO4(1.4 L) was added. The mixture was warmed to 22-25 °C and stirred for 30 minutes. The aqueous phase was separated and the organic phase washed with water (700 mL). The aqueous phase was decanted and the organic phase concentrated to provide the title compound.
Method 2
[00180] A solution of 3,7-dipropyl-3,7-diazabicyclo[3.3.1]nonane (183 g, 0.87 mol) in
MTBE (300 mL) was added to the solution of N-t-butyloxycarbonyl-3- azabicyclo [3.3.0] octane from Example 1, Method 2 in a flask fitted with a mechanical stirrer, an addition funnel, a ReactIR probe, and a thermometer and the mixture was cooled to -75 to
-72 0C. A solution of sec-butyllithium (510 mL, 1.6 M) was added, keeping the reaction temperature below -70 °C, until the absorbance at 1698 cm"1 had disappeared and the absorbance at 1654 cm"1 ceased to increase. The solution was stirred for 3 hours at -75 to -72
°C. The reaction mixture was sparged with 10% CO2 in N2 keeping the reaction temperature e ow - /υ "U. lne sparge was stoppe w en t e a sor ance or 2 appears in the eact spectrum (2339 cm"1). The mixture was warmed to 0-5 °C and a solution of 30 wt% NaHSO4 (1.4 L) was added and the mixture was warmed to 22-25 °C then stirred 30 minutes. The phases were separated and the aqueous phase was checked to make sure the pH was lower than 3. The organic phase was washed with water (700 mL) then concentrated to 300 mJL Ethyl acetate (1.7 L) was added and the mixture concentrated to 300 mL twice to give a solution of the title compound in ethyl acetate.
Example 3: (S)-l,2,3,4-tetrahydronaphthalen-l-aminium (lS,3aR,6aS)-2-(f- butoxycarbonyl)octahydrocyclopenta[c]pyrrole-l-carboxylate (9a)

8 9a
Method 1
[00181] Ethyl acetate (2.3 L) was added to the residue of Example 2, method 1, and the mixture filtered through a pad of Celite®. (S)-1 ,2,3,4-tetrahydro-l-naphthylamine (56.7 g, 0.385 mol) was added and the solution was stirred for 3-4 hours at 22-25 0C. The mixture was filtered and the solids were rinsed with ethyl acetate (200 mL). The solids were dried at 20-30 0C under vacuum for 4 hours to give 99.02 g of product (73% yield, 90% ee by chiral HPLC).
[00182] To a 3-necked RBF fitted with a temperature contoller, a mechanical stirrer, a reflux condenser, and a nitrogen bubbler, was charged the (S)-l,2,3,4-tetrahydro-l- naphthylammonium salt (88.98g, 0.22 mol), ethyl acetate (712 mL), and 2-propanol (666 mL). The mixture was warmed to 70-75 0C with stirring. The mixture was stirred for 15-30 minutes, then cooled to -5 to -10 °C over 1 hour. The resultant slurry was filtered and the solids were rinsed with cold ethyl acetate (180 mL). The solids were dried in vacuo at 35-40 °C to give 7.37 g of a white solid (83% yield, 98% ee).
Method 2
[00183] The ethyl acetate solution of racemic N-t-butyloxycarbonyl-3- azabicyclo[3.3.0]octane-2-carboxylic acid from Example 2, Method 2, was added to a solution of (S)-l,2,3,4-tetrahydro-l-naphthylamine (56.7 g, 0.385 mol) in ethyl acetate (300 mL). The mixture was strirred for 3-4 hours at 22-25 °C, then filtered, and the solids washed with ethyl acetate (200 mL). The product was dried at 20-30 °C under vacuum for 4 hours to give the title compound (99.02 g, 36% yield) with a 95 to 5 diasteromer ratio. [00184] A mixture of the salt as prepared above (89.0 g), ethyl acetate, and 2-propanol was warmed to 70-75 0C until complete dissolution. The mixture was cooled to -5 to -10 0C over two hours and stirred for 3-4 hours. The mixture was filtered and the product dried at 35-40 0C to give the title compound (73.7g, 83% yield, >99.5% ee).
Example 4: (R)-l-phenylethanaminium (lS,3aR,6aS)-2-(f- butoxycarbonyl)octahydrocyclopenta[c]pyrrole-l-carboxylate (9b)

8 9b
[00185] To a soution of racemic N-t-butyloxycarbonyl-3-azabicyclo[3.3.0]octane-2- carboxylic acid (4.66 g) in ethyl acetate (100 mL) was added (R)-α-methylbenzylamine (56.7 g) and the solution was stirred for 16 hr at 22-25 °C. The mixture was filtered and the solids were rinsed with ethyl acetate. The solids were dried at 20-30 °C under vacuum for 4 hours to give 1.47 g of product (43%, 82% ee, 92:8 ratio of exorendo diastereomers).
Example 5: (lS,3aR,6aS)-tf-butyl octahydrocyclopenta[c]pyrrole-l-carboxylate, t- butylester, oxalate

9a
Method 1
[00186] A mixture of the (S)- 1 ,2,3 ,4-tetrahydro- 1 -naphthylammonium salt prepared as in
Example 3, Method 1 (81.7 g, 0.203 mol), /-butyl methyl ether (400 mL) and 5% NaHSO4- H2O (867 mL, 0.304 mol) was stirred for 30 minutes until all solids were dissolved. The organic phase was washed with water (334 mL) then concentrated to 259 mL. /-Butyl methyl ether (334 mL) was added and the solution was concentrated again to 259 mL. The addition- concentration process was repeated twice more. After the final concentration, t-BuOH (158 mL) and dimethylaminopyridine (5.04 g, 41.3 mmol) were added. A solution of BoC2O (67.6 g, 0.31 mol) in t-butylmethyl ether (52.0 mL) was added. After stirring for 5 hours at ambient temperature, /-butyl methyl ether (158 mL) and 5% aqueous NaHSO4-H2O (260 mL) were added and the resultant mixture was stirred. The organic phase was washed with 5% aqueous NaCl (twice, 260 mL each). The organic phase was concentrated to 320 mL, and tetrahydrofuran (320 mL) was added. The organic phase was concentrated again to 320 mL, and tetrahydrofuran (320 mL) was added. After concentrating to 320 mL once more, methane sulfonic acid (80.1 g, 0.62 mol) was added and the solution was stirred at ambient temperature for 4.5 hours. The reaction mixture was added to a 30% aqueous solution of K2CO3 (571 mL) and stirred. The aqueous phase was extracted with isopropyl acetate (320 mL). The combined organic phases were concentrated to 320 mL, and isopropyl acetate (320 mL) was added. The organic solution was concentrated again to 320 mL. The organic phase was washed with water (320 mL). Isopropyl acetate (320 mL) was added to the organic phase and the solution was concentrated to 192 mL. Isopropyl acetate (320 mL) was added a second time, and the organic solution was concentrated to 192 mL. A solution of oxalic acid (24.1 g, 267 mmol) in isopropyl acetate (448 mL) was added to the orgam'c solution over 2 hours. The mixture was stirred for 2-4 hours, and the slurry was filtered. The white solids were rinsed with isopropyl acetate (100 mL) and dried at 35-40 0C under vacuum to yield 52.6 g of the title compound (85% yield).
Method 2
[00187] A mixture of (S)- 1 ,2,3,4-tetrahydro- 1 -naphthylammonium salt as prepared by the method of Example 3, Method 2 (148 g, 0.609 mol), t-butyl methyl ether (726 mL) and 5% NaHSO4-H2O (1.58 L, 0.913 mol) was stirred until all of the solids had dissolved. The phases were separated and the organic phase washed with water (726 mL). The organic phase was concentrated to about 400 mL. t-Butyl methyl ether (726 mL) was added and the mixture concentrated to 590 mL. The addition of t-butyl methyl ether and concentration was repeated to give a final volume of 350 mL. Dimethylaminopyridine (8.42 g, 68.9 mmol) and t-butyl alcohol(260 mL) were added, followed by addition of a solution of BoC2O (112 g, 0.52 mol) in MTBE (88 mL) over 0.5 hour. The mixture was stirred for 5 hours at 22-25 0C.
A solution of 5% sodium bisulfate in water was added and the mixture stirred for 0.5 hour. The organic phase was washed with 5% sodium chloride (twice, 440 mL each) and concentrated to 270 mL. Tetrahydrofuran (540 mL) was added and the mixture concentrated to 270 mL; this procedure was repeated twice more to give a final volume of 270 mL. Methane sulfonic acid (67 mL) was added over 0.5 hour while maintaining a temperature of lower than 30 °C and the mixture stirred at 22-25 °C for 12 hours. The mixture was added to a 30% aqueous solution of pottassium carbonate (478 mL) while maintaining a temperature of 22-25 °C. The mixture was filtered, the phases separated and the aqueous phase extracted with isopropyl acetate (twice, 540 mL each). The organic phase was concentrated to 270 mL, then twice evaporated with isopropyl acetate (540 ml) to give a final volume of 540 mL. The organic phase was washed with water (twice, 540 mL), then twice evaporated with isopropyl acetate (320 mL) to give a final volume of 320 mL. Additional isopropyl acetate (429 mL) was added followed by addition of a solution of oxalic acid (40.4 g, 0.448 mol) in t- butylmethyl ether (321 mL) over 2 hours maintaining a temperature of 22-25 0C. The mixture was stirred for 3 hours at 22-25 0C then filtered. The filter cake was washed with isopropyl acetate (100 mL) and the prouduct dried at 35-40 °C under vacuum to give the title compound as a white solid (88.4g, 81%).
Example 6: (lS,3aR,6aS)-*-butyl 2-((S)-2-(benzyloxycarbonylamino)-3,3- dimethylbutanoyl)octahydrocyclopenta[c]pyrrole-l-carboxylate (27)

Method 1
[00188] A 3-L 3-neck round bottomed flask equipped with an overhead stirrer, condenser, thermocouple, and nitrogen outlet was purged with nitrogen for several minutes. In a separate flask, sulfuric acid (46.2 mL, 0.867 mol) was diluted with 442 mL of water. The solution was allowed to cool slightly. Cbz-L-tert-Leucine dicyclohexylamine salt (330.0 g, 0.739 mol) was charged to the reaction flask. t-Butyl methyl ether (1620 mL) was added to the reactor, and the mixture was stirred to suspend the salt. The acid solution prepared above was added to the reactor over about 10 minutes, keeping the temperature at 20±5 °C. The mixture was stirred at room temperature for approximately 1 hour, then diluted slowly with water (455 mL). Agitation was stopped, and the layers were allowed to settle. The lower (aqueous) phase was withdrawn to provide 1100 mL colorless solution of pH 1. To the organic phase remaining in the flask was charged additional water (200 mL). The mixture was stirred at room temperature for approximately lhour. Agitation was stopped, and the layers were allowed to settle. The lower (aqueous) phase was withdrawn to provide 50OmL colorless solution of pH 2. The organic phase was heated to about 35 °C, diluted with DMF (300 mL), and concentrated under reduced pressure to the point at which distillation slowed significantly, leaving a concentrate of about 500 mL. The concentrate was transferred without rinsing to a 1-L Schott bottle. The concentrate, a clear colorless solution, weighed 511.6 g. Based on solution assay analysis and the solution weight, the solution contained 187.2 g (0.706 mol) Cbz-L-tørt-Leucine.
[00189] To a 5-L 4-neck round bottomed flask equipped with an overhead stirrer, thermocouple, addition funnel and nitrogen inlet were charged HOBT»H2O (103.73 g, 0.678 mol, 1.20 molar eq.), EDC-HCl (129.48 g, 0.675 mol, 1.20 molar eq.) and DMF (480 mL). The slurry was cooled to 0-5 0C. A 36.6 wt% solution of the acid of Cbz-L-tert-Leucine in DMF (491.3 g, 0.745 mol, 1.32 molar eq.) was added over 47 minutes to the reaction mixture while keeping the temperature at 0-5 °C. The reaction mixture was stirred for 1 hour and 27 minutes. A solution of 3-azabicyclo(3.3.0)octane-2-carboxylic acid /-butyl ester in isopropyl acetate (28.8 wt%, 414.3 g, 0.564 mol) was added over 53 minutes while keeping the reaction temperature at 0-5.1 0C. The reaction mixture was warmed to 20±5 °C over about lhour. 4- Methylmorpholine (34.29 g, 0.339 mol, 0.60 molar eq.) was added over 5 minutes. The reaction mixture was agitated for 16 hours then isopropyl acetate (980 mL) was added to the reaction solution. A solution of histamine*2HCl (41.58 g, 0.226 mol, 0.40 molar eq.) in water (53.02 g) was added to the reaction mixture within 4 minutes, followed by 4- methylmorpholine (45.69 g, 0.45 mol, 0.80 molar eq.). The reaction mixture was sampled after 3.5 hours. Water (758 mL) was added, the mixture stirred for about 20 minutes, then allowed to settle for 11 minutes. The phases were separated. The aqueous phase was extracted with isopropyl acetate (716 mL) and the organic phases were combined. IN aq. HCl was prepared by adding 37 wt% hydrochloric acid (128.3 mL) to water (1435 ml). The organic phase was washed for about 20 minutes with the IN hydrochloric acid. A 10wt% aq. K2CO3 solution was prepared by dissolving K2CO3 (171 g, 1.23 mol, 2.19 molar eq.) in water (1540 mL). The organic phase was washed with the 10 wt% aq. K2CO3 solution for about 20minutes. The final clear, very slightly yellow organic solution, weighing 1862.1 g, was sampled and submitted for solution assay. Based on the solution assay and the weight of the solution, the solution contained 238.3 g (0.520 mol) of product of the title compound. 1H NMR (DMSO-d6, 500 MHz): δ 7.37 ppm (5 H, s), 7.25-7.33 ppm (1 H, m), 5.03 ppm (2 H3 s), 4.17 ppm (1 H, d), 3.98 ppm (1 H, d), 3.67-3.75 ppm (2 H, m), 2.62-2.74 ppm (1 H, m), 2.48-2.56 ppm (1 H, m), 1.72-1.89 ppm (2 H, m), 1.60-1.69 ppm (1 H, m), 1.45-1.58 ppm (2 H, m), 1.38 ppm (9 H, s), 1.36-1.42 ppm (1 H, m), 0.97 ppm (9 H, s).
Method 2
[00190] A solution of potassium carbonate (73.3 g) in water (220 mL) was added to a suspension of (IS, 2S,5R) 3-azabicyclo[3.3.0]octane-2-carboxylic, t-butylester, oxalate (80.0 g,) in isopropyl acetate (400 mL) while maintaining a temperature of about 20 0C. The mixture was stirred for 0.5 hour, tha phases separated and the organic phase washed with 25% w/w aqueous potassium carbonate (80 mL) to give a slution of the free base. In a separate flask, aqueous sulfuric acid (400 mL, 0.863 M) was added to a suspension of Cbz-t- leucine dicyclohexylamine salt (118.4g) in /-butylmethyl ether (640 mL) while maintaing a temperature of about 20 0C. The mixture was stirred for 0.5 hour, the phases separated and the organic phase washed with water (200 mL). The phase were separated and N- methylmorpholine (80 mL) was added to the organic phase which was concentrated under reduced pressure at 40 0C to 80 mL to give the free acid as a solution in N-methyl morpholine. This solution was added to a mixture of EDC* HCl (50.8 g) HOBt hydrate (40,6 g) in N-methylmorpholine (280 mL) at 0-10 0C. The mixture was stirred for 1 hour at about 5 0C. The solution of 3-azabicyclo[3.3.0]octane-2-carboxylic, t-butylester from above was added at 0-20 0C followed by N-methylmorpholine (32 mL). The mixture was stired for 6 hour then diluted with isopropyl acetate (600 mL) followed by IN HCl (400 mL). After stirring 0.5 hour, the phases were separated and the organic phase washed with 25% w/w aqueous potassium carbonate (400 mL) and water (80 mL). The mixture was stirred for about 1 hour and the phases separated to give a solution of the title compound in isopropyl acetate.
Method 3
[00191] (IS, 2S,5R) 3-azabicyclo[3.3.0]octane-2-carboxylic, /-butylester, oxalate (1.0 eq.) was suspended in isopropyl acetate (6 vol.) and a solution of potassioum carbonate (3.0 eq.) in water (3.5 vol.) was added at 20-250C. The mixture was stirred for 3 hours then the phases separated. The organic phase was washed with water (2 vol.). [00192] Cbz-Meucine dicyclohexylamine salt (1.05 eq.) was suspended in isopropyl acetate (6 vol.) and sulfuric acid (1.3 eq.) in water (5 vol.) was added at 20-250C. The mixture was stirred for 30 minutes, the phases separated, and the organic phase washed twice with water (2.5 vol each).
[00193] The two solutions from above were combined and then cooled to 0-50C. HOBt hydrate (1.1 eq.) and EDC (1.1 eq.) were suspended in the mixture and the mixture stirred for 6 hours. The mixture was washed with water (5 vol.) and the resulting organic phase treated with L-lysine (1 eq.) and-N-methylmorpholine (NMM) (2 eq.) at 20-250C to destroy excess activated ester. The mixture was then washed with 5% potassium carbonate (5 vol.), IN hydrochloric acid (5 vol.), 5% potassium carbonate (5 vol.) and twice with water (5 vol. each) to give a solution of the title compound in isopropyl acetate.
Example 7: (lS,3aR,6aS)-*-butyI 2-((S)-2-amino-3,3-dimethyIbutanoyl)- octahydrocyclopenta[c]pyrrole-l-carboxylate (28)

Method 1
[00194] A 1 L Buchi hydrogenator was purged with nitrogen three times. A 307.8 g portion of a 12.8 wt% solution of (lS,3aR,6aS)-t-butyl 2-((S)-2-(benzyloxycarbonylamino)-3,3- dimethylbutanoyl)octahydrocyclopenta[c]pyrrole-l-carboxylate (as prepared by the method of Example 6, Method 1) in isopropyl acetate (39.39 g, 0.086 mol) was charged to the reactor. Isopropyl acetate (100 mL) was added to the reactor. A slurry of 50% water and wet 20% Pd(OH)2/carbon (3.97 g) in isopropyl acetate (168 mL) was prepared and charged to the reactor and agitation was started. The reactor was pressurized to 30 psig with nitrogen gas and vented down to atmospheric pressure. This was repeated twice. The reactor was pressurized to 30 psig with hydrogen and vented down to atmospheric pressure. This was repeated twice. The reactor was pressurized to 30 psig with hydrogen and stirred at ambient temperature for 1 hour. The mixture was filtered using a Buchner funnel with a Whatman # 1 filter paper to remove catalyst. The filter cake was washed with isopropyl acetate (80 mL). The procedure was repeated twice more using 617 g and 290.6 g of the 12.8 wt% solution of the starting Cbz compound. The material from the three hydrogenations were combined and distilled under reduced pressure (28" Hg). The resultant solution (468.68 g) was assayed for the title compound (23.2%, 98.9 % purity).
1H NMR (DMSO-d6, 500 MHz): δ 3.96 ppm (1 H, d), 3.67 ppm (1 H, dd), 3.53 ppm (1 H, dd), 3.19 ppm (1 H5 s), 2.66-2.75 ppm (1 H, m), 2.49-2.53 ppm (1 H, m), 1.75-1.92 ppm (2 H, m), 1.66-1.74 ppm (1 H, m), 1.48-1.60 ppm (4 H, m), 1.38 ppm (9 H, s), 1.36-1.42 ppm (1 H5 m), 0.91 ppm (9 H5 s)
Method 2
[00195] The solution of the Cbz derivative 27 from Example 6, Method 2, was added to 20% Pd(OH)2/water (50%, 12.2 g) in a hydrogenation apparatus. The apparatus was pressurized to 30 psi with hydrogen then stirred for 2 hr at about 20 0C. The mixture was filtered to remove the catalyst, the filter cake washed with isopropyl acetate (160 mL). The combined filtrates were evaporated with about 4 volumes of heptane at 40 0C 2 to 3 times to remove the isopropyl acetate. The resultant slurry was cooled to 0 0C, filtered and the product dried under vacuum to give the title compound (78.8 g, 98.3% purity).
Method 3
[00196] Asolution of (1 S,3aR,6aS)-t-butyl 2-((S)-2-amino-353-dimethylbutanoyl)- octahydrocyclopenta[c]pyrrole-l-carboxylate in isopropyl acetate from Example 6, Method 35 was added to 20% Pd(OH)2 (2 wt% loading, 50% wet) and the mixture was hydrogenated at 2 bar and 20-25 0C for 2 hours. The catalyst was removed by filtration and washed with isopropyl acetate (2 vol.). The filtrate was concentrated to 10 vol. under reduced pressure at 40 0C to give a solution of the title compound in isopropyl acetate.
Example 8: (lS,3aR,6aS)-*-butyl 2-((S)-2-((S)-2-(benzyloxycarbonylamino)-2- cycIohexylacetamido)-3,3-dimethyIbutanoyl)octahydrocyclopenta[c]pyrrole-l- carboxylate (30)

Method 1 [00197] To a 3 L 3-neck round bottomed flask equipped with an overhead stirrer, thermocouple, addition funnel, nitrogen outlet and ice/water bath was charged HOBt*H2O (51.74 g; 0.338 mol, 1.05 molar eq.), EDCΗC1 (64.8 g; 0.338 mol, 1.05 molar eq.) followed by DMF (197.1 g, 208.8 mL) and agitation was started. The slurry was cooled to 0-5 0C, then a solution of the acid 29 (98.45 g; 0.338 mol, 1.05 molar eq.) in DMF (172.4 g; 182.9 mL) was prepared and charged to the addition funnel. This was added over about 30 minutes to the batch, maintaining the temperature at 0-5 0C. Once addition was complete the reaction mixture was agitated at 0-5 °C for 2 hours. The solution of the amine 28 in isopropyl acetate (450 g solution; containing 104.4 g of acid 29, 0.322mol) was charged to an addition funnel and added drop wise over lhour maintaining the temperature at 0-5 0C. Sample analysis indicated incomplete reaction and additional EDC hydrochloride (3.89 g) was added. After 3 hours, analysis of a sample showed 1.8% amine 28 remained. A slurry of HOBT»H2O (2.59 g; 0.0169 mol), and EDC»HC1 (3.24 g; 0.0169 mol) was prepared in DMF (10.44 mL) and cooled to 0-5 0C . A solution of acid 29 (4.92 g; 0.169 mol) in DMF (10.44 mL) was prepared and added to the slurry of EDC»HC1 and HOBT in DMF over 30minutes, maintaining the reaction temperature at 0-5 0C. The mixture was stirred for 1 hour at 0-5 0C then added to the original mixture maintaining 0-5 0C. The mixture was stirred for 14 hours at about 25 0C. A solution of histamine»2HCl (11.84 g; 0.064 mol) in water (8.9 mL) was prepared and added to the reaction mixture over 5-10 minutes. A charge of 4- methylmorpholine (13.01 g; 0.129 mol) was added to the batch over about 10 minutes, maintaining the batch temperature at 20±5 0C. The reaction mixture was diluted with isopropyl acetate (443 mL), followed by water (585 mL). A solution of potassium carbonate (57.8 g) in water (585 mL) was added and the mixture was stirred for 0.5 hour. The layers were separated the aqueous layer was extracted twice with isopropyl acetate (twice, 235 mL each). The combined organic phases were washed with 18 % aqueous HCl in water (585 mL), then NaHCO3 (43.25 g) in water (585 mL). The layers were separated to give a light yellow solution of product 30 in isopropyl acetate weighing 1159.3 g (1275 mL) containing 16.0 w/w % 30 in isopropyl acetate.
1H NMR (DMSO-d6, 500 MHz): δ 7.74 (IH, d), 7.36 (5H, m), 7.34-7.26 (IH, m), 5.01 (2H, s), 4.51 (IH, d), 4.02 (IH, t), 3.96 (IH, d), 3.73 (IH, m), 3.66 (IH, m), 3.68 (IH, m), 2.53 (IH, m), 1.86-1.76 (2H, m), 1.70-1.30 (1OH, m), 1.39 (9H, s), 1.15-0.85 (5H, m), 0.96 (9H, s).
Method 2
[00198] A solution of Cbz acid 29 (59.62 g) in N-methylpyrrolidone (126 mL) was added to a suspension of EDCHCL (39.23 g) HOBt hydrate (31.34 g) in N-methylpyrrolidone (221 mL) while maintaining a temperature of about 0 0C. After the addition, the mixture was stirred for 1.5 hours at about 0 0C. A solution of the amine 28 (63.24 g, as prepared in Example 7, Method 2) in isopropyl acetate (632 mL) was added to the mixture maintaining a temperature of about 0 0C. After the addition the mixture was allowed to warm to room temperature and stirred for 5 hours. A solution of potassium carbonate (20.17g) in water (316 mL) was added while maintaining a temperature of about 20 0C. The mixture was vigorously stirred for 0.5 hour. The phases were separated and the organic phase vigorously stirred with potassium carbonate (105.3 g) in water (316 mL). The organic phase was separated and washed with IN HCl (316 mL), and then water (158 mL) to give a 12.7% w/w solution of the title compound 30 in isopropyl acetate.
Method 3 I
[00199] To a solution of (1 S,3aR,6aS)-t-butyl 2-((S)-2-amino-3,3-dimethylbutanoyl)- octahydrocyclopenta[c]pyrrole-l-carboxylate (1 eq) in isopropyl acetate (10 vol) was added NMP (5 vol) followed by EDC (1.15 eq), HOBT hydrate (1.0 eq) and (S)-2- (benzyloxycarbonylamino)-2-cyclohexylacetic acid (29, 1.05 eq) and the suspension was stirred at 20-250C for 4 hr. The mixture was washed with 5% potassium carbonate (5 vol). A mixture of glycine (1 eq), NMM (2 eq) and water (1 vol) was added and the mixture stirred for 4 hr. The mixture was then washed with 5% potassium carbonate (5 vol), IN hydrochloric acid (5 vol), 5% potassium carbonate (5 vol) and twice with water (5 vol each) to give a solution of the title compound in isopropyl acetate.
Example 9: (lS,3aR,6aS)-fert-butyl 2-((S)-2-((S)-2-amino-2-cyclohexylacetamido)-3,3- dimethylbutanoyl)octahydrocyclopenta[c]pyrrole-l-carboxylate (31)

Method 1
[00200] A 60-gallon hasteloy hydrogenating reactor was charged with a solution of the Cbz peptide 30 (15.1 kg) in isopropyl acetate (109 kg). This solution was reduced under vacuum at 50 0C to 68 L. The mixture was then cooled to 25±5 °C and MeOH (15.4 kg) added. This mixture was drained into a container and the reactor was dried. To the dried reactor was charged Pd(OH)2/C (20%, 1.51 kg). The solution containing the Cbz peptide 30 was added to W 2
the reactor and blanketed with hydrogen (30 psi). The reaction was stirred at 20±5 °C and at 150-220 rpm for 2 hours. After completion, a slurry of activated carbon (0.97 kg) in isopropyl acetate (6.8 kg) was added batch and the mixture stirred for 15 minutes. The mixture was filtered over Celite® (2.0 kg) via Sparkler filter and through a 0.1 -um cartridge filter. The reactor was rinsed with isopropyl acetate (33.0 kg) and the rinse was combined with the reaction mixture. The system was rinsed additionally with a mixture of isopropyl acetate (25.6 kg) and MeOH (5.73 kg). The combined organics were reduced under vacuum at 65 °C to 30 L. The solution was cooled to 20-30 °C and heptane added (30.8 kg). Distillation was instituted again and the mixture reduced to 30 L. This procedure was repeated for a total of 4 heptane additions (as above) and solvent reductions (as above). The mixture was cooled to 0-5 0C and the product filtered and washed with heptane (12.6 kg). The wet solid (14.0 kg) was dried under vacuum at 15-20 0C to constant weight to give the title compound (10.17 kg).
1H NMR (DMSOd6, 500 MHz): δ 7.97 (IH, d), 4.49 (IH5 d), 3.96 (IH, d), 3.76 (IH, m), 3.67 (IH, m), 3.05 (IH, d), 2.70 (IH, m), 2.53 (IH, m), 1.87-1.77 (2H, m), 1.7-1.3 (1OH, m), 1.39 (9H, s), 1.2-0.85 (5H, m), 0.96 (9H, s).
Method 2
[00201] The solution of compound 30 from Example 8, Method 1, was added to 50% wet 20 wt% Pd(OH)2 on carbon (3.16 g) in a pressure reactor. The reactor was pressurized at 30 psi with hydrogen and the mixture stirred for about 1 hour. The catalyst was filtered, the filter washed with isopropyl acetate and the combined organics distilled to about 65 mL. The mixture was evaporated with heptane (316 mL) several times until analysis indicates <0.5% isopropyl acetate. The resultant slurry is diluted to about 320 mL then warmed to reflux. The solution was slowly cooled to about 5 0C, the suspension stirred for 1 hour then filtered. The filter cake was washed with about 65 mL of heptane and the product dried under vacuum at 300C to give the title compound (80.16 g) as a white solid.
Method 3
[00202] The solution of (1 S,3aR,6aS)-t-butyl 2-((S)-2-((S)-2-(benzyloxycarbonylamino)-2- cyclohexylacetamido)-3,3-dimethylbutanoyl)octahydrocyclopenta[c]pyrrole- 1 -carboxylate in isopropyl acetate from Example 9, Method 3, was added to 20% Pd(OH)2 (2 wt% loading, 50% wet) and the mixture hydrogenated at 2 bar and 20-25 0C for 2 hour. The catalyst was removed by filtration and washed with isopropyl acetate (1 vol.). The solvent was exchanged by distillation twice with heptane (8.6 vol.) at reflux. The mixture was cooled to 78 0C over 1 hour, then to 22 0C over 2 hours. After 1 hour at 22 0C the suspension was filtered and the cake washed with heptane (3.2 vol.) and the product dried under vacuum at 30 0C with a nitrogen purge to give the title compound.
Example 10: (lS,3aR,6aSH-butyl 2-((S)-2-((S)-2-cycIohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3-dimethylbutanoyl)octahydrocycIopenta[c]pyrrole-l- carboxylate (33)

Method 1
[00203] To a 10OmL round bottomed flask was added pyrazine-2-carboxylic acid 32 (1.6070 g, 12.95 mmol) and DMF (4 mL). The slurry was stirred at 20-25 0C. Meanwhile, a solution of CDI was prepared by combining CDI (2.1012 g, 12.96 mmol, 1 molar eq.) and DMF (8.80 g, 9.3 mL) in a 25 mL flask. Mild heating (30 °C) aided in dissolution. The CDI solution was cooled to 20-25 0C and added to the slurry of pyrazine-2-carboxylic acid. Stirring was continued for 1.5 hours to assure complete activation of the acid as carbon dioxide was produced as a byproduct. Meanwhile, the amine 31 (5.0002 g, 10.78 mmol) was dissolved in DMF (14.15 g, 15 mL) with mild warming to 30 °C aided in the dissolution of the material. This solution was cooled to 20-25 °C. The activated pyrazine solution was also cooled to about 15 0C. The solution of compound 31 was added to the activated pyrazine carboxylic acid while maintaining the temperature at 30 0C for about 1 hour. The solution was allowed to cool to 20-25 0C then added to a solution of potassium carbonate (0.25 g) in water (100 mL) at 0 0C. The mixture was filtered and washed with water (four times, 50 mL each). The filter cake was dried under vacuum beginning at 20-25 °C and warmed to 30 0C after 24hours until the cake was constant weight to give the title compound (5.99 g). 1H NMR (DMSO-d6j 500 MHz): δ 9.19 ppm (1 H, d, J =1.3 Hz), 8.90 ppm (1 H, d, J = 2.5 Hz), 8.76 ppm (1 H, dd, J = 2.4 Hz, 1.5 Hz), 8.50 ppm (1 H, d, J = 9.2 Hz)5 8.22 ppm (1 H, d, J = 9.0 Hz), 4.68 ppm (1 H, dd, J = 9.1 Hz, 6.6 Hz)5 4.53 ppm (1 H, d, J = 9.0 Hz), 3.96 ppm (1 H5 d, J = 4.2 Hz), 3.73 ppm (1 H5 dd, J = 10.5 Hz, 7.5 Hz), 3.68 ppm (1 H, dd, J = 10.6 ppm, 3.4 ppm), 2.68-2.74 ppm (1 H, m), 2.52-2.58 ppm (1 H, m), 1.70-1.88 ppm (3 H, m), 1.51-1.69 ppm (7 H, m), 1.31-1.44 ppm (2 H, m), 1.39 ppm (9 H, s), 1.00-1.19 ppm (4 H5 m), 0.97 ppm (9 H, s), 0.91-0.97 ppm (1 H, m).
Method!
[00204] Oxalyl chloride (11.29 mL) was added to a solution of pyrazine-2-carboxylic acid 32 and N-methylmorpholine (59.28 mL) in methylene chloride (150 mL) at about 30 0C. The mixture was stirred for 0.5 hour, then a solution of the amine 31 (50.0 g) in methylene chloride (150 mL) was added at about 30 0C. After 0.5 hour, the mixture was washed with water (250 mL). The aqueous phase was extracted with methylene chloride (100 mL) to give a solution of the title compound in methylene chloride which was used directly in the next step (Example 11, Method 2).
Example 11: (lS)-2-((S)-2-((S)-2-cyclohexyI-2-(pyrazine-2-carboxamido)acetamido)-3,3- dimethylbutanoyl)octahydrocyclopenta[c]pyrrole-l-carboxylic acid (34)

Method 1
[00205] Concentrated HCl (150 g, 0.015 mol, 1.2 molar eq.) was slowly added at 0 °C to a stirred solution of the pyrazinyl peptide 33 (50.0 g) in formic acid (100. Og). After 3.3 hours, the reaction mixture was diluted with 166.5 g of ice water. Methylene chloride (100 mL) was added and the reaction was stirred for 10 minutes to dissolve the product. The phases were separated and the aqueous layer extracted with methylene chloride (100 mL). The combined organic phases were washed with water (75 mL) then concentrated to about 1/3 volume at 50 0C, 1 atm. Toluene (100 mL) was added at room temperature and the homogeneous solution was evaporated under vacuum at <56 0C to about 1/3 volume. The mixture was cooled to 20- 25 0C as a precipitate formed. Heptane (75 mL) was slowly added and the slurry stirred for 10-15 minutes. The slurry was filtered and the filter cake was washed with heptane (50 mL). The solids were dried under vacuum at 20-25 °C to give the title compound (15.19 g).
Method 2 [00206] The methylene chloride solution of the starting compound 33 from Example 10, Method 2, was cooled to 0-5 0C then concentrated HCl (200 mL) was added while maintaining a temperature of <10 0C. The mixture was stirred for 3 hours, then diluted with water (200 mL) while maintaining a temperature of < 10 0C. The phases were separated and the aqueous phase extracted with methylene chloride (100 mL). The combined organic phases were washed with water (100 mL) and the aqueous wash phase extracted with methylene chloride. The combined organic extracts were refluxed under an inverse Dean- Stark trap to azeotrope water. The mixture was concentrated by distillation to a minimum volume then diluted with toluene (500 mL) then concentrated by distillation at atmospheric pressure to 250 mL. The mixture was slowly cooled to 20 0C over about 6 hours. The resultant slurry was filtered, the filter cake washed with toluene (100 mL) then dried at about 45 0C in a vacuum oven to provide the title compound (64.7 g) as a pale yellow powder containing about 17 % toluene.
Example 12: (lS,3aR,6aS)-2-((S)-2-((S)-2-cyclohexyl-2-(pyrazine-2- carboxamido)acetamido)-3,3-dimethylbutanoyl)-N-((3S)-l-(cyclopropylamino)-2- hydroxy-l-oxohexan-3-yl)octahydrocyclopenta[c]pyrrole-l-carboxamide (35)

Method 1
[00207] A 500 mL 3-neck round bottomed flask equipped with an overhead stirrer, condenser, thermocouple, and nitrogen outlet was purged with nitrogen for several minutes. The peptide-acid 34 (25.0 g, 0.049 mol), EDC-HCl (10.35 g, 0.054 mol, 1.1 molar eq.), and HOBt-H2O (8.27 g, 0.054 mol, 1.1 molar eq.) were charged to the flask followed by 175 mL of methylene chloride. The mixture was stirred at room temperature for lhour then added over 20 minutes to a suspension of hydroxyamide-amine 18 (11.1 g, 0.054 mol, 1.1 molar eq.) in methylene chloride (75 mL) while maintaining a temperature below 10 °C. Upon W 2
complete addition, N-methylmorpholine (5.94 mL, 0.054 mol, 1.1 molar eq.) was added in 2 portions. The mixture was allowed to warm to room temperature and stirred for 3 hours. The reaction was quenched by the addition OfNaHCO3 (8.0 g) in 200 mL of water. The phases were separated and the organic layer washed with water (175 mL), 0.5 N aq. HCl (200 mL), water (three times, 200 mL each) and saturated NaCl (200 mL) to give a 16% by weight methylene chloride solution of the title compound 35 of 100 A% purity (molar yield 100%).
Method 2
[00208] N-methylmorpholine (38.19 mL, 347.3 mmol) was added to a mixture of the peptide-acid 34 (100.0 g, 89.2 wt%, 173.7 mmol), HOBt hydrate (26.79 g, 87.6 wt%, 173.7 mmol), EDCI (36.62 g, 191.04 mmol), and the hydroxyamide-amine 18 in methylene chloride over 30 minutes while maintaining a temperature of 0-5 0C. After the addition, the mixture was warmed to 20 0C and stirred for 5 hours. The mixture was then diluted with water (500 mL) and stirred for about 0.5 hour. The phases were separated and the organic phase washed with IN HCl (500 mL), 5 wt% aqueous sodium bicarbonate (500 mL) to give a solution of the title compound in methylene chloride, 98.5% AUC purity, 95% solution yield.
Method 3
[00209] Peptide acid 34 (1.00 eq.), EDCI (1.10 eq.), HOBt hydrate (1.00 eq.), and hydroxyamine 18«HC1 (1.05 eq.) were suspended in CH2Cl2 (5 vol.) and the mixture was cooled to 0-5 0C. NMM (2.0 eq) was added over 30-60 minutes while maintaining the reaction temperature below 5°C. The reaction mixture was warmed to 20-25 °C over 30 minutes and stirred for additional 5 hours. The reaction was washed with water (5 vol.), IN HCl (5 vol), and 5 wt% aqueous NaHCO3 (5 vol.) to provide a solution of the title compound in CH2Cl2.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE

