FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review

Oritavancin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(3S, 6R, 7R, 22R, 23S, 26S, 36R, 38aR) -22 – (3-Amino-2 ,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyloxy) -3 – (carbamoylmethyl ) -10,19-dichloro-44-[2-O-[3 - (4'-chlorobiphenyl-4-ylmethylamino) -2,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyl] – beta-D-glucopyranosyloxy] -
| CAS No. | 171099-57-3 |
| CBNumber: | CB92451283 |
| Molecular Formula: | C86H97Cl3N10O26 |
| Formula Weight: | 1793.12 |
Also known as NDISACC-(4-(4-chlorophenyl)benzyl)A82846B and LY333328,N-(4-(4-chlorophenyl)benzyl)A82846B
Abbott (Supplier), Lilly (Originator), InterMune (Licensee)
The medicines company—
the Oritavancin Program Results.pdf
 phx.corporate-ir.net/External.File?item…t=1Jul 2, 2013 - Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
phx.corporate-ir.net/External.File?item…t=1Jul 2, 2013 - Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014.
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
About Oritavancin
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.

Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
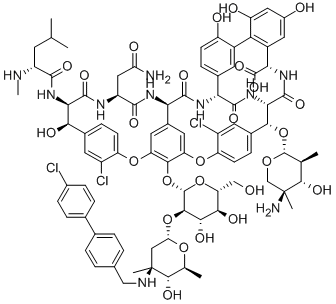 oritavancin
oritavancin
The 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
Results have been presented (in 2003) but possibly not yet published from two pivotal Phase 3 clinical trials testing the efficacy of daily intravenous oritavancin for the treatment of complicated skin and skin-structure infections (cSSSI) caused by Gram-positive bacteria. The primary endpoints of both studies were successfully met, with oritavancin achieving efficacy with fewer days of therapy than the comparator agents (vancomycin followed by cephalexin). In addition, oritavancin showed a significantly improved safety profile with a 19.2 percent relative reduction in the overall incidence of adverse events versus vancomycin/cephalexin (p<0.001) in the second and larger pivotal trial.[10]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
Regulatory submissions
USA
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the US FDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
Europe
June 2008, Targanta’s Marketing Authorization Application (MAA) for oritavancin was submitted and accepted for review by the European Medicines Agency (EMEA),[16] but the company later withdrew the application in Aug 2009.[17]
About The Medicines Company
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
 oritavancin
oritavancin
Drug substance
Oritavancin diphosphate
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=oritavancin
- LY 333328 diphosphate
- LY333328 diphosphate
- Oritavancin diphosphate
- UNII-VL1P93MKZN
- 192564-14-0 CAS NO
INTRODUCTION
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.
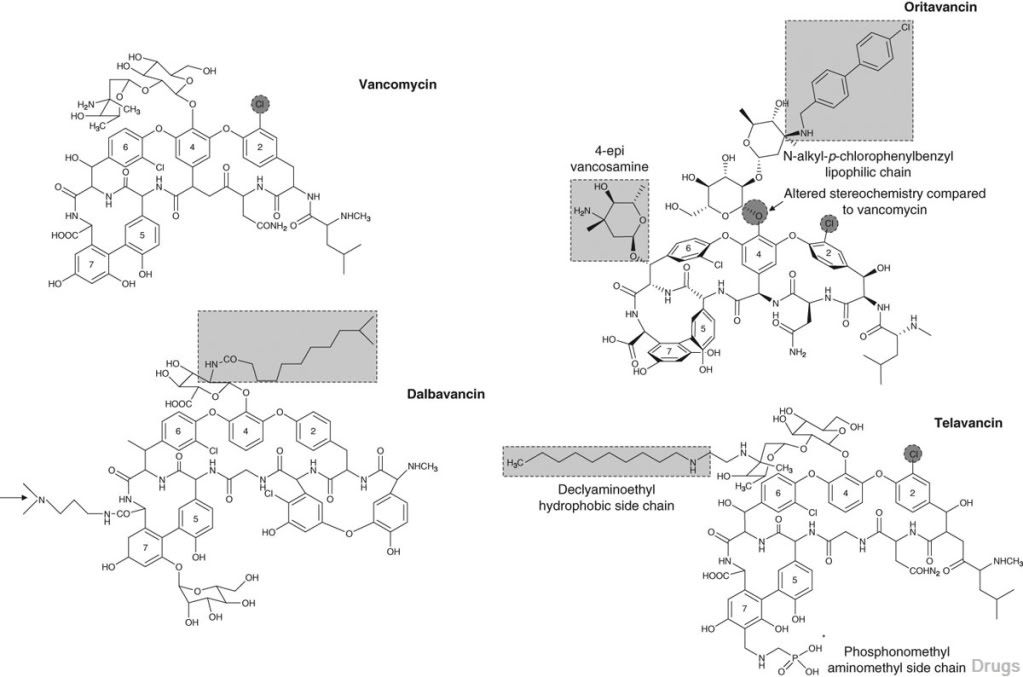
vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:

R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602


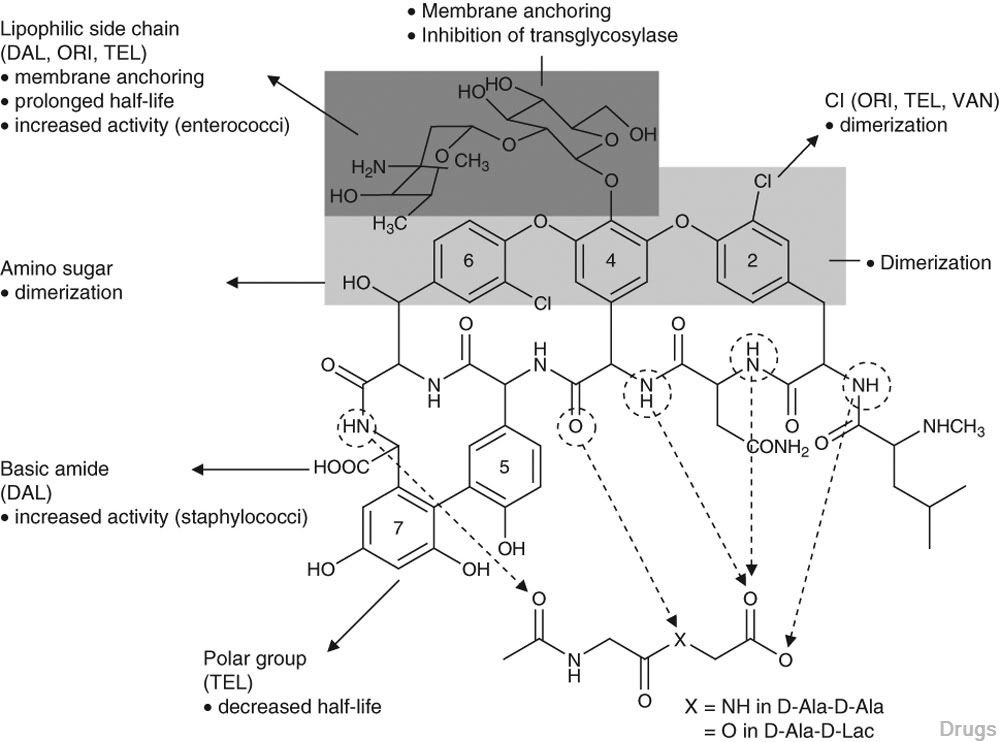
Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin (U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
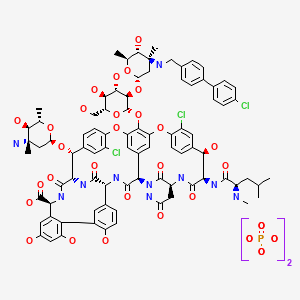 ORITAVANCIN DIPHOSPHATE
ORITAVANCIN DIPHOSPHATE
SYNTHESIS

LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
J Antibiot1996, 49, (6) :575-81
(3S,6R,7R,22R,23S,26S,36R,38aR)-3-(Carbamoylmethyl)-10,19-dichloro-7,28,30,32-tetrahydroxy-6-(N-methyl-D-leucylamido)-2,5,24,38,39-pentaoxo-22-(L-vancosaminyloxy)-44-[2-O-(L-vancosaminyl)-beta-D-glucopyranosyloxy]-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-1H,22H-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,36-dimetheno-[1,6,9]oxadiazacyclohexadecino[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylic acid; A82846B (I)
4′-chloro[1,1'-biphenyl]-4-carbaldehyde (II)
4′-chloro[1,1'-biphenyl]-4-carbaldehyde (II)
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
…………………..
EXAMPLE 4
Preparation of Compound 229
A three liter 3-necked flask was fitted with a
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
………………….
Oritavancin (also termed N-(4-(4-chlorophenyl)benzyl)A82846B and LY333328) has the following Formula III:

References
- Targanta Revives Oritavancin: Next Weapon Against cSSSI? BioWorld Today, November 26, 2007
- “Biotechs pick up slack in antibiotics development”. 17 May 2011.
- http://www.farm.ucl.ac.be/Full-texts-FARM/Domenech-2009-1.pdf ”Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization” 2009
- Scheinfeld, N (2007). “A comparison of available and investigational antibiotics for complicated skin infections and treatment-resistant Staphylococcus aureus and enterococcus“.J Drugs Dermatol. 6 (4): 97–103. PMID 17373167.
- 2007 ICAAC Posters: E-1612 “In Vitro Activity Profile of Oritavancin against a Broad Spectrum of Aerobic and Anaerobic Bacterial Pathogens”/E -1613 “In Vitro Activity Profile of Oritavancin (ORI) Against Organisms Demonstrating Key Resistance Profiles to Other Antimicrobial Agents”/E-1614 “In vitro Time Kill Studies of Oritavancin against Drug-resistant Isolates ofStaphylococcus aureus and Enterococci”/E-1615 “Anti-Enterococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1616 “Anti-Streptococcal Activity Profile of Oritavancin, a Potent Lipoglycopeptide under Development for Use Against Gram-Positive Infections”/E-1617 “In Vitro Activity Profile of Oritavancin (ORI) Against Resistant Staphylococcal Populations From a Recent Surveillance Initiative”/E-1620 “Pharmacokinetic Concentrations of Oritavancin Kill Stationary-Phase and Biofilm Staphylococcus aureus In Vitro.” / Targanta Press Release September 19, 2007
- ICAAC 2007 Posters: “In Vitro Susceptibility of Genotypically Distinct Clostridium difficileStrains to Oritavancin” and “Activity of Metronidazole, Vancomycin and Oritavancin Against Epidemic Clostridium difficile Spores” / Targanta Press Release September 19, 2007
- ASM 2007 Poster: “Efficacy of Oritavancin in a Murine Model of Bacillus anthracis Spore Inhalation Anthrax” / Targanta Press Release May 24, 2007
- Belley; McKay, GA; Arhin, FF; Sarmiento, I; Beaulieu, S; Fadhil, I; Parr Jr, TR; Moeck, G (2010).“Oritavancin Disrupts Membrane Integrity of Staphylococcus aureus and Vancomycin-Resistant Enterococci To Effect Rapid Bacterial Killing”. Antimicrobial agents and chemotherapy 54(12): 5369–71. doi:10.1128/AAC.00760-10. PMC 2981232. PMID 20876372.
- Zhanel et al. (2012). “Oritavancin: Mechanism of Action”. Clin Infect Dis.doi:10.1093/cid/cir920.
- ICAAC 2003 Late-breaker poster: “Phase III Trial Comparing 3-7 days of Oritavancin vs. 10-14 days of Vancomycin/Cephalexin in the Treatment of Patients with Complicated Skin and Skin Structure Infections (cSSSI)” / InterMune Press Release September 15, 2003
- ClinicalTrials.gov NCT00514527
- Comparison of the Efficacy and Safety of Oritavancin Front-Loaded Dosing Regimens to Daily Dosing: An Analysis of the SIMPLIFI Trial. May 2011. doi:10.1128/AAC.00029-11.
- “Drugs.com, Targanta Submits Oritavancin New Drug Application”. Retrieved 2008-02-12.
- “FDA News, Targanta to Get FDA Decision by December”. Retrieved 2008-04-10.
- http://www.fiercebiotech.com/press-releases/fda-issues-complete-response-letter-oritavancin Dec 2008.
- “Pharmaceutical Business Review, EMEA accepts Targanta’s oritavancin MAA for review”. Retrieved 2008-06-26.
- http://www.nelm.nhs.uk/en/NeLM-Area/News/2009—August/24/European-application-for-investigational-antibiotic-oritavancin-withdrawn-/
- http://onlinelibrary.wiley.com/doi/10.1111/j.1574-6976.2003.tb00628.x/pdf
- http://www.pjps.pk/wp-content/uploads/pdfs/26/5/Paper-30.pdf
- Antimicrobial Agents and Chemotherapy, 2003 , vol. 47, 5 p. 1700 – 1706
- Antimicrobial Agents and Chemotherapy, 1999 , vol. 43, 1 p. 115 – 120
- Antimicrobial Agents and Chemotherapy, 1997 , vol. 41, 10 p. 2165 – 2172
- Tetrahedron, 2004 , vol. 60, 47 p. 10611 – 10618………… NMRhttp://www.sciencedirect.com/science/article/pii/S0040402004015108
Cooper, R.D.G.; Snyder, N.J.; Zweifel, M.J.; et al.; Reductive alkylation of glycopeptide antibiotics: Synthesis and antibacterial activity. J Antibiot 1996, 49, 6, 575-81.
Fromtling, R.A.; Castaer, J.; LY-333328. Drugs Fut 1998, 23, 1, 17.
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0817797; JP 1999502534; WO 9630401 .
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0667353; EP 1016670; EP 1031576 .
| EP0435503A1 * | Dec 11, 1990 | Jul 3, 1991 | Eli Lilly And Company | Improvements in or relating to gylcopeptide derivatives |
| US4639433 * | Aug 14, 1985 | Jan 27, 1987 | Eli Lilly And Company | Glycopeptide derivatives |
| US4698327 * | Apr 18, 1986 | Oct 6, 1987 | Eli Lilly And Company | Novel glycopeptide derivatives |
| US20040106590 * | Aug 29, 2003 | Jun 3, 2004 | Barry Eisenstein | Methods and reagents for treating infections of clostridium difficile and diseases associated therewith |
| US20050197333 | Dec 22, 2004 | Sep 8, 2005 | Van Duzer John H. | Rifamycin analogs and uses thereof |
| US20070014849 | Sep 20, 2006 | Jan 18, 2007 | Daniela Jabes | Use of ramoplanin to treat diseases associated with the use of antibiotics |
| US20030176327 * | Oct 18, 2002 | Sep 18, 2003 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040147441 | Aug 25, 2003 | Jul 29, 2004 | Leach Timothy S. | Methods and reagents for preventing bacteremias |
| WO1999010006A1 | Aug 18, 1998 | Mar 4, 1999 | Lilly Co Eli | Therapy for staphylococcus aureus |
| WO2000066144A2 | Apr 19, 2000 | Nov 9, 2000 | Lilly Co Eli | Monthly doses of glycopeptide antibiotics for treatment of streptococcus pneumoniae infections |
| WO2008097364A2 | Sep 24, 2007 | Aug 14, 2008 | Targanta Therapeutics Corp | Use of oritavancin for prevention and treatment of anthrax |
| WO1998052592A1 * | May 5, 1998 | Nov 26, 1998 | Lilly Co Eli | Urea and thiourea derivatives of glycopeptides |
| WO2002036612A1 * | Nov 2, 2001 | May 10, 2002 | Univ Cambridge Tech | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements |
| WO2007138999A1 | May 25, 2007 | Dec 6, 2007 | Shionogi & Co | Glycopeptide antibiotic derivative |
| WO2009081958A1 | Dec 25, 2008 | Jul 2, 2009 | Shionogi & Co | Glycosylated glycopeptide antibiotic derivative |
| EP2314599A1 | Nov 24, 2005 | Apr 27, 2011 | National University Corporation Nagoya University | Glycopeptide antibiotic monomer derivatives |
| US5919756 * | May 1, 1997 | Jul 6, 1999 | Eli Lilly And Company | Amides |
| US5919771 * | Apr 30, 1998 | Jul 6, 1999 | Eli Lilly And Company | Urea and thiourea derivatives of glycopeptides |
| US7078380 | Nov 2, 2001 | Jul 18, 2006 | Cambridge University Technical Services Limited | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane associating elements |
| US8481696 | Dec 25, 2008 | Jul 9, 2013 | Shionogi & Co., Ltd. | Glycosylated glycopeptide antibiotic derivatives |


No comments:
Post a Comment