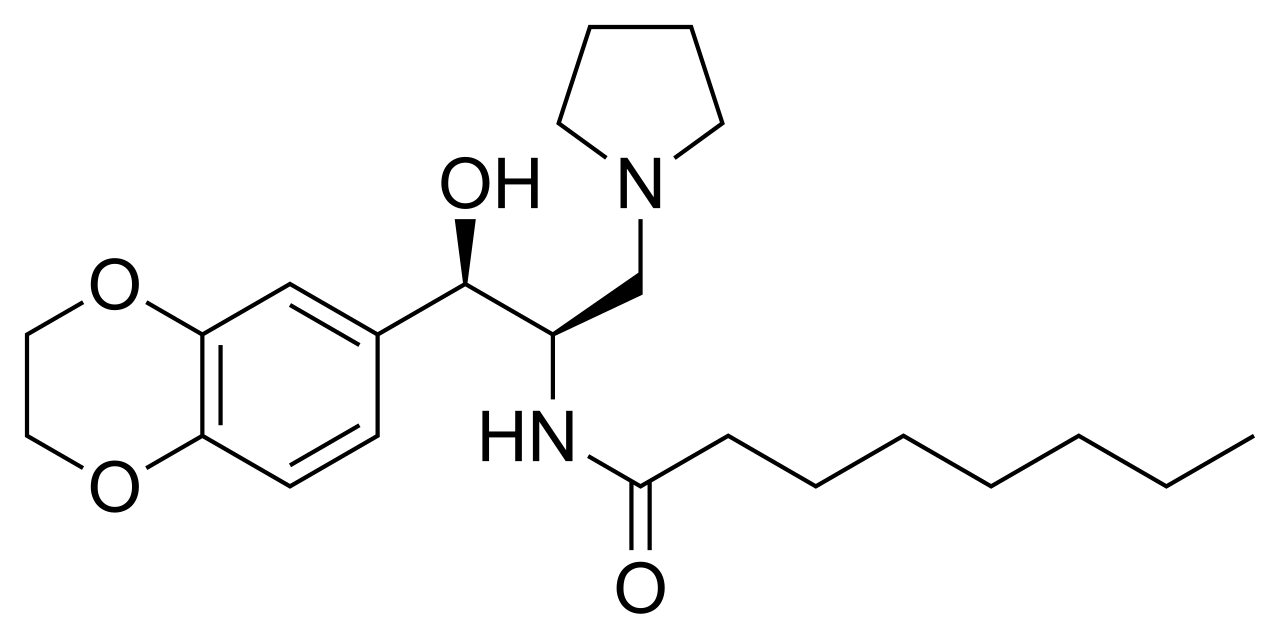
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
Eliglustat (INN, USAN;[1] trade name Cerdelga) is a treatment for Gaucher’s diseasedeveloped by Genzyme Corp that was approved by the FDA August 2014.[2] Commonly used as the tartrate salt, the compound is believed to work by inhibition ofglucosylceramide synthase.[3][4]
In March 2015, eliglustat tartrate was approved in Japan for the treatment of Gaucher disease. Eliglustat tartrate was described specifically within the US FDA’s Orange Booked listed US6916802, which is set to expire in April 2022.
In May 2015, the Orange Book also listed that eliglustat tartrate had Orphan Drug Exclusivity and New Chemical Entity exclusivity until 2019 and 2021, respectively.
it having been developed and launched as eliglustat tartrate by Genzyme (a wholly owned subsidiary of Sanofi), under license from the University of Michigan.
Eliglustat tartrate is known to act as inhibitors of glucosylceramide synthase and glycolipid, useful for the treatment of Gaucher’s disease type I and lysosome storage disease.
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
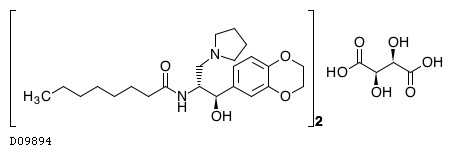
Eliglustat tartrate (USAN)
CAS:928659-70-5
February 15, 2013
Genzyme , a Sanofi company (EURONEXT: SAN and NYSE: SNY), today announced positive new data from the Phase 3 ENGAGE and ENCORE studies of eliglustat tartrate, its investigational oral therapy for Gaucher disease type 1. The results from the ENGAGE study were presented today at the 9th Annual Lysosomal Disease Network WORLD Symposium in Orlando, Fla. In conjunction with this meeting, Genzyme also released topline data from its second Phase 3 study, ENCORE. Both studies met their primary efficacy endpoints and together will form the basis of Genzyme’s registration package for eliglustat tartrateThe data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease”The company is developing eliglustat tartrate, a capsule taken orally, to provide a convenient treatment alternative for patients with Gaucher disease type 1 and to provide a broader range of treatment options for patients and physicians. Genzyme’s clinical development program for eliglustat tartrate represents the largest clinical program ever focused on Gaucher disease type 1 with approximately 400 patients treated in 30 countries.“The data presented at this year’s WORLD symposium reinforce our confidence that eliglustat tartrate may become an important oral option for patients with Gaucher disease,” said Genzyme’s Head of Rare Diseases, Rogerio Vivaldi MD. “We are excited about this therapy’s potential and are making excellent progress in our robust development plan for bringing eliglustat tartrate to the market.”ENGAGE Study Results:In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.
Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
WO 2015059679
| Process for the preparation of eliglustat free base – comprising the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat. | |
| Dr Reddy’s Laboratories Ltd | |
| New crystalline eliglustat free base Form R1 and a process for its preparation are claimed. Also claimed is a process for the preparation of eliglustat free base which comprises the reaction of S-(+)-phenyl glycinol with phenyl-alpha-bromoacetate to obtain 5-phenylmorpholin-2-one, which is further converted to eliglustat.Further eliglustat oxalate, its crystalline form, and a process for the preparation of crystalline eliglustat oxalate, are claimed. | |
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy.
Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence:
(i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate,
(ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone,
(iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine,
(iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
Example 1 : Preparation of 5-phenyl morpholine-2-one hydrochloride
To a (S) + phenyl glycinol (100g) add N, N-diisopropylethylamine (314ml) and acetonitrile (2000ml) under nitrogen atmosphere at room temperature. It was cooled to 10- 15° C. Phenyl bromoacetate (172.4g) dissolved in acetonitrile (500ml) was added to the above solution at 15° C over a period of 30 min. The reaction mixture is allowed to room temperature and stirred for 16-20h. Progress of the reaction was monitored by thin layer chromatography. After completion of the reaction, the reaction mixture was concentrated under reduced pressure at a water bath
temperature less than 25° C to get a residue. The residue was dissolved in ethyl acetate (1000ml) and stirred for 1 h at 15-20°C to obtain a white solid. The solid material obtained was filtered and washed with ethyl acetate (200ml). The filtrate was dried over anhydrous sodium sulphate (20g) and concentrated under reduced pressure at a water bath temperature less than 25° C to give crude compound (1000g) as brown syrup. The Crude brown syrup is converted to HCI salt by using HCI in ethyl acetate to afford 5-phenyl morpholine-2-one hydrochloride (44g) as a white solid. Yield: 50%, Mass: m/z = 177.6; HPLC (% Area Method): 90.5%
Example 2: Preparation of (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4] dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one.
5-phenyl morpholine-2-one hydrochloride (100g) obtained from above stage 1 is dissolved in toluene (2500ml) under nitrogen atmosphere at 25-30°C. 1 ,4-benzodioxane-6-carboxaldehyde (185.3g) and sodium sulphate (400g) was added to the above solution and the reaction mixture was heated at 100-105°C for 72h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature less than 25° C to get a residue. The residue was cooled to 10°C, ethyl acetate (2700ml) and 50% sodium bisulphate solution (1351 ml) was added to the residue and stirred for 1 h at 10°C to obtain a white solid. The obtained white solid was filtered and washed with ethyl acetate. The separated ethyl acetate layer was washed with water (1000ml), brine (1000ml) and dried over anhydrous sodium sulphate. The organic layer was concentrated under reduced pressure at a water bath temperature of 45-50°C to get a crude material. The obtained crude material is triturated with diethyl ether (1500ml) to get a solid material which is filtered and dried under vacuum at room temperature for 2-3h to afford (1 R,3S,5S,8aS)-1 ,3-Bis-(2′,3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (148g) as a yellow solid. Yield: 54%, Mass: m/z = 487.7; HPLC (% Area Method): 95.4 %
Example 3: Preparation of (2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 ”^henyl-ethy^
(1 R,3S,5S,8aS)-1 !3-Bis-(2′!3′-dihydro-benzo[1 ,4]dioxin-6′-yl)-5-phenyl-tetrahydro-oxazolo[4,3-c][1 ,4]oxazin-8-one (70g) obtained from above stage 2 was dissolved in chloroform (1400ml) at room temperature. It was cooled to 0-5°C and pyrrolidone (59.5ml) was added at 0-5°C over a period of 30 minutes. The reaction mixture was allowed to room temperature and stirred for 16-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was concentrated under reduced pressure at a water bath temperature of 40-45°C to obtain a crude. The obtained crude was dissolved in methanol (1190ml) and 1 N HCI (1 190ml) at 10-15° C, stirred for 10 minutes and heated at 80-85°C for 7h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C.The aqueous layer was extracted with ethyl acetate and the organic layer was washed with 1 N HCI (50ml). The aqueous layer was basified with saturated sodium bicarbonate solution up to pH 8-9 and extracted with ethyl acetate (3x70ml). The combined organic layers was washed with brine (100ml), dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50-55°C to afford (2S,3R,1″S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (53g) as a yellow foamy solid. Yield: 90%, Mass: m/z = 412.7, HPLC (% Area Method): 85.1 %
Example 4: Preparation of (1 R,2R,1 “S)-1-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol.
(2S,3R,1 “S)-3-(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)-3-hydroxy-2-(2″-hydroxy-1 “-phenyl-ethylamino)-1 -pyrrolidin-1 -yl-propan-1 -one (2.5g) obtained from above stage 3 dissolved in Tetrahydrofuran (106ml) was added to a solution of Lithium aluminium hydride (12.2g) in tetrahydrofuran (795ml) at 0°C and the reaction mixture was heated at 60-65°C for 10h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 5- 10°C and quenched in saturated sodium sulphate solution (100ml) at 5-10°C. Ethyl acetate was added to the reaction mass and stirred for 30-45 min. The obtained solid is filtered through celite bed and washed with ethyl acetate. Filtrate was dried over anhydrous sodium sulphate and concentrated under reduced pressure at a water bath temperature of 50°C to afford (1 R,2R, 1″S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (43.51 g) as a yellow gummy liquid. The crude is used for the next step without further purification. Yield: 85%, Mass: m/z = 398.7, HPLC (% Area Method): 77 %
Example 5: Preparation of (1 R, 2R)-2-Amino-1-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol.
(1 R,2R,1 “S)-1 -(2′,3′-(Dihydro-benzo[1 ,4]dioxin-6′-yl)2-hydroxy-2-(2″-hydroxy-1 ‘-phenyl-ethylamino)-3-pyrrolidin-1 -yl-propan-1 -ol (40g) obtained from above stage 4 was dissolved in methanol (400ml) at room temperature in a 2L hydrogenation flask. Trifluoroacetic acid (15.5ml) and 20% Pd (OH) 2 (40g) was added to the above solution under nitrogen atmosphere. The reaction mixture was hydrogenated under H2, 10Opsi for 16-18h at room temperature. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was filtered through celite bed and washed with methanol (44ml) and water (44ml). Methanol was concentrated under reduced pressure at a water bath temperature of 50-55°C and the aqueous layer was washed with ethyl acetate. The aqueous layer was basified with 10M NaOH till the PH reaches 12-14 and then extracted with dichloromethane (2x125ml). The organic layer was dried over anhydrous sodium sulphate (3gm) and concentrated under reduced pressure at a water bath temperature of 45°C to obtain a gummy liquid. The gummy liquid was triturated with methyl tertiary butyl ether for 1 h to get a white solid, which is filtered and dried under vacuum at room temperature to afford (1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (23g) as a white solid. Yield: 82.3%, Mass (m/zj: 278.8, HPLC (% Area Method): 99.5%, Chiral HPLC (% Area Method): 97.9%
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
……………………………..
……………………………..
Nmr predict
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-1h.png)
13 C NMR
![N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide NMR spectra analysis, Chemical CAS NO. 491833-29-5 NMR spectral analysis, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-09-06/001/571/702/491833-29-5-13c.png)
CAS NO. 491833-29-5, N-[(1R,2R)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-pyrrolidin-1-ylpropan-2-yl]octanamide
C-NMR spectral analysis
………………..
Compound 7
(1R,2R)-Nonanoic acid[2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide

This compound was prepared by the method described for Compound 6 using Nonanoic acid N-hydroxysuccinimide ester. Analytical HPLC showed this material to be 98.4% pure. mp 74–75° C.
1H NMR (CDCl3) δ 6.86–6.76 (m, 3H), 5.83 (d, J=7.3 Hz, 1H), 4.90 (d, J=3.3 Hz, 1H), 4.24 (s, 4H), 4.24–4.18 (m, 1H), 2.85–2.75 (m, 2H), 2.69–2.62 (m, 4H), 2.10 (t, J=7.3 Hz, 2H), 1.55–1.45 (m, 2H), 1.70–1.85 (m, 4H), 1.30–1.15 (m, 10H), 0.87 (t, J=6.9 Hz, 3H) ppm.
Intermediate 4(1R,2R)-2-Amino-1-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-3-pyrrolidin-1-yl-propan-1-ol

Intermediate 3 (5.3 g, 13.3 mmol) was dissolved in methanol (60 mL). Water (6 mL) and trifluoroacetic acid (2.05 m/L, 26.6 mmol, 2 equivalents) were added. After being placed under nitrogen, 20% Palladium hydroxide on carbon (Pearlman’s catalysis, Lancaster or Aldrich, 5.3 g) was added. The mixture was placed in a Parr Pressure Reactor Apparatus with glass insert. The apparatus was placed under nitrogen and then under hydrogen pressure 110–120 psi. The mixture was stirred for 2–3 days at room temperature under hydrogen pressure 100–120 psi. The reaction was placed under nitrogen and filtered through a pad of celite. The celite pad was washed with methanol (100 mL) and water (100 mL). The methanol was removed by rotoevaporation. The aqueous layer was washed with ethyl acetate three times (100, 50, 50 mL). A 10 M NaOH solution (10 mL) was added to the aqueous layer (pH=12–14). The product was extracted from the aqueous layer three times with methylene chloride (100, 100, 50 mL). The combined organic layers were dried with Na2SO4, filtered and rotoevaporated to a colorless oil. The foamy oil was vacuum dried for 2 h. Intermediate 4 was obtained in 90% yield (3.34 g).
Intermediate 3(1R,2R,1″S)-1-(2′,3′-Dihydro-benzo[1,4]dioxin-6′-yl)-2-(2″-hydroxy -1′-phenyl-ethylamino)-3-pyrrolidin-1-yl-propan-1-ol

To a 3-neck flask equipped with a dropping funnel and condenser was added LiAlH4(Aldrich, 1.2 g, 31.7 mmol, 2.5 equivalents) and anhydrous THF (20 mL) under nitrogen. A solution of Intermediate 2 (5.23 g, 12.68 mmol) in anhydrous THF (75 mL) was added dropwise to the reaction over 15–30 minutes. The reaction was refluxed under nitrogen for 9 hours. The reaction was cooled in an ice bath and a 1M NaOH solution was carefully added dropwise. After stirring at room temperature for 15 minutes, water (50 mL) and ethyl acetate (75 mL) was added. The layers were separated and the aqueous layer was extracted twice with ethyl acetate (75 mL). The combined organic layers were washed with saturated sodium chloride solution (25 mL). After drying with Na2SO4 the solution was filtered and rotoevaporated to yield a colorless to yellow foamy oil. Intermediate 3 was obtained in 99% yield (5.3 g).
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| N-[(1R,2R)-1-(2,3-Dihydro-1,4-benzodioxin-6-yl)-1-hydroxy-3-(1-pyrrolidinyl)-2-propanyl]octanamide | |
| CLINICAL DATA | |
| TRADE NAMES | Cerdelga |
| LEGAL STATUS |
|
| IDENTIFIERS | |
| CAS REGISTRY NUMBER | 491833-29-5 |
| ATC CODE | A16AX10 |
| PUBCHEM | CID 23652731 |
| CHEMSPIDER | 28475348 |
| CHEBI | CHEBI:82752 |
| CHEMICAL DATA | |
| FORMULA | C23H36N2O4 |
| MOLECULAR MASS | 404.543 g/mol |
- https://download.ama-assn.org/resources/doc/usan/x-pub/eligustat.pdf
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm410585.htm
- Lee, L.; Abe, A.; Shayman, J. A. (21 May 1999). “Improved Inhibitors of Glucosylceramide Synthase”. Journal of Biological Chemistry 274(21): 14662–14669.doi:10.1074/jbc.274.21.14662.
- Shayman, JA (Aug 1, 2010). “ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease.”.Drugs of the future 35 (8): 613–620.PMID 22563139.
| WO2008150486A2* | May 30, 2008 | Dec 11, 2008 | Genzyme Corp | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
| WO2009045503A1* | Oct 3, 2008 | Apr 9, 2009 | Genzyme Corp | Method of treating polycystic kidney diseases with ceramide derivatives |
| WO2010014554A1* | Jul 27, 2009 | Feb 4, 2010 | Genzyme Corporation | Glucosylceramide synthase inhibition for the treatment of collapsing glomerulopathy and other glomerular disease |
| WO2010039256A1* | Oct 2, 2009 | Apr 8, 2010 | Genzyme Corporation | 2-acylaminopropoanol-type glucosylceramide synthase inhibitors |
………………..
SWEDEN


Alfred Nobel had the unpleasant surprise of reading his own obituary, titled The merchant of death is dead, in a French newspaper.



 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
No comments:
Post a Comment