
MILTEFOSINE
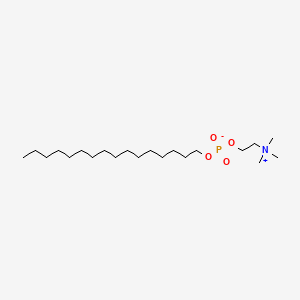
2-(hexadecoxy-oxido-phosphoryl)oxyethyl-trimethyl-azanium
58066-85-6
Hexadecylphosphocholine, Miltex, HDPC, HePC, Hexadecylphosphorylcholine, 58066-85-6, Miltefosina, Miltefosinum, Impavido
Molecular Formula: C21H46NO4P Molecular Weight: 407.568002
March 19, 2014 -- The U.S. Food and Drug Administration today approved Impavido (miltefosine) to treat a tropical disease called leishmaniasis.
Leishmaniasis is a disease caused by Leishmania, a parasite which is transmitted to humans through sand fly bites. The disease occurs primarily in people who live in the tropics and subtropics. Most U.S. patients acquire leishmaniasis overseas.
Impavido is an oral medicine approved to treat the three main types of leishmaniasis: visceral leishmaniasis (affects internal organs), cutaneous leishmaniasis (affects the skin) and mucosal leishmaniasis (affects the nose and throat). It is intended for patients 12 years of age and older. Impavido is the first FDA-approved drug to treat cutaneous or mucosal leishmaniasis.
“Today’s approval demonstrates the FDA’s commitment to making available therapeutic options to treat tropical diseases,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
The FDA granted Impavido fast track designation, priority review, and orphan product designation. These designations were granted because the drug demonstrated the potential to fill an unmet medical need in a serious disease or condition, the potential to be a significant improvement in safety or effectiveness in the treatment of a serious disease or condition, and is intended to treat a rare disease, respectively. With this approval, Impavido’s manufacturer, Paladin Therapeutics, is awarded a Tropical Disease Priority Review Voucher under a provision included in the Food and Drug Administration Amendments Act of 2007 that aims to encourage development of new drugs and biological products for the prevention and treatment of certain tropical diseases.
Impavido’s safety and efficacy were evaluated in four clinical trials. A total of 547 patients received Impavido and 183 patients received either a comparator drug or a placebo. Results from these trials demonstrated that Impavido is safe and effective in treating visceral, cutaneous and mucosal leishmaniasis.
The labeling for Impavido includes a boxed warning to alert patients and health care professionals that the drug can cause fetal harm and therefore should not be given to pregnant women. Health care professionals should advise women to use effective contraception during and for five months after Impavido therapy.
The most common side effects identified in clinical trials were nausea, vomiting, diarrhea, headache, decreased appetite, dizziness, abdominal pain, itching, drowsiness and elevated levels of liver enzymes (transaminases) and creatinine.
Paladin Therapeutics is based in Montreal, Canada
Miltefosine (INN, trade names Impavido and Miltex) is a phospholipid drug. Chemically it is a derivative of alkylphosphocholinecompounds discovered in the early 1980s. It was developed in the late 1980s as an anticancer drug by German scientists Hansjörg Eibl and Clemens Unger.[2] Simultaneously but independently it was found that the drug could kill Leishmania parasites, and since the mid-1990s successful clinical trials were conducted. The drug became the first (and still the only prescribed) oral drug in the treatment ofleishmaniasis. It is now known to be a broad-spectrum antimicrobial drug, active against pathogenic bacteria and fungi,[1][3] as well as human trematode Schistosoma mansoni and its vector host, the snail Biomphalaria alexandrina.[4] It can be administered orally and topically.
In the target cell, it acts as an Akt inhibitor. Therefore, it is also under investigation as a potential therapy against HIV infection.[5][6]
Phospholipid group alkylphosphocholine were known since the early 1980s, particularly in terms of their binding affinity with cobra venom.[7]In 1987 the phosholids were found to potent toxins on leukemic cell culture.[8] Initial in vivo investigation on the antineoplastic activity showed positive result, but then only at high dosage and at high toxicity.[9] At the same time in Germany, Hansjörg Eibl, at the Max Planck Institute for Biophysical Chemistry, and Clemens Unger, at the University of Göttingen, demonstrated that the antineoplastic activity of the phospholipid analogue miltefosine (at the time known as hexadecylphosphocholine) was indeed tumour-specific. It was highly effective against methylnitrosourea-induced mammary carcinoma, but less so on transplantable mammary carcinomas and autochthonous benzo(a)pyrene-induced sarcomas, and relatively inactive on Walker 256 carcinosarcoma and autochthonous acetoxymethylmethylnitrosamine-induced colonic tumors of rats.[10][11] It was subsequently found that miltefosine was strucrally unique among lipds having anticancer property in that it lacks the glycerol group, is highly selective on cell types and acts through different mechanism.[12][13]
In the same year as the discovery of the acticancer property, miltefosine was reported by S. L. Croft and his team at the London School of Hygiene and Tropical Medicine as having antileishmanial effect as well. The compound was effective against Leishmania donovani amastigotes in cultured mouse peritoneal macrophages at a dose of 12.8 mg/kg/day in a five-day course.[14] However priority was given to the development of the compound for cutaneous metastases of breast cancer. In 1992 a new research was reported in which the compound was highly effective in mouse against different life cycle stages of different Leishmania species, and in fect more potent than the conventional sodium stibogluconate therapy by a factor of more than 600.[15] Results of the first clinical trial in humans were reported from Indian patients with chronic leishmaniasis with high degree of success and safety.[16] This promising development promulgated a unique public–private partnership collaboration between ASTA Medica (later Zentaris GmbH), the WHO Special Programme for Research and Training in Tropical Diseases, and the Government of India. Eventually, several successful Phase II and III trials led to the approval of miltefosine in 2002 as the first and only oral drug for leishmaniasis.[1]
Miltefosine is registered and used by Zentaris GmbH in India, Colombia and Germany for the treatment of visceral and cutaneous leishmaniasis, and is undergoing clinical trials for this use in several other countries, such as Brazil[17] and Guatemala.[18]
Miltefosine is a phosphocholine analogue that was originally launched in 1993 by Baxter Oncology for the treatment of cancer. In 2003, Zentaris (formerly part of Asta Medica) launched the drug for the oral treatment of visceral leishmaniasis. Zentaris has also brought the product to market for the treatment of cutaneous leishmaniasis. Jado Technologies is conducting phase II clinical trials for the treatment of antihistamine resistant urticaria. Clinical trials had been ongoing for several indications, including the treatment of cutaneous mastocytosis or cutaneous involvement of systemic mastocytosis. Jado is investigating topical and oral versions of the compound in phase II trials in several allergy indications.
Miltefosine is effective against promastigotes and intracellular amastigotes, which survive and multiply in phagolysosomal compartments of macrophages and make up the two stages of the leishmania lifecycle. Although the exact mechanism of action of the drug has not been determined, it may exert its therapeutic effect through inhibition of phospholipid metabolism. Another theory suggests that miltefosine may interfere with leishmaniacal membrane signal transduction, lipid metabolism and glycosylphosphatidylinositol anchor biosynthesis. The drug is well absorbed in the gastrointestinal tract after a single oral administration and is widely distributed throughout the body.
Miltefosine was originally developed under a collaboration between the Indian government, the German biopharmaceutical company Zentaris, and the Tropical Disease Research (TDR) programme, co-sponsored by the World Health Organization and the United Nations Development Programme (UNDP). Subsequent to the product's approval, Zentaris partnered with various organizations for its distribution. In February 2004, Roche and Zentaris entered into a marketing agreement, pursuant to which Roche agreed to support Zentaris in the registration process and to market miltefosine in Brazil.
Several medical agents have some efficacy against visceral or cutaneous leishmaniasis, however a 2005 survey concluded that Miltefosine is the only effective oral treatment for both forms of leishmaniasis.[19]
Miltefosine is being investigated by researchers interested in finding treatments for infections which have become resistant to existing drugs. Animal and in vitro studies suggest it may have broad anti-protozoal and anti-fungal properties:
- Animal studies suggest miltefosine may also be effective against Trypanosoma cruzi, the parasite responsible for Chagas' disease.[20]
- Several studies have found the drug to be effective against Cryptococcus neoformans, Candida, Aspergillus and Fusarium.[21]
- An in vitro study found that miltefosine is effective against metronidazole-resistant variants of Trichomonas vaginalis, a sexually transmitted protozoal disease.[22]
- Hexadecyltrimethylammonium bromide, a compound structurally similar to miltefosine, was recently found to exhibit potent in vitro activity against Plasmodium falciparum.[23]
- Miltefosine is being made available in the United States through the CDC for emergency use under an expanded access IND protocol for treatment of free-living amoeba (FLA) infections:Primary amoebic meningoencephalitis caused by Naegleria fowleri and Granulomatous Amebic Encephalitis caused by Balamuthia mandrillaris, and Acanthamoeba species.[24][25]
Investigatory usage against HIV infection
Miltefosine targets HIV infected macrophages, which play a role in vivo as long-lived HIV-1 reservoirs. The HIV protein Tat activates pro-survival PI3K/Akt pathway in primary human macrophages. Miltefosine acts by inhibiting the PI3K/Akt pathway, thus removing the infected macrophages from circulation, without affecting healthy cells.[5] It significantly reduces replication of HIV-1 in cocultures of human dendritic cells (DCs) and CD4(+) T cells, which is due to a rapid secretion of soluble factors and is associated with induction of type-I interferon (IFN) in the human cells.[26]
In leishmanisis the recommended dose as oral monotherapy is 2.5 mg/kg/day for a total of 28 days. However, due to frequent commercial shortage of the 10 mg capsule, dosages are often altered. For example, the Indian government recommends 100 mg/day miltefosine for patients with a body weight ≥25 kg (corresponding to ∼1.7–4 mg/kg/day) and 50 mg/day for body weights <25 kg (corresponding to ∼2–5.5 mg/kg/day).[1] Even up to 150 mg/day for 28 days was found to be quite safe.[27]
The main side effects reported with miltefosine treatment are nausea and vomiting, which occur in 60% of patients. Adverse effect is more severe in women and young children. The overall effects are quite mild and easily reverse.[28] It is embryotoxic and fetotoxic in rats and rabbits, and teratogenic in rats but not in rabbits. It is therefore contraindicated for use during pregnancy, andcontraception is required beyond the end of treatment in women of child-bearing age.[29]
 | miltefosine (1-hexadecylphosphoryl-choline, HePC); Calbiochem 475841 |
Compounds o f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
f the general formula I belonging to the class of phospholipids (X is O and R2 is a group of formula II), e.g. alkyloxy phospholipids (Y is O) and the corresponding alkylthio derivatives (Y is S), can be prepared as described in the literature (Bittman, R.; J. Med. Chem. 1997, 40, 1391-1395; Reddy, K. C.; Tetrahedron Lett. 1994, 35, 2679-2682; Guivisdalsky, P. N.; J. Med. Chem. 1990, 33, 2614-2621 and references cited therein) or by standard variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO and SCO, respectively) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
Compounds of the general formula I belonging to the class of phosphonolipids (X is a direct bond and R2 is a group of formula II), e.g alkyloxy phosphonolipids (Y is O and R2 is a group of formula II) and the corresponding alkylthio derivatives (Y is S) can be prepared as published by Bittman et al. (Bittman, R.; J. Med. Chem. 1993, 36, 297-299; Bittman, R.; J. Med. Chem.1994, 37, 425-430 and references cited therein) or by synthetic variations of the procedures described therein. Synthesis of the corresponding ester and thioester analogues (Y is OCO or SCO) can be accomplished by standard acylation of the hydroxy or thio precursor materials.
SEE
Antitumor ether lipids: An improved synthesis of ilmofosine and an enantioselective synthesis of an ilmofosine analog
Tetrahedron Lett 1994, 35(17): 2679
Tetrahedron Lett 1994, 35(17): 2679
AND
Hexadecylphosphocholine, a new antineoplastic agent: Cytotoxic properties in leukaemic cells
J Cancer Res Clin Oncol 1986, 111: 24
J Cancer Res Clin Oncol 1986, 111: 24
References
- Dorlo, T. P. C.; Balasegaram, M.; Beijnen, J. H.; de Vries, P. J. (2012). "Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis". Journal of Antimicrobial Chemotherapy 67 (11): 2576–2597. doi:10.1093/jac/dks275.PMID 22833634.
- Eibl, H; Unger, C (1990 Sep). "Hexadecylphosphocholine: a new and selective antitumor drug.". Cancer Treatment Reviews 17 (2-3): 233–42. PMID 2272038.
- Almeida Pachioni, JD; Magalhães, JG; Cardoso Lima, EJ; Moura Bueno, LD; Barbosa, JF; Malta de Sá, M; Rangel-Yagui, CO (2013). "Alkylphospholipids - a promising class of chemotherapeutic agents with a broad pharmacological spectrum.". Journal of Pharmacy & Pharmaceutical sciences : a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques 16 (5): 742–59. PMID 24393556.
- Eissa, Maha M; El Bardicy, Samia; Tadros, Menerva (2011). "Bioactivity of miltefosine against aquatic stages of Schistosoma mansoni, Schistosoma haematobium and their snail hosts, supported by scanning electron microscopy". Parasites & Vectors 4 (1): 73.doi:10.1186/1756-3305-4-73. PMC PMC3114006. PMID 21569375.
- ^ Jump up to:a b Chugh P, Bradel-Tretheway B, Monteiro-Filho CM, et al. (2008). "Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy". Retrovirology 5 (1): 11. doi:10.1186/1742-4690-5-11. PMC 2265748. PMID 18237430.
- "Parasitic Drug Shows HIV-Fighting Promise". AIDSmeds.com. 2008-02-01. Retrieved 2008-02-02.
- Teshima, K; Ikeda, K; Hamaguchi, K; Hayashi, K (1983). "Bindings of cobra venom phospholipases A2 to micelles of n-hexadecylphosphorylcholine.". Journal of Biochemistry 94(1): 223–32. PMID 6619110.
- Fleer, EA; Unger, C; Kim, DJ; Eibl, H (1987). "Metabolism of ether phospholipids and analogs in neoplastic cells.". Lipids 22 (11): 856–61. PMID 3444378.
- Berger, MR; Petru, E; Schmähl, D (1987). "Therapeutic ratio of mono or combination bacterial lipopolysaccharide therapy in methylnitrosourea-induced rat mammary carcinoma.". Journal of Cancer Research and Clinical Oncology 113 (5): 437–45. PMID 3624299.
- Muschiol, C; Berger, MR; Schuler, B; Scherf, HR; Garzon, FT; Zeller, WJ; Unger, C; Eibl, HJ; Schmähl, D (1987). "Alkyl phosphocholines: toxicity and anticancer properties.". Lipids 22 (11): 930–4. PMID 3444388.
- Berger, MR; Muschiol, C; Schmähl, D; Eibl, HJ (1987). "New cytostatics with experimentally different toxic profiles.". Cancer treatment Reviews 14 (3-4): 307–17. PMID 3440252.
- Hilgard, P; Stekar, J; Voegeli, R; Engel, J; Schumacher, W; Eibl, H; Unger, C; Berger, MR (1988). "Characterization of the antitumor activity of hexadecylphosphocholine (D 18506).".European Journal of Cancer & Clinical Oncology 24 (9): 1457–61. PMID 3141197.
- Eibl, H; Unger, C (1990 Sep). "Hexadecylphosphocholine: a new and selective antitumor drug.". Cancer Treatment Reviews 17 (2-3): 233–42. PMID 2272038.
- Croft, S.L.; Neal, R.A.; Pendergast, W.; Chan, J.H. (1987). "The activity of alkyl phosphorylcholines and related derivatives against Leishmania donovani". Biochemical Pharmacology 36 (16): 2633–2636. doi:10.1016/0006-2952(87)90543-0.
- Kuhlencord, A; Maniera, T; Eibl, H; Unger, C (1992). "Hexadecylphosphocholine: oral treatment of visceral leishmaniasis in mice.". Antimicrobial Agents and Chemotherapy 36(8): 1630–1634. doi:10.1128/AAC.36.8.1630. PMC PMC192021. PMID 1329624.
- Sundar, Shyam; Rosenkaimer, Frank; Makharia, Manoj K; Goyal, Ashish K; Mandal, Ashim K; Voss, Andreas; Hilgard, Peter; Murray, Henry W (1998). "Trial of oral miltefosine for visceral leishmaniasis". The Lancet 352 (9143): 1821–1823. doi:10.1016/S0140-6736(98)04367-0.PMID 9851383.
- Cristina, Márcia; Pedrosa, Robert (September 2005). "Hospital de Doenças Tropicais testa droga contra calazar". Sapiência (in Portuguese) (Fundação de Amparo à Pesquisa do Estado do Piauí). Archived from the original on 2006-08-22. Retrieved 2006-09-01.
- Soto J, Berman J (2006). "Treatment of New World cutaneous leishmaniasis with miltefosine.". Trans R Soc Trop Med Hyg 100: S34. doi:10.1016/j.trstmh.2006.02.022.PMID 16930649.
- Berman, J. (2005). "Clinical status of agents being developed for leishmaniasis". Expert Opinion on Investigational Drugs 14 (11): 1337–1346. doi:10.1517/13543784.14.11.1337.PMID 16255674.
- Saraiva V, Gibaldi D, Previato J, Mendonça-Previato L, Bozza M, Freire-De-Lima C, Heise N (2002). "Proinflammatory and cytotoxic effects of hexadecylphosphocholine (miltefosine) against drug-resistant strains of Trypanosoma cruzi.". Antimicrob Agents Chemother 46 (11): 3472–7. doi:10.1128/AAC.46.11.3472-3477.2002. PMC 128733. PMID 12384352.
- Widmer F, Wright L, Obando D, Handke R, Ganendren R, Ellis D, Sorrell T (2006)."Hexadecylphosphocholine (miltefosine) has broad-spectrum fungicidal activity and is efficacious in a mouse model of cryptococcosis.". Antimicrob Agents Chemother 50 (2): 414–21. doi:10.1128/AAC.50.2.414-421.2006. PMC 1366877. PMID 16436691.
- Blaha C, Duchêne M, Aspöck H, Walochnik J (2006). "In vitro activity of hexadecylphosphocholine (miltefosine) against metronidazole-resistant and -susceptible strains of Trichomonas vaginalis". J. Antimicrob. Chemother. 57 (2): 273–8.doi:10.1093/jac/dki417. PMID 16344287.
- Choubey V, Maity P, Guha M, et al. (February 2007). "Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism". Antimicrob. Agents Chemother. 51 (2): 696–706. doi:10.1128/AAC.00919-06.PMC 1797733. PMID 17145794.
- Naegleria fowleri - Primary Amebic Meningoencephalitis (PAM)
- Brain-Eating Amoeba: How One Girl Survived
- Garg, Ravendra; Tremblay, Michel J. (October 2012). "Miltefosine represses HIV-1 replication in human dendritic cell/T-cell cocultures partially by inducing secretion of type-I interferon".Virology 432 (2): 271–276. doi:10.1016/j.virol.2012.05.032. PMID 22704066.
- Sundar, Shyam; Jha, T.K.; Thakur, C.P.; Bhattacharya, S.K.; Rai, M. (2006). "Oral miltefosine for the treatment of Indian visceral leishmaniasis". Transactions of the Royal Society of Tropical Medicine and Hygiene 100 (Suppl 1): S26–S33. doi:10.1016/j.trstmh.2006.02.011.PMID 16730038.
- S.D. Seth (2008). "Drug therapy of leishmaniasis". In S.D. Seth. Textbook of Pharmacology. Elsevier India. p. 31. ISBN 9788131211588.
- Sindermann, H.; Engel, J. (December 2006). "Development of miltefosine as an oral treatment for leishmaniasis". Transactions of the Royal Society of Tropical Medicine and Hygiene 100 (Suppl 1): S17–S20. doi:10.1016/j.trstmh.2006.02.010. PMID 16730362.
| US8211875 | 7-4-2012 | LOCAL TREATMENT OF NEUROFIBROMAS |
| US2011263531 | 10-28-2011 | METHODS FOR THE TREATMENT AND AMELIORATION OF ATOPIC DERMATITIS |
| US7998945 | 8-17-2011 | Methods for the treatment and amelioration of atopic dermatitis |
| US2007264206 | 11-16-2007 | Mucosal formulation |
| US2007167408 | 7-20-2007 | NOVEL ALKYL PHOSPHOLIPID DERIVATIVES WITH REDUCED CYTOTOXICITY AND USES THEREOF |
 an animation to soothe ones eye
an animation to soothe ones eye
No comments:
Post a Comment