
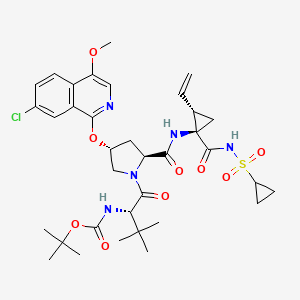
ASUNAPREVIR
630420-16-5 CAS
THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES
1. Cyclopropanecarboxamide, N-[(1,1-dimethylethoxy)carbonyl]-3-methyl-L-valyl-(4R)-4-[(7-chloro-4-methoxy-1-isoquinolinyl)oxy]-L-prolyl-1-amino-N-(cyclopropylsulfonyl)-2-ethenyl-, (1R,2S)-
2. 1,1-dimethylethyl [(1S)-1-{[(2S,4R)-4-(7-chloro-4methoxyisoquinolin-1-yloxy)-2-({(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)pyrrolidin-1-yl]carbonyl}-2,2-dimethylpropyl]carbamate
MOLECULAR FORMULA C35H46ClN5O9S
MOLECULAR WEIGHT 748.3
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION ...........BMS-650032
CAS REGISTRY NUMBER 630420-16-5
Asunaprevir (formerly BMS-650032) is an experimental drug candidate for the treatment of hepatitis C. It is undergoing development by Bristol-Myers Squibb and is currently inPhase III clinical trials.[1]
In 2013, the company Bristol-Myers Squibb received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and BMS-791325.
Asunaprevir is an inhibitor of the hepatitis C virus enzyme serine protease NS3.[2]
Asunaprevir is being tested in combination with pegylated interferon and ribavirin, as well as in interferon-free regimens with other direct-acting antiviral agents includingdaclatasvir[3][4][5]
Asunaprevir is an antiviral agent originated by Bristol-Myers Squibb undergoing the registration in Japan for the treatment of chronic hepatitis C virus infection in combination with daclatasvir in patients who are non-responsive to interferon plus ribavirin and interferon based therapy ineligible naive/intolerant
- "A Phase 3 Study in Combination With BMS-790052 and BMS-650032 in Japanese Hepatitis C Virus (HCV) Patients". ClinicalTrials.gov.
- C. Reviriego (2012). Drugs of the Future 37 (4): 247–254.doi:10.1358/dof.2012.37.4.1789350.
- Preliminary Study of Two Antiviral Agents for Hepatitis C Genotype 1. Lok, A et al. New England Journal of Medicine. 366(3):216-224. January 19, 2012.
- "Bristol-Myers' Daclatasvir, Asunaprevir Cured 77%: Study". Bloomberg. Apr 19, 2012.
- AASLD: Daclatasvir plus Asunaprevir Rapidly Suppresses HCV in Prior Null Responders. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Bioorganic and Medicinal Chemistry Letters, 2011 , vol. 21, 7 pg. 2048 - 2054
WO 2003099274, WO 2003099274, WO 2009085659
6-20-2012
|
Crystalline
forms of
N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N-
((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide
| |
4-25-2012
|
Hepatitis C Virus Inhibitors
| |
3-30-2011
|
HEPATITIS C VIRUS INHIBITORS
| |
11-12-2008
|
Hepatitis C virus inhibitors
| |
2-8-2006
|
Hepatitis C virus inhibitors
|
Hepatitis C virus (HCV) is a major human pathogen, infecting an estimated 170 million persons worldwide—roughly five times the number infected by human immunodeficiency virus type 1. A substantial fraction of these HCV infected individuals develop serious progressive liver disease, including cirrhosis and hepatocellular carcinoma.
Presently, the most effective HCV therapy employs a combination of alpha-interferon and ribavirin, leading to sustained efficacy in 40 percent of patients. Recent clinical results demonstrate that pegylated alpha-interferon is superior to unmodified alpha-interferon as monotherapy. However, even with experimental therapeutic regimens involving combinations of pegylated alpha-interferon and ribavirin, a substantial fraction of patients do not have a sustained reduction in viral load. Thus, there is a clear and unmet need to develop effective therapeutics for treatment of HCV infection.


http://www.google.com/patents/US8338606
........................
Compound 277
Compound
277 was prepared by following Scheme 2 of Example 269 except that 3-
(4-chloro-phenyl)-3-methoxy-acrylic acid was used in place of 2-
trifluormethoxycinnamic acid in step 1.
Step 1:
Modifications: 4.24 g 3-(4-chloro-phenyl)-3-methoxy-acrylic acid used, 130 mg product obtained (3% yield) Product:

Data: 1H NMR(400 MHz, CD3OD)
δ ppm 3.96 (s, 3 H), 7.19 (dd, 7=8.80, 2.45 Hz, 1 H), 7.28 (d, 7=2.45
Hz, 1 H), 7.34 (s, 1 H), 8.25 (d, 7=9.05 Hz, 1 H); MS: (M+H)+ 210.
Step 2:
Modifications: 105 mg 7-chloro-4-methoxy-2H-isoquinolin-l-one used, 60 mg product obtained (71% yield). Product:

Data: Η NMR (400 Hz, CDC13)
δ ppm 4.05 (s, 3 H), 7.67 (dd, 7=8.80, 1.96 Hz, 1 H), 7.80 (s, 1 H),
8.16 (d, 7=9.05 Hz, 1 H), 8.24 (d, 7=1.96 Hz, 1 H); MS: (M+H)+ 229.
Step 3:
Modifications:
46 mg l,7-dichloro-4-methoxy-isoquinoline and 113 mg { l-[2-(l-
cyclopropanesulfonylaminocarbonyl-2-vinyl-cyclopropylcarbamoyl)-4-hydroxy-
pyrrolidine-1 -carbon yl]-2,2-dimethyl-propyl} -carbamic acid
tert-butyl ester used, 50 mg product obtained (31% yield). Product:

Compound 277
Data: 1H NMR (400 Hz, CD3OD)
δ ppm 1.06 (m, 11 H), 1.16 (s, 9 H), 1.24 (m, 2 H), 1.44 (dd, 7=9.54,
5.38 Hz, 1 H), 1.88 (dd, 7=8.07, 5.62 Hz, 1 H), 2.28 (m, 2 H), 2.59 (dd,
7=13.69, 6.85 Hz, 1 H), 2.94 (m, 1 H), 4.00 (s, 3 H), 4.05 (d, 7=11.74
Hz, 1 H), 4.19 (s, 1 H), 4.43 (d, 7=11.49 Hz, 1 H), 4.56 (dd, 7=10.03,
6.85 Hz, 1 H), 5.12 (d, 7=11.49 Hz, 1 H), 5.30 (d, 7=17.12 Hz, 1 H),
5.76 (m, 2 H), 7.57 (s, 1 H), 7.67 (d, 7=8.56 Hz, 1 H), 8.04 (s, 1 H),
8.08 (d, 7=8.80 Hz, 1 H); MS: (M+H)+ 749.
..............
https://www.google.co.in/patents/US6995174?dq=WO+2003099274&ei=1DW5Uoa0C4GTrgfy84HgBQ&cl=en


..................
WO 2003099274

https://www.google.co.in/patents/US6995174?dq=WO+2003099274&ei=fje5Us3WBo3JrQfcsoHgAw&cl=en
.........................
https://www.google.co.in/patents/US20090202476?dq=WO+2009085659&ei=dzy5UpL_LMXXrQewxYG4Dw&cl=en


Preparation of Compound C
DMSO
(264 ml) was added to a mixture of Compound A (6 g, 26.31 mmol, 1.0 eq,
96.5% potency), Compound B (6.696 g, 28.96 mmol, 1.1 eq) and KOtBu
(8.856 g, 78.92 mmol, 3 eq) under nitrogen and stirred at 36° C. for 1
h. After cooling the dark solution to 16° C., it was treated with water
(66 ml) and EtOAc (132 ml). The resulting biphasic mixture was acidified
to pH 4.82 with 1N HCl (54 ml) at 11.2-14.6° C. The phases were
separated. The aqueous phase was extracted once with EtOAc (132 ml). The
organic phases were combined and washed with 25% brine (2×132 ml). Rich
organic phase (228 ml) was distilled at 30-40° C./50 mbar to 37.2 ml. A
fresh EtOAc (37.2 ml) was added and distilled out to 37.2 ml at 30-35°
C./50 nm bar. After heating the final EtOAc solution (37.2 ml) to 50°
C., heptane ((37.2 ml) was added at 46-51° C. and cooled to 22.5° C.
over 2 h. It was seeded with 49 mg of Compound C and held at 23° C. for
15 min to develop a thin slurry. It was cooled to 0.5° C. in 30 min and
kept at 0.2-0.5° C. for 3 h. After the filtration, the cake was washed
with heptane (16.7 ml) and dried at 47° C./80 mm/15.5 h to give Compound
C as beige colored solids (6.3717 g, 58.9% corrected yield, 99.2%
potency, 97.4 AP).
Preparation of Compound E
DIPEA
(2.15 ml, 12.3 mmol, 1.3 eq followed by EDAC (2 g, 10.4 mmol, 1.1 eq)
were added to a mixture of Compound C (4 g, 9.46 mmol, 97.4% potency,
98.5 AP), Compound D (4.568 g, 11.35 mmol, 1.20 eq), HOBT-H2O (0.86 g,
4.18 mmol, 0.44 eq) in CH2Cl2 (40
ml) at 23-25° C. under nitrogen. The reaction was complete after 3 h at
23-25° C. It was then washed with 1N HCl (12 ml), water (12 ml) and 25%
brine (12 ml). MeOH (80 ml) was added to the rich organic solution at
25° C., which was distilled at atmospheric pressure to ˜60 ml to
initiate the crystallization of the product. The crystal slurry was then
cooled from 64° C. to 60° C. in 5 min and stirred at 60° C. for 1 h. It
was further cooled to 24° C. over 1.5 h and held at 24° C. for 2 h.
After the filtration, the cake was washed with MeOH (12 ml) and dried at
51° C./20-40 nm i/18 h to give Compound E (5.33 g, 89% yield, 97.7%
potency, 99.1 AP).
Preparation of Compound F
5-6N
HCl in IPA (10.08 ml, 50.5 mmol, Normality: 5N) was added in four
portions in 1 h to a solution of Compound E (8 g, 12.6 mmol, 97.7%
potency, 99.1 AP) in IPA (120 ml) at 75° C. After stirring for 1 h at
75° C., the resulting slurry was cooled to 21° C. in 2 h and stirred at
21° C. for 2 h. It was filtered and the cake was washed with IPA (2×24
ml). The wet cake was dried at 45° C./House vacuum/16 h to give Compound
F as an off-white solid (6.03 g, 84.5% yield, 98.5% potency, 100 AP).
Preparation of Compound (I)
DIPEA
(9.824 ml) followed by HATU (7.99 g) were added to a stirred mixture of
Compound F (10 g, 99.2% potency, 99.6 AP) and Compound G (4.41 g) in CH2Cl2 (100
ml) at 2.7-5° C. under nitrogen. The resulting light brown solution was
stirred at 0.2-3° C. for 1.5 h, at 3-20° C. in 0.5 h and at 20-23° C.
for 15.5 h for a reaction completion. It was quenched with 2N HCl (50
ml) at 23° C. and stirred for 20 min at 23-24° C. The biphasic mixture
was polish filtered through diatomaceous earth (Celite®) (10 g) to
remove insoluble solids of HOAT and HATU. The filter cake was washed
with 20 ml of CH2Cl2. After separating the organic
phase from the filtrates, it was washed with 2N HCl (5×50 ml) and water
(2×50 ml). The organic phase (115 ml) was concentrated to ˜50 ml, which
was diluted with absolute EtOH (200 proof, 100 ml) and concentrated
again to ˜50 ml. Absolute EtOH (50 ml) was added to bring the final
volume to 100 ml. It was then warmed to 50° C. to form a clear solution
and held at 50° C. for 35 min. The ethanolic solution was cooled from 50
to 23° C. over 15 min to form the crystal slurry. The slurry was
stirred at 23 CC for 18 h, cooled to 0.3° C. over 30 min and kept at
0.2-0.3° C. for 2 h. After the filtration, the cake was washed with cold
EtOH (2.7° C., 2×6 ml) and dried at 53° C./72 mm/67 h to give Compound
(I) in Form T1F-1/2 as an off white solid (10.49 g, 80.7% yield, 99.6
AP).https://www.google.co.in/patents/US20090202476?dq=WO+2009085659&ei=dzy5UpL_LMXXrQewxYG4Dw&cl=en
.........extra info
Hepatitis
C virus (HCV) infection is the principal cause of chronic liver disease
that can lead to cirrhosis, carcinoma and liver failure.1
More than 200 million people worldwide are chronically infected by this
virus. Currently, the most effective treatment for HCV infection is
based on a combination therapy of injectable pegylated interferon-α (PEG
IFN-α) and antiviral drug ribavirin. This treatment, indirectly
targeting the virus, is associated with significant side effects often
leading to treatment discontinuation in certain patient populations.2
In addition, this treatment regimen cures only less than 50% of
patients infected with genotype-1 which is the predominant genotype
(while genotype 1a is most abundant in the US, the majority of sequences
in Europe and Japan are from genotype 1b).3
Limited efficacy and adverse side effects of current treatment, and
high prevalence of infection worldwide highlight an urgent need for more
effective, convenient, and well-tolerated treatments.4
HCV
NS3 serine protease plays a critical role in the HCV replication by
cleaving downstream sites (with the assistance of the cofactor NS4A)
along the HCV viral polyprotein to produce functional proteins.
Recently, NS3/4A protease inhibitors have emerged as a promising
treatment for HCV infection.5
There are two distinct classes of NS3 protease inhibitors in clinical
development. The first class is comprised of serine-trap inhibitors,
exemplified by VX-950 (telaprevir)6 and SCH-503034 (boceprevir).7 The second class is represented by reversible noncovalent inhibitors such as macrocyclic inhibitors BILN-2061 (ciluprevir),8 ITMN-191 (danoprevir),9 TMC-43535010 and MK-7009 (vaniprevir).11 Due to concern over cardiac issues in animals treated with macrocyclic BILN-2061,12 newer acyclic inhibitors have recently been developed exemplified by BI-20133513 and BMS-650032.14 However, a rapid development of viral resistance has been observed for patients treated with HCV NS3 protease inhibitors.15
Therefore, the discovery of new NS3 protease inhibitors with novel
binding paradigm and thus potentially differentiated resistance profile
is highly desirable.
References and notes
- Rev. Med. Virol., 13 (2003), p. 57
- Hepatology, 36 (2002), p. S237
- Am. J. Med., 117 (2004), p. 344
- For a recent review on HCV anti-viral agents, see: Expert Opin. Invest. Drugs, 19 (2010), p. 63
- Curr. Opin. Pharmacol., 8 (2008), p. 522
- For a recent review on HCV NS3/4A protease inhibitors, see: Curr. Opin. Invest. Drugs, 10 (2009), p. 821
- Expert Rev. Anti. Infect. Ther., 7 (2009), p. 537
- Infect. Disord. Drug Targets, 6 (2006), p. 3
- Acc. Chem. Res., 41 (2008), p. 50
- J. Med. Chem., 47 (2004), p. 1605
- Antimicrob. Agents Chemother., 52 (2008), p. 4432
- Bioorg. Med. Chem. Lett., 18 (2008), p. 4853
- J. Med. Chem., 53 (2010), p. 2443
- Gastroenterology, 127 (2004), p. 1347
- J. Med. Chem., 53 (2010), p. 6466
- (a)Chemical and Engineering News (April 12, 2010 issue), 88, pp 30–33.
- (b)Perrone, R.K.; Wang, C.; Ying, W.; Song, A.I. WO 2009085659
- Sci. Transl. Med., 2 (2010), p. 30ra32


THANKS AND REGARD'S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
No comments:
Post a Comment