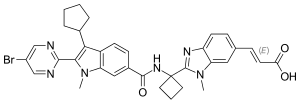
DELEOBUVIR
(2E)-3-(2-{1-[2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-1-methyl-1H-indole-6-carboxamido]cyclobutyl}-1-methyl-1H-benzimidazol- 6-yl)prop-2-enoic acid
1221574-24-8 CAS please check may be sodium salt??
cas no as per below ref ……863884-77-9 (free acid)
PHASE 3
BI-207127NA
BI-207127 (free acid)
BI-207127 (free acid)
BI-207127 is a novel HCV RNA polymerase inhibitor in phase III clinical development at Boehringer Ingelheim for the treatment of hepatitis C.
| Company | Boehringer Ingelheim GmbH |
| Description | Oral non-structural protein 5B (NS5B) RNA-dependent polymerase inhibitor |
| Molecular Target | HCV NS5B polymerase |
| Mechanism of Action | Viral polymerase inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Indication | Hepatitis C virus (HCV) |
| Partner |
Deleobuvir (formerly BI 207127) is an experimental drug for the treatment of hepatitis C. It is being developed by Boehringer-Ingelheimand is currently in Phase II trials. It is a non-nucleoside hepatitis C virus NS5B polymerase inhibitor. Deleobuvir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including faldaprevir.
Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of deleobuvir, faldaprevir, and ribavirin performed well in HCV genotype 1b patients.[1] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.Deleobuvir (BI 207127) is an investigational oral nonnucleoside inhibitor of hepatitis C virus (HCV) NS5B RNA polymerase. Antiviral activity, virology, pharmacokinetics, and safety were assessed in HCV genotype 1-infected patients receiving 5 days’ deleobuvir monotherapy. In this double-blind phase 1b study, treatment-naive (TN; n = 15) and treatment-experienced (TE; n = 45) patients without cirrhosis received placebo or deleobuvir at 100, 200, 400, 800, or 1,200 mg every 8 h (q8h) for 5 days. Patients with cirrhosis (n = 13) received deleobuvir at 400 or 600 mg q8h for 5 days. Virologic analyses included NS5B genotyping and phenotyping of individual isolates. At day 5, patients without cirrhosis had dose-dependent median HCV RNA reductions of up to 3.8 log10 (with no placebo response); patients with cirrhosis had median HCV RNA reductions of approximately 3.0 log10. Three patients discontinued due to adverse events (AEs). The most common AEs were gastrointestinal, nervous system, and skin/cutaneous tissue disorders. Plasma exposure of deleobuvir was supraproportional at doses ≥ 400 mg q8h and approximately 2-fold higher in patients with cirrhosis than in patients without cirrhosis. No virologic breakthrough was observed. NS5B substitutions associated with deleobuvir resistance in vitro were detected in 9/59 patients; seven encoded P495 substitutions, including P495L, which conferred 120- to 310-fold-decreased sensitivity to deleobuvir. P495 variants did not persist in follow-up without selective drug pressure. Deleobuvir monotherapy was generally well tolerated and demonstrated dose-dependent antiviral activity against HCV genotype 1 over 5 days.
These results were confirmed in the SOUND-C3 study, presented at the 2013 APASL Liver Conference, which found that 16 week triple therapy with deleobuvir + faldaprevir + ribavirin gave 95% SVR12 in HCV genotype 1b patients but poor virological response in genotype 1a.[2]
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- S Zeuzem, J-F Dufour, M Buti, V Soriano, R Buynak, P Mantry, J Taunk, JO Stern, R Vinisko, J-P Gallivan, WO Bocher and FJ Mensa.“Interferon-free treatment with faldaprevir, deleobuvir (BI 207127) and ribavirin in SOUND-C3: 95% SVR12 in HCV GT-1b”. 23rd Conference of the Asian Pacific Association for the Study of the Liver (APASL) 6–9 June 2013. Retrieved 12 Sep 2013.
PATENTS
WO 2013147750
| WO 2013147749 |
WO 2012041771
WO 2012044520
WO 2012016995
WO 2005080388
……………………………………………………
PATENT
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010059667A1 | Nov 18, 2009 | May 27, 2010 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition of a potent hcv inhibitor for oral administration |
| WO2011005646A2 | Jul 1, 2010 | Jan 13, 2011 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition for a hepatitis c viral protease inhibitor |
| WO2012041771A1 * | Sep 23, 2011 | Apr 5, 2012 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US4211771 | Feb 13, 1978 | Jul 8, 1980 | Robins Ronald K | Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide |
| US6063772 | Jun 15, 1998 | May 16, 2000 | Icn Pharmaceuticals, Inc. | Specific modulation of Th1/Th2 cytokine expression by ribavirin in activated T-lymphocytes |
| US6277830 | Jul 7, 1999 | Aug 21, 2001 | Schering Corporation | 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon |
| US6323180 | Aug 5, 1999 | Nov 27, 2001 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6403564 | Oct 14, 1999 | Jun 11, 2002 | Schering Corporation | Ribavirin-interferon alfa combination therapy for eradicating detectable HCV-RNA in patients having chronic hepatitis C infection |
| US7141574 | Jul 18, 2002 | Nov 28, 2006 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| US7514557 | May 23, 2005 | Apr 7, 2009 | Boehringer Ingelheim International Gmbh | Process for preparing acyclic HCV protease inhibitors |
| US7582770 | Feb 18, 2005 | Sep 1, 2009 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7585845 | May 20, 2004 | Sep 8, 2009 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US7642352 | Feb 10, 2006 | Jan 5, 2010 | Boehringer Ingelheim International Gmbh | Process for preparing 2,3-disubstituted indoles |
| US20090087409 | Nov 26, 2008 | Apr 2, 2009 | Boehringer Ingelheim (Canada) Ltd. | Viral Polymerase Inhibitors |
| US20100068182 | Sep 16, 2009 | Mar 18, 2010 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US20100093792 | Sep 15, 2009 | Apr 15, 2010 | Boehringer Ingelheim International Gmbh | Crystalline forms of a potent hcv inhibitor |
* Cited by examiner
NON-PATENT CITATIONS
| Ref | ||
|---|---|---|
| 1 | BALAGOPAL GASTROENTEROLOGY vol. 139, 2010, pages 1865 – 1876 | |
| 2 | BERG ET AL. HEPATOL vol. 52, no. S1, 2010, | |
| 3 | * | DOMINIQUE LARREY ET AL: “Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin“, JOURNAL OF HEPATOLOGY, vol. 57, no. 1, 7 March 2012 (2012-03-07), pages 39-46, XP55040240, ISSN: 0168-8278, DOI: 10.1016/j.jhep.2012.02.015 |
| 4 | G. CAIRNS GENE VARIANT THAT HELPS HEPATITIS C TREATMENT MAY HINDER HIV TREATMENT, [Online] 2011, Retrieved from the Internet: <URL:http://www.bhiva.org/Ncws.aspx?NewsID=a7503829-94b9-4d2f-bd91-ld2fbaad6c8d> | |
| 5 | GE ET AL. NATURE vol. 461, 2009, pages 399 – 401 | |
| 6 | GHANY; MARC ET AL.: ‘An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases‘ HEPATOLOGY vol. 54, no. 4, 2011, pages 1433 – 44 | |
| 7 | * | LIZ HIGHLEYMAN: “AASLD: All-Oral Combination of BI 201335, BI 207127 and Ribavirin Shows Good Efficacy at 12 Weeks“, INTERNET CITATION, [Online] 1 December 2011 (2011-12-01), pages 1-3, XP002684260, Retrieved from the Internet: URL:www.hivandhepatitis.com/hepatitis-c/he patitis-c-topics/hcv-treatment/3371-aasld- all-oral-combination-of-bi-201335-bi-20712 7-and-ribavirin-shows-good-efficacy-at-12- weeks> [retrieved on 2012-09-27] |
| 8 | * | POL S ET AL: “SVR AND PHARMACOKINETICS OF THE HCV PROTEASE INHIBITOR BI201335 WITH PEGIFN/RBV IN HCV GENOTYPE-1 PATIENTS WITH COMPENSATED LIVER CIRRHOSIS AND NON-RESPONSE TO PREVIOUS PEGIFN/RBV“, JOURNAL OF HEPATOLOGY, vol. 54, no. Suppl. 1, March 2011 (2011-03), page S486, XP55038942, & 46TH ANNUAL MEETING OF THE EUROPEAN-ASSOCIATION-FOR-THE-STUDY-OF-THE- LIVER (EASL); BERLIN, GERMANY; MARCH 30 -APRIL 03, 2011 ISSN: 0168-8278 |
| 9 | S. M. BIRGE ET AL. J. PHARM. SCI. vol. 66, 1977, pages 1 – 19 | |
| 10 | * | STEFAN ZEUZEM ET AL: “Efficacy of the Protease Inhibitor BI 201335, Polymerase Inhibitor BI 207127, and Ribavirin in Patients With Chronic HCV Infection“, GASTROENTEROLOGY, ELSEVIER, PHILADELPHIA, PA, vol. 141, no. 6, 1 December 2011 (2011-12-01), pages 2047-2055, XP002664706, ISSN: 0016-5085, DOI: 10.1053/J.GASTRO.2011.08.051 |
| 11 | SULKOWSKI MS ET AL. HEPATOL vol. 50, 2009, page 2A | |
| 12 | SULKOWSKI MS ET AL. J HEPATOL vol. 52, no. 1, 2010, pages S462 – S463 | |
| 13 | WHITE PW ET AL. ANTIMICROB AGENTS CHEMOTHER vol. 54, no. 11, 2010, pages 4611 – 4618 | |
| 14 | WHO COLLABORATIVE STUDY GROUP. VOX SANG vol. 76, 1999, pages 149 – 158 | |
| 15 | * | ZEUZEM STEFAN ET AL: “STRONG ANTIVIRAL ACTIVITY AND SAFETY OF IFN-SPARING TREATMENT WITH THE PROTEASE INHIBITOR BI 201335, THE HCV POLYMERASE INHIBITOR BI 207127 AND RIBAVIRIN IN PATIENTS WITH CHRONIC HEPATITIS C“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 52, no. Suppl, 1 October 2010 (2010-10-01), pages 876A-877A, XP009154421, ISSN: 0270-9139 |
| 16 | * | ZEUZEM STEFAN ET AL: “VIROLOGIC RESPONSE TO AN INTERFERON-FREE REGIMEN OF BI201335 AND BI207127, WITH AND WITHOUT RIBAVIRIN, IN TREATMENT-NAIVE PATIENTS WITH CHRONIC GENOTYPE-1 HCV INFECTION: WEEK 12 INTERIM RESULTS OF THE SOUND-C2 STUDY“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 54, no. Suppl. 1, 1 November 2011 (2011-11-01), page 1436A, XP009163087, ISSN: 0270-9139, DOI: 10.1002/HEP.24666 [retrieved on 2011-09-30] |
…………………………………………………………
The following……

having the chemical name: (E)-3-[2-(l-{ [2-(5-Bromo-pyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carbonyl]-amino}-cyclobutyl)-3-methyl-3H-benzimidazol-5-yl]- acrylic acid, is known as a selective and potent inhibitor of the HCV NS5B RNA- dependent RNA polymerase and useful in the treatment of HCV infection. Compound (2) falls within the scope of HCV inhibitors disclosed in U.S. Patents 7,141,574 and
7,582,770, and US Application Publication 2009/0087409. Compound (2) is disclosed specifically as Compound # 3085 in U.S. Patent 7,582,770. Compound (2), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (2) include the crystalline forms, in particular the crystalline sodium salt form which is prepared as herein described.
It is known in the art that particular HCV subtypes and patient subgenotypes may respond differently to HCV therapy. HCV Genotype la is traditionally more difficult to treat and are less responsive to antiviral therapy than Genotype lb. See, e.g., Ghany, Marc et al. “An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases”, Case No.: 09-0592-PCT
Hepatology, 54(4): 1433-44 (2011)). In addition, and particularly with interferon-based therapy, specific single nucleotide polymorphisms (SNPs) located on the long arm of chromosome 19 within the gene cluster of IL-28B (Interleukin (IL) 28B, (also called lambda interferon), of the patient undergoing therapy can directly effect the
responsiveness of that patient to the antiviral therapy. In particular, patients having a non- CC genotype of SNP rsl2979860 or a non-TT genotype of rs 8099917 are traditionally more difficult to treat and are less responsive in terms of a sustained virological response (SVR) than patients having the CC or TT genotype.. The SNP that was most strongly associated with SVR in the genome-wide analysis was rs 12979860 followed by rs 8099917. See, e.g., Ge et al., Nature, 461 :399-401 (2009) and Balagopal,
Gastroenterology, 139: 1865-1876 (2010). See G. Cairns, “Gene variant that helps hepatitis C treatment may hinder HIV treatment”, 2011, at:
http://www.bhiva.org^ Thus, there is a need in the art for therapies that are effective against even the more difficult-to-treat patient subpopulations, particularly those exhibiting HCV subtype la and the non-CC IL28B subgenotype, as well as those exhibiting compensated liver disease.
Examples
I. Methods for Preparing Compound (1)
Methods for preparing amorphous Compound (1) and a general description of
pharmaceutically acceptable salt forms can be found in US Patents 6,323,180, 7,514,557 and 7,585,845. Methods for preparing additional forms of Compound (1), in particular the crystalline sodium salt form, can be found in U.S. Patent Application Publication No. 2010/0093792.
II. Formulations of Compound (1) Case No. : 09-0592-PCT
One example of a pharmaceutical formulation of Compound (1) include an oral solution formulation as disclosed in WO 2010/059667. Additional examples include capsules containing a lipid-based liquid formulation, as disclosed in WO 201 1/005646. III. Methods for Preparing Compound (2)
Methods for preparing amorphous Compound (2) can be found in U.S. Patents 7, 141 ,574 and 7,582,770, and US Application Publication 2009/0087409.
The following Example provides the method for preparing an additional form of
Compound (2), the sodium salt form, that may be used in the present invention.
Example 1 – Preparation of Compound (2) Sodium Salt
Step 1. Synthesis of Isopropyl 3-Cyclopentyl-l-methyl-lH-indole-6-carboxylate

Because of the instability of brominated product, methyl 3 -cyclopentyl- 1 -methyl- 1Η- indole-6-carboxylate needed to be converted into the more stable isopropyl 3-cyclopentyl- l-methyl- lH-indole-6-carboxylate via a simple and high yielding operation. The conversion worked the best with stoichiometric amounts of solid lithium isopropoxide. Use of 0.1 eq lithium isopropoxide led to longer reaction times and as a result to more hydrolysis by-product, while lithium isopropoxide solution in THF caused a problematic isolation and required distillation of THF.
Procedure: Case No.: 09-0592-PCT
The mixture of methyl 3 -cyclopentyl- 1 -methyl- lH-indole-6-carboxylate (50.0 g, 0.194 mol) and lithium isopropoxide (16.2 g, 95%, 0.233 mol) in 2-propanol was stirred at 65+5 °C for at least 30 min for complete trans-esterification. The batch was cooled to 40+5 °C and water (600 g) was added at a rate to maintain the batch temperature at 40+5°C. After addition, the mixture was cooled to 20-25 °C over 2+0.5 h and held at 20-25 °C for at least 1 h. The batch was filtered and rinsed with 28 wt% 2-propanol in water (186 g), and water (500 g). The wet cake was dried in vacuo (< 200 Torr) at 40-45 °C until the water content was < 0.5% to give isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (52.7 g, 95% yield) in 99.2 A% (240 nm).
The starting material methyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate can be prepared as described in Example 12 of U.S. Patent 7,141,574, and in Example 12 of U.S. Patent 7,642,352, both herein incorporated by reference.
Step 2. Synthesis of Isopropyl 2-Bromo-3-cyclopentyl-l-methyl-lH-indole-6- carboxylate

This process identified the optimal conditions for the synthesis of 2-bromo-3-cyclopentyl- l-methyl-lH-indole-6-carboxylate via bromination of the corresponding 3 -cyclopentyl- 1- methyl-lH-indole-6-carboxylate with bromine. It’s very important to control the reaction temperature and to quench the reaction mixture with a mixture of aqueous sodium thiosulfate and 4-methylmorpholine to minimize the formation of the dibromo- and 2- indolone impurities. Further neutralization of the crude product with NaOH in isopropanol greatly increases the stability of the isolated product. Case No.: 09-0592-PCT
Procedure:
The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (50.0 g, 0.175 mol) and acetonitrile (393 g) was cooled to -6+3 °C. Bromine (33.6 g, 0.210 mol) was added while the batch was maintained at -6+3°C. The resulting slurry was stirred at – 6+3°C for at least 30 min. When HPLC showed > 94 % conversion (the HPLC sample must be quenched immediately with aqueous 4-methylmorpholine/sodium thiosulfate solution), the mixture was quenched with a solution of sodium thiosulfate (15.3 g) and 28.4 g 4-methylmorpholine in water (440 g) while the temperature was maintained at -5+5 °C. After it was stirred at 0+5 °C for at least 2 h, the batch was filtered and rinsed with 85 wt methanol/water solution (415 g), followed by water (500 g), and dried until water content is < 30%. The wet cake was suspended in 2-propanol (675 g), and heated to 75+5 °C. The resulting hazy solution was treated with 1.0 M aqueous sodium hydroxide solution (9.1 g) and then with 135.0 g water at a rate to maintain the batch at 75+5°C. The suspension was stirred at 75+5°C for at least 30 min, cooled to 15+2 °C over 30-40 min, and held at 15+2 °C for at least 1 h. The batch was filtered, rinsed with 75 wt% 2-propanol/water solution (161 g), and dried in vacuo (<200 Torr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3-cyclopentyl-l -methyl- lH-indole-6-carboxylate as a solid (55.6 g, 87 % yield ) in 99.5 A% (240 nm) and 97.9 Wt%. Alternative Procedure:
The mixture of isopropyl 3-cyclopentyl-l-methyl-lH-indole-6-carboxylate (84 g, 0.294 mol) and isopropyl acetate (1074 g) was cooled to between -10-0 °C. Bromine (50 g, 0.312 mol) was added while the batch was maintained at -10 – 0 °C. The resulting slurry was stirred at the same temperature for additional 30 min and quenched with a pre-cooled solution of sodium thiosulfate pentahydrate (13 g) and triethylamine (64.5 g) in water (240 g) while the temperature was maintained at 0-10 °C. The mixture was heated to 40 – 50 °C and charged with methanol (664 g). After it was stirred at the same temperature for at least 0.5 h, the batch was cooled to 0 – 10 °C and stirred for another 1 hr. The precipitate was filtered, rinsed with 56 wt% methanol/water solution (322 g), and dried in vacuo (<200 Case No. : 09-0592-PCT
Torr) at 50-60 °C until the water content was < 0.4% to give isopropyl 2-bromo-3- cyclopentyl-l -methyl- lH-indole-6-carboxylate as a beige solid (90-95 g, 80-85 % yield ).
Step 3a,b. Preparation of compound I by one-pot Pd-catalyzed borylation- Suzuki coupling reaction

To a clean and dry reactor containing 20.04 g of isopropyl 2-bromo-3-cyclopentyl- l- methyl- lH-indole-6-carboxylate, 1.06 g of Pd(TFP)2Cl2(3 mol%) and 0.76 g of tri(2- furyl)phosphine (6 mol%) was charged 8.35 g of triethylamine (1.5 equivalent), 39.38 g of CH3CN at 23+10 °C under nitrogen or argon and started agitation for 10 min. 9.24 g of 4,4,5, 5-tetramethyl-l ,3,2-dioxaborolane was charged into the reactor. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for 6h until the reaction completed. The batch was cooled to 30+5 °C and quenched with a mixture of 0.99 g of water in 7.86 g of
CH3CN. 17.24 g of 5-bromo-2-iodopyrimidine and 166.7 g of degassed aqueous potassium phosphate solution (pre-prepared from 46.70 g of K3PO4 and 120 g of H20) was charged subsquently under argon or nitrogen. The content was heated to reflux (ca. 76-77 °C) for 2 h until the reaction completed. 4.5 g of 1-methylimidazole was charged into the reactor at 70 °C. The batch was cooled to 20+3 °C over 0.5h and hold at 20+3 °C for at least lh. The solid was collected by filtration. The wet cake was first rinsed with 62.8 g of 2-propanol, Case No. : 09-0592-PCT
followed by 200 g of H20. The solid was dried under vacuum at the temperature below 50 °C.
Into a dry and clean reactor was charged dried I, 10 wt Norit SX Ultra and 5 V of THF. The content was heated at 60+5 °C for at least 1 h. After the content was cooled to 35+5 °C, the carbon was filtered off and rinsed with 3 V of THF. The filtrate was charged into a clean reactor containing 1-methylimidazole (10 wt % relative to I). After removal of 5 V of THF by distillation, the content was then cooled to 31 ±2 °C. After the agitation rate was adjusted to over 120 rpm, 2.5 V of water was charged over a period of at least 40 minutes while maintaining the content temperature at 31 + 2 °C. After the content was agitated at 31 + 2 °C for additional 20 min, 9.5 V of water was charged into the reactor over a period of at least 30 minutes at 31 + 2 °C. The batch was then cooled to about 25 + 3 °C and stirred for additional 30 minutes. The solid was collected and rinsed with 3 V of water. The wet product I was dried under vacuum at the temperature below 50 °C (19.5 g, 95 wt , 76% yield).
Alternative Procedure:
To a clean and dry reactor containing 40 g of isopropyl 2-bromo-3-cyclopentyl- l-methyl- lH-indole-6-carboxylate (0.1 10 mol), 0.74 g of Pd(OAc)2 (3.30 mmol, 3 mol% equiv.) and 3.2 g of tri(2-furyl)phosphine (13.78 mmol, 12.5 mol% equiv.) was charged 16.8 g of triethylamine (1.5 equivalent), 100 mL of acetonitrile at 25 °C under nitrogen or argon. 20.8 g of 4,4,5, 5-tetramethyl- l ,3,2-dioxaborolane was charged into the reactor within 30 min. The mixture was heated to reflux (ca. 81 -83 °C) and stirred for over 5 hrs until the reaction completed. The batch was cooled to 20 °C and quenched with a mixture of 2.7 g of water in 50 mL of CH3CN. The batch was warmed to 30 °C, stirred for 1 hr and transferred to a second reactor containing 34.4 g of 5-bromo-2-iodopyrimidine in 100 mL of acetonitrile. The reactor was rinsed with 90 mL of acetonitrile. To the second reactor was charged with degassed aqueous potassium phosphate solution (pre-prepared from 93.2 g of K3PO4 and 100 g of H20) under argon or nitrogen. The content was heated to reflux (ca. 80 °C) for over 3 h until the reaction completed. 9.2 g of 1 -methylimidazole was charged into the reactor at 70 °C and the mixture was stirred for at least 10 min. The aqueous phase was removed after phase separation. 257 g of isopropanol was charged at 70 Case No.: 09-0592-PCT
°C. The batch was cooled slowly to 0 °C and hold for at least 1 h. The solid was collected by filtration. The wet cake was rinsed twice with 2-propanol (2 x 164 g) and dried under vacuum at the temperature below 50 °C to give I as a yellow to brown solid (26 g, 75% yield).
Step 4. Hydrolysis of I to II

I (20 g) and l-methyl-2-pyrrolidinone (NMP) (113 g) were charged into a clean reactor under nitrogen. After the batch was heated to 50-53 °C with agitation, premixed aq. NaOH (5.4 g of 50% aq. NaOH and 14.3 g of water) was introduced into the reactor. The resulting mixture was stirred at 50-53 °C for about 10 hrs until the reaction completed. A premixed aq. HOAc (60 g of water and 9.0 g of HOAc) was added over 0.5 h at 45 ±5 °C to reach pH 5.5- 7.5. The batch was cooled to 20+5 °C and then kept for at least 1.0 h. The solid product was collected and rinsed with 80 g of NMP/water (1 :3 volume ratio) and then 60 g of water. The product was dried under vacuum at the temperature below 50 °C to give II as a pale yellow powder (19 -20 g, purity > 99.0 A% and 88.4 wt%, containing 5.4 wt% NMP). The yield is about 93-98%.
Notes: The original procedure used for the hydrolysis of I was carried out with aq. NaOH (2.5 eq) in MeOH/THF at 60 °C. Although it has been applied to the preparation of II on several hundred grams scale, one disadvantage of this method is the formation of 5-MeO pyrimidine during hydrolysis (ca. 0.4 A%), which is extremely difficult to remove in the subsequent steps. In addition, careful control has to be exerted during crystallization. Case No.: 09-0592-PCT
Otherwise, a thick slurry might form during acidification with HO Ac. The use of NMP as solvent could overcome all aforementioned issues and give the product with desired purity.
Alternative Process
To a reactor was charged I (71 g), isopropanol (332 g), aqueous NaOH (22 g, 45 wt ) and water (140 g) at ambient temperature. The mixture was heated to reflux (80 °C) and stirred for at least 3 hrs until the reaction completed. The batch was cooled to 70 °C and charged a suspension of charcoal (3.7 g) in isopropanol (31 g). The mixture was stirred at the same temperature for over 10 min and filtered. The residue was rinsed with isopropanol (154 g). Water (40 g) was charged to the filtrate at 70 – 80 °C, followed by slow addition of 36% HC1 solution (20 g) to reach pH 5- 6. The batch was stirred for over 30 min at 70 °C, then cooled to 20 °C over 1 hr and kept for at least 1.0 h. The solid product was collected and rinsed with 407 g of isopropanol/water (229 g IPA, 178 g H20). The product was dried under vacuum at 80 °C for over 5 hrs to give II as a white powder (61 g, 95% yield).
Notes on Steps 5 to 8 below:
A concise and scalable 4-step process for the preparation of the benzimidazole
intermediate V was developed. The first step was the preparation of 4-chloro-2-(methyl)- aminonitrobenzene starting from 2,4-dichloronitrobenzene using aqueous methyl amine in DMSO at 65 °C. Then, a ligandless Heck reaction with n-butyl acrylate in the presence of Pd(OAc)2, ‘PrzNEt, LiCl, and DMAc at 110 °C was discovered.
Step 5: SNAr reaction of (5-chloro-2-nitrophenyl)-methylamine

To a solution of (5-chloro-2-nitrophenyl)-methylamine (40 g, 208.3 mmol, 1 equiv) in DMSO (160 mL) was added 40% MeNH2solution in water (100 mL, 1145. 6 mmol, 5.5 eq) slowly keeping the temperature below 35 °C. The reaction was stirred at r.t. until the Case No.: 09-0592-PCT
complete consumption of the starting material (>10 h). Water (400 mL) was added to the resulting orange slurry and stirred at r.t. for additional 2 h. The solid was filtered, rinsed with water (200 mL) and dried under reduced pressure at 40 °C. (5-chloro-2-nitrophenyl)- methylamine (36.2 g, 93% yield, 94 A% purity) was isolated as a solid.
Step 6: Heck Reaction of (5-chloro-2-nitrophenyl)-methylamine

DMAc (5 vol), 1 10 °C, 7-22 h To a mixture of 4-chloro-2-methylaminonitrobenzene (50.0 g, 268.0 mmol, 1.0 eq),
Pd(OAc)2 (0.30 g, 1.3 mmol, 0.005 eq) and LiCI (11.4 g 268.0 mmol, 1.0 eq) in DMAc (250 mL) was added ‘Pr2NEt (56 mL, 321.5 mmol, 1.2 eq) followed by n-butyl acrylate (40 mL, 281.4 mmol, 1.05 eq) under nitrogen. The reaction mixture was stirred at 110 °C for 12 h, then cooled to 50 °C. 1 -methylimidazole (10.6 mL, 134.0 mmol, 0.5 eq) was added and the mixture was stirred for 30 min before filtering and adding water (250 mL). The resulting mixture was cooled to r.t. over 1 h. The resulting solid was filtered and washed with water and dried to yield n-butyl 3-methylamino-4-nitrocinnamate (71.8 g, 96 %, 99.2 A% purity).
Step 7: Reduction of n-butyl (3-methylamino-4-nitro)-cinnamate

III Case No.: 09-0592-PCT
To a reactor was charged n-butyl 3-methylamino-4-nitrocinnamate (70.0 g, mmol, 1.0 eq) , Raney Ni (4.9 g, ~20wt% H20), charcoal “Norit SX Ultra” (3.5 g), toluene (476 mL) and MeOH (224 mL). The reactor was charged with hydrogen (4 bar) and the mixture was stirred at 20- 25 °C for about 2 hrs until the reaction was completed. The reaction mixture was filtered and rinsed the filter residue with toluene (70 mL). To the combined filtrates were added “Norit SX Ultra” charcoal (3.5 g). The mixture was stirred at 50 °C for 1.0 hr and filtered. The filtrate was concentrated under reduced pressure to remove solvents to 50% of the original volume. The remained content was heated to 70 °C and charged slowly methyl cyclohexane (335 mL) at the same temperature. The mixture was cooled to about 30 – 40 °C and seeded with III seed crystals, then slowly cooled the suspension to— 10 °C. The solid was filtered and rinsed with methyl cyclohexane in three portions (3 x 46 mL). The wet cake was dried in vacuo at 40 °C to give III (53.3 g, 215 mmol, 86%).
Step 8: Preparation of benzimidazole V
DCC

To reactor-1 was charged III (35 g, 140.95 mmol) in toluene (140 g). The mixture was heated to 50 °C to obtain a clear solution. To a second reactor was charged IV (36.4 g, 169.10 mmol) and toluene (300 g), followed by addition of a solution of dicyclohexyl carbodimide (11.6 g, in 50% toluene, 28.11 mmol) at 0 – 10 °C. The mixture was stirred at the same temperature for 15 min, then charged parallelly with the content of reactor-1 and the solution of dicyclohexyl carbodimide (52.4 g, in 50% toluene, 126.98 mmol) within 1 hr while maintaining the batch temperature at 0 – 10 °C. The mixture was agitated at the same temperature for 3 hrs, and warmed to 25 °C for another 1 hr. Once III was consumed, toluene (-300 mL) was distilled off under reduced pressure at 70 – 80 °C. n-Butanol (200 g) was added, followed by 3 M HCI solution in n-butanol (188 g) while maintaining the Case No.: 09-0592-PCT
temperature at 70 – 80 °C (Gas evolution, product precipitates). After stirring for over 30 min. at 70 – 80 °C, the mixture was cooled to 20 – 30 °C over 1 hr. The precipitate was filtered and washed with acetone (172 g) and toluene (88 g). The wet cake was dried in vacuo at -60 °C to give V toluene solvate as off white solid (60 – 72 g, 85 – 95% yield). Compound V could be used directly for the next step or basified prior to next step to obtain the free base compound VI used in the next step.
Step 9. Synthesis of (E)-Butyl 3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l- hydroxy-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate VII

5) MeOH/H20
Notes:
The conversion of the acid into acid chloride was achieved using inexpensive thionyl chloride in the presence of catalytic amount of NMP or DMF. An efficient crystallization was developed for the isolation of the desired product in high yield and purity.
Procedure (using free base VI):
To the suspension of 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (see Step 4) (33.36 g, 90.0 wt %, containing -0.2 equiv of NMP from previous step,75.00 mmol) in THF (133.4 g) was added thionyl chloride (10.71 g). The mixture was stirred at 25+5 °C for at least 1 h. After the conversion was completed as determined by HPLC (as derivative of diethylamine), the mixture was cooled to 10+5 °C and N,N-diisopropylethylamine (378.77 g, 300 mmol) below 25 °C. A solution of (E)-butyl 3-(2-(l-aminocyclobutyl)-l-methyl-lH-benzo[if|imidazol-6-yl)acrylate VI (25.86 g, 97.8 Wt%, 77.25 mmol) dissolved in THF (106.7 g) was added at a rate to maintain the Case No.: 09-0592-PCT
temperature of the content < 25 °C. The mixture was stirred at 25+5 °C for at least 30 min for completion of the amide formation. The mixture was distilled at normal pressure to remove ca. 197 mL (171.5 g) of volatiles (Note: the distillation can also be done under reduced pressure). The batch was adjusted to 40+5 °C, and MeOH (118.6 g) was added. Water (15.0 g) was added and the mixture was stirred at 40+5 °C until crystallization occurred (typically in 30 min), and held for another 1 h. Water (90 g) was charged at 40+5 °C over 1 h, and the batch was cooled to 25+5 °C in 0.5 h, and held for at least 1 h. The solid was filtered, rinsed with a mixture of MeOH (39.5 g), water (100 g), and dried in vacuo (< 200 Torr) at 50+5 °C to give (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3- cyclopentyl- 1 -methyl- lH-indole-6-carboxamido)cyclobutyl)- 1 -methyl- 1H- benzo[if|imidazol-6-yl)acrylate VII (51.82 g, 96.6 % yield) with a HPLC purity of 98.0 A% (240 nm) and 99.0 Wt%.
Alternative Process (using compound V from Step 8)
To reactor 1 was charged 2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6- carboxylic acid II (33.6 g), toluene (214 g) and N-methylpyrrolidone (1.37 g). The mixture was heated to 40 °C, then added a solution of thionyl chloride (13 g) in toluene (17 g). The mixture was stirred at 40 °C for at least 0.5 h and cooled to 30 °C. To a second reactor was charged with compound V (the bis-HCl salt toluene solvate from Step 8) (39.4 g), toluene (206 g) and N,N-diisopropylethylamine (70.8 g) at 25 °C. The content of reactor 1 was transferred to reactor 2 at 30 °C and rinsed with toluene (50 g). The mixture was stirred at 30 °C for another 0.5 h, then charged with isopropanol (84 g) and water (108 g) while maintained the temperature at 25 °C. After stirring for 10 min, remove the aqueous phase after phase cutting. To the organic phase was charged isopropanol (43 g), water (54 g) and stirred for 10 min. The aqueous phase was removed after phase cutting. The mixture was distilled under reduced pressure to remove ca.250 mL of volatiles, followed by addition of methyl tert-butyl ether (MTBE, 238 g). The batch was stirred at 65 °C for over 1 hr, then cooled to 20 C over 1 hr and held for another 1 hr at the same temperature. The solid was filtered, rinsed with MTBE (95 g), and dried in vacuo at 80 °C to give (E)-butyl 3-(2-(l-(2- Case No.: 09-0592-PCT
(5-bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl-lH-indole-6-carboxamido)cyclobutyl) methyl- lH-benzo[if|imidazol-6-yl)acrylate VII as a beige solid (50 g, 90 % yield).
Step 10. Synthesis of (E)-3-(2-(l-(2-(5-Bromopyrimidin-2-yl)-3-cyclopentyl-l-methyl- lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[</]imidazol-6-yl)acrylic acid (Compound (1))

Notes:
In this process, hydrolysis of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl- l-methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[d]imidazol-6- yl)acrylate was carried out in mixture of THF/MeOH and aq NaOH. Controlled acidification of the corresponding sodium salt with acetic acid is very critical to obtain easy-filtering crystalline product in high yield and purity.
Procedure:
To the suspension of (E)-butyl 3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6- yl)acrylate VII (489.0 g, 91.9 Wt%, 633.3 mmol) in THF (1298 g) and MeOH (387 g) was added 50% NaOH (82.7 g, 949.9 mmol), followed by rinse with water (978 g). The mixture was stirred between 65-68 C for about 1 h for complete hydrolysis. The resulting solution was cooled to 35 C, and filtered through an in-line filter (0.5 micron), and rinsed with a pre-mixed solution of water (978 g) and MeOH (387 g). The solution was heated to Case No.: 09-0592-PCT
60 +4 C, and acetic acid (41.4 g, 689 mmol) was added over 1 h while the mixture was well agitated. The resulting suspension was stirred at 60 ±4 C for 0.5 h. Another portion of acetic acid (41.4 g, 689 mmol) was charged in 0.5 h, and batch was stirred at 60 ±4 C for additional 0.5 h. The batch was cooled to 26 ±4 C over 1 h and held for 1 h. The batch was filtered, rinsed with a premixed solution of water (1956 g) and MeOH (773.6 g), dried at 50 C under vacuum to give (E)-3-(2-(l-(2-(5-bromopyrimidin-2-yl)-3-cyclopentyl-l- methyl-lH-indole-6-carboxamido)cyclobutyl)-l-methyl-lH-benzo[(i]imidazol-6-yl)acrylic acid (1) (419.0 g, 95 % yield) with > 99.0 A% (240 nm) and 94.1 Wt% by HPLC. Step 11. Formation of Compound (1) Sodium Salt (Type A)

To a reactor were charged Compound (1) (150 g, mmol), THF (492 mL), H20 (51 mL) and 45% aqueous NaOH solution (20.4 g, mmol). The mixture was stirred for >1 hr at -25 °C to form a clear solution (pH = 9 -11). To the solution was charged a suspension of Charcoal (1.5 g) and H20 (27 mL). The mixture was stirred at -35 °C for >30 min and filtered. The filter was rinsed with THF (108 mL) and H20 (21 mL). The filtrate was heated to 50 °C and charged with methyl ethylketone (MEK) (300 mL). The mixture was seeded with Compound (1) sodium salt MEK solvate (Type A) seeds (0.5 g) and stirred for another 1 hr at 50 °C. To the mixture was charged additional MEK (600 mL). The resultant mixture was stirred for another 1 hr at 50 °C and then cooled to 25 °C. The precipitate was filtered and rinsed with MEK twice (2 x 300 mL). The wet cake was dried in vacuum at 80 °C to give Compound (1) sodium salt (Type A) (145.6 g, 94%). Case No.: 09-0592-PCT
The Compound (1) sodium salt (Type A) MEK solvate seeds used in the above process step can be manufactured by the above process except without using seeds and without drying of the solvate.
No comments:
Post a Comment