
TOFOGLIFLOZIN
CSG-452
R-7201
RG-7201
R-7201
RG-7201
CAS..1201913-82-7
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol hydrate (1:1)
THERAPEUTIC CLAIM Treatment of diabetes mellitus
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3′R,4′S,5′S,6′R)-
2. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol monohydrate
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3′R,4′S,5′S,6′R)-
2. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2'-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3′R,4′S,5′S,6′R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2'-[2H]pyran]-3′,4′,5′-triol monohydrate
MOLECULAR WEIGHT 404.5
SPONSOR Chugai Pharmaceuticals
CODE DESIGNATION CSG452
CODE DESIGNATION CSG452
Tofogliflozin (USAN, codenamed CSG452) is an experimental drug for the treatment ofdiabetes mellitus and is being developed by Chugai Pharma in collaboration with Kowa andSanofi.[1] It is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. As of September 2012, the drug is in Phase III clinical trials.[2][3]
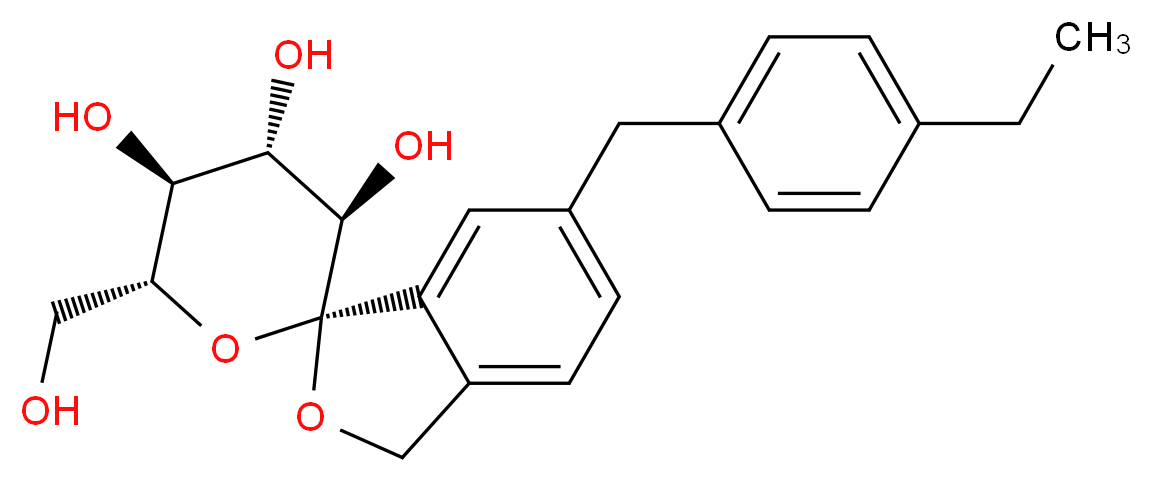
Tofogliflozin is the name of the monohydrate, which is the form used as a drug. The active moiety or anhydrous form (ChemSpider ID: 28530778, CHEMBL2110731) has the chemical formula C22H26O6 and a molecular mass of 386.44 g/mol.[4]
SGLT2 inhibitors inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. Glucopyranosyl-substituted benzene derivative are described in the prior art as SGLT2 inhibitors, for example in
WO 01/27128, WO 03/099836, WO 2005/092877, WO 2006/034489,
WO 2006/064033, WO 2006/117359, WO 2006/117360,
WO 2007/025943, WO 2007/028814, WO 2007/031548,
WO 2007/093610, WO 2007/128749, WO 2008/049923, WO 2008/055870, WO 2008/055940.
The glucopyranosyl-substituted benzene derivatives are proposed as inducers of urinary sugar excretion and as medicaments in the treatment of diabetes.
The term “canagliflozin” as employed herein refers to canagliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2005/012326 and WO 2009/035969 for example. Preferred hydrates, solvates and crystalline forms are described in the patent applications WO 2008/069327 for example.
atigliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/007517 for example.
ipragliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
tofogliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2007/140191 and WO 2008/013280 for example.
remogliflozin and prodrugs of remogliflozin, in particular remogliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods of its synthesis are described in the patent applications EP 1213296 and EP 1354888 for example.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
luseoghflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

ertugliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

and is described for example in WO 2010/023594.
The compound of the formula

is described for example in WO 2008/042688 or WO 2009/014970.
Dapagliflozin

Remogliflozin and Remogliflozin Etabonate

Sergliflozin and Sergliflozin Etabonate

1-Chloro-4-(β-D-glucopyranos-1-yl)-2-(4-ethyl-benzyl)-benzene

(1S)-1,5-anhydro-1-[5-(azulen-2-ylmethyl)-2-hydroxyphenyl]-D-glucitol

(1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-D-glucitol

Thiophen Derivatives of the Formula (7-1)

1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene

Spiroketal Derivatives of the Formula (9-1)

The compound is described for example in WO 03/099836. Crystalline forms are described for example in WO 2008/002824.
The compound is described for example in EP 1354888 A1.
The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
The compound is described in WO 2006/034489.
The compound (4-(Azulen-2-ylmethyl)-2-(β-D-glucopyranos-1-yl)-1-hydroxy-benzene) is described in WO 2004/013118 and WO 2006/006496. The crystalline choline salt thereof is described in WO 2007/007628.
The compound is described in WO 2004/080990 and WO 2005/012326. A cocrystal with L-proline is described in WO 2007/114475.
wherein R denotes methoxy or trifluoromethoxy. Such compounds and their method of production are described in WO 2004/007517, DE 102004063099 and WO 2006/072334.
The compound is described in WO 2005/012326. A crystalline hemihydrate is described in WO 2008/069327.
wherein R denotes methoxy, trifluoromethoxy, ethoxy, ethyl, isopropyl or tert. butyl. Such compounds are described in WO 2007/140191 and WO 2008/013280.
- Chugai Pharmaceutical: Development Pipeline
- Nagata, T.; Fukazawa, M.; Honda, K.; Yata, T.; Kawai, M.; Yamane, M.; Murao, N.; Yamaguchi, K.; Kato, M.; Mitsui, T.; Suzuki, Y.; Ikeda, S.; Kawabe, Y. (2012). “Selective SGLT2 inhibition by tofogliflozin reduces renal glucose reabsorption under hyperglycemic but not under hypo- or euglycemic conditions in rats”. AJP: Endocrinology and Metabolism 304 (4): E414–E423. doi:10.1152/ajpendo.00545.2012.PMID 23249697. edit
- Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. (2012). “Discovery of Tofogliflozin, a NovelC-Arylglucoside with anO-Spiroketal Ring System, as a Highly Selective Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes”. Journal of Medicinal Chemistry 55 (17): 7828–7840.doi:10.1021/jm300884k. PMID 22889351. edit
- Statement on a nonproprietary name adopted by the USAN council: Tofogliflozin.
PATENTS
papers
Chinese Chemical Letters, 2013 , vol. 24, 2 pg. 131 – 133
Journal of Medicinal Chemistry, 2012 , vol. 55, 17 pg. 7828 – 7840
nmr
WO 2011074675
WO 2011074675

1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
MS (ESI +): 387 [M +1] +.
second set
J. Med. Chem., 2012, 55 (17), pp 7828–7840
DOI: 10.1021/jm300884k
1H NMR (400 MHz, CD3OD) δ: 1.20 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42–3.47 (1H, m), 3.63–3.67 (1H, m), 3.75–3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.3 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07–7.14 (4H, m), 7.17–7.23 (3H, m).
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2.
MS (ESI): 387 [M + H]+. HRMS (ESI), m/z calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801.
……………………………
prepn
[Example 1] (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro- -6′-(hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran] -3 ‘, 4′, one of the preparation step [compound of formula (IX)] 5′-triol Preparation of methanol (2 – hydroxymethyl-phenyl – bromo-4)

To the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.1kg) in – bromoterephthalic was added at below 30 ℃ solution (7.5kg, 30.6mol) of the acid, and the mixture was stirred for 1 hour at 25 ℃. Then cooled to 19 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). In addition to methanol (15.0kg) in the mixture was kept for a while.
Again, to the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.0kg) in – was added at below 30 ℃ solution (7.5kg, 30.6mol) of bromo terephthalic acid, and the reaction was carried out for 1 hour at 25 ℃. Then cooled to 18 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). After addition of methanol (15.0kg) in the mixture is combined with the reaction mixture obtained in the previous reaction, and then the solvent was distilled off under reduced pressure. After addition of methanol (36kg) residue was obtained, and the solvent was evaporated under reduced pressure. Furthermore, (54 ℃ dissolved upon confirmation) which was dissolved by warming was added to methanol (36kg) to the residue. After cooling to room temperature the solution was stirred for 30 minutes added water (60kg). After addition of water (165kg) In addition to this mixture was cooled to 0 ℃, and the mixture was stirred for one hour. Centrifuge the obtained crystals were washed twice with water (45kg), and dried for 2 hours under reduced pressure to give (11.8kg, 54.4mol, 89% yield) of the title compound.
1 H-NMR (DMSO-d 6) δ: 4.49 (4H, t, J = 5.8Hz), 5.27 (1H, t, J = 5.8Hz), 5.38 (1H, t, J = 5.8Hz), 7.31 (1H, d, J = 7.5Hz), 7.47 (1H, d, J = 7.5Hz), 7.50 (1H, s).
Preparation of benzene (ethoxy methyl – methyl – - methoxy-1 1) – bromo-1 ,4 – 2:2 process bis

(- Bromo-4 – 2-hydroxyethyl methyl phenyl) in tetrahydrofuran (57kg) in the solution (8.0kg, 36.9mol) of methanol, I added (185.12g, 0.74mol) of pyridinium p-toluenesulfonate. After cooling to -15 ℃ below the mixture, 2 – was added at -15 ℃ or less (7.70kg, 106.8mol) methoxy propene, and the mixture was stirred 1 h at -15 ~ 0 ℃. Was added aqueous potassium carbonate (25 wt%, 40kg) and the reaction mixture was warmed to room temperature and separate the organic layer was added toluene (35kg). After washing with water (40kg) The organic layer was evaporated under reduced pressure. Was dissolved in toluene (28kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 1.42 (6H, s), 1.45 (6H, s), 3.24 (3H, s), 3.25 (3H, s), 4.45 ( 2H, s), 4.53 (2H, s), 7.28 (1H, dd, J = 1.5,8.0 Hz), 7.50 (1H, d, J = 8.0Hz), 7. 54 (1H, d, J = 1.5Hz).
MS (ESI +): 362 [M +2] +.
MS (ESI +): 362 [M +2] +.
Preparation of on – (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy)-6 – trimethylsilyloxy methyl – tetrahydropyran-2: Step 3

Glucono -1,5 – - D-(+) in tetrahydrofuran (70kg) in the solution (35.8kg, 353.9mol) of N-methylmorpholine (7.88kg, 44.23mol) and lactone, chlorotrimethylsilane ( was added at 40 ℃ less 29.1kg, and 267.9mol), and the mixture was stirred for 2 hours at 30 ~ 40 ℃ resulting mixture. Was cooled to 0 ℃ the reaction mixture was added toluene (34kg) water (39kg), and the organic layer was separated. Twice sodium dihydrogen phosphate aqueous solution (5 wt%, 39.56kg) in, washed once with water (39kg) the organic layer the solvent was evaporated under reduced pressure. Was dissolved in toluene (34.6kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74- 3.83 (3H, m), 3.90 (1H, t, J = 8.0Hz), 3.99 (1H, d, J = 8.0Hz), 4.17 (1H, dt, J = 2 .5,8.0 Hz).
Step 4: (1S, 3′R, 4′S, 5′S, 6′R) -3 ‘, 4′, 5 ‘, 6′-tetrahydro -6,6′ – bis (hydroxymethyl) – spiro [ (3H), 2'-[2H] pyran] -3 ‘, 4′, 5′-Preparation of triol isobenzofuran-1

(Methyl – - – methoxy 1-ethoxy-methyl) – bromo-1 ,4 – 2 prepared in step 2 bis cooled to below -10 ℃ toluene solution of benzene, hexane solution to (15 wt% n-butyl lithium , was added at below 0 ℃ 18.2kg, and 42.61mol), and the mixture was stirred 1.5 h at 5 ℃ resulting mixture. (10.5kg, 40.7mol), was added tetrahydrofuran (33.4kg) then magnesium bromide diethyl ether complex in the mixture, and the mixture was stirred for 1 hour at 25 ℃. Was added at below -10 ℃ toluene solution of the on – tris (trimethylsilyloxy) -6 – - 3,4,5 cooled to -15 ℃ below the mixture prepared in step 3 trimethylsilyloxy methyl – tetrahydropyran-2 was. After stirring 0.5 h at -15 ℃ or less, poured into 20% aqueous ammonium chloride solution to (80kg) of this solution, and the organic layer was separated. After washing with water (80kg) and the organic layer obtained, and the solvent was evaporated under reduced pressure. I was dissolved in methanol (43kg) residue was obtained. Was stirred for 1 hour at 20 ℃ was added (1.4kg, 7.4mol) and p-toluenesulfonic acid monohydrate in the mixture. Thereafter, it was stirred for another hour and cooled to 0 ℃, centrifuged crystals obtained was washed with methanol (25kg), and dried for 8 hours at reduced pressure under 40 ℃, (5.47kg, yield the title compound I got 50%) rate.
1 H-NMR (DMSO-d 6) δ :3.20-3 .25 (1H, m) ,3.41-3 .45 (1H, m) ,3.51-3 .62 (4H, m) , 4.39 (1H, t, J = 6.0Hz) ,4.52-4 .54 (3H, m), 4.86 (1H, d, J = 4.5Hz), 4.93 (1H, d, J = 5.5Hz), 4.99 (1H, d, J = 12.5Hz), 5.03 (1H, d, J = 12.5Hz), 5.23 (1H, t, J = 5 .8 Hz) ,7.24-7 .25 (2H, m), 7.29 (1H, dd, J = 1.5,8.0 Hz).
Step 5: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(methoxycarbonyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3′ , 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2'-[2H] pyran isobenzofuran] spiro

(1S, 3′R, 4′S, 5′S, 6′R) – tetrahydro -6,6 ‘- bis (hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran ] -3 ‘, 4′, 5′-triol 4 (5.3kg, 17.8mol) and – dissolved in acetonitrile (35kg) (13.7kg, 112.1mol) a chloroformate, in the solution of dimethylaminopyridine I was added at 12 ℃ or less (10.01kg, 105.9mol) methyl. Heated to 20 ℃, After stirring for 1 h, was added ethyl acetate (40kg) and water (45kg), and the organic layer was separated and the mixture. Once (45.4kg) aqueous solution consisting of (9.01kg) sodium chloride and potassium hydrogen sulfate (1.35kg), sodium chloride aqueous solution (weight 10%, 44.5kg), sodium chloride aqueous solution (the organic layer was washed successively 20% by weight, in 45.0kg), and the solvent was evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (18kg) and the residue obtained was then evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (13.2kg) again and the residue obtained was obtained as ethylene glycol dimethyl ether solution of the title compound. I was used as it was in the six step.
1 H-NMR (CDCl 3) δ: 3.54 (3H, s), 3.77 (6H, s), 3.811 (3H, s), 3.812 (3H, s), 4.23 ( 1H, dd, J = 2.8,11.9 Hz), 4.32 (1H, dd, J = 4.0,11.9 Hz) ,4.36-4 .40 (1H, m), 5.11 -5.24 (5H, m), 5.41 (1H, d, J = 9.8Hz), 5.51 (1H, t, J = 9.8Hz), 7.25 (1H, d, J = 7.5Hz), 7.42 (1H, d, J = 7.5Hz), 7.44 (1H, s).
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
Step 6: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3 ’4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2'-[2H] pyran isobenzofuran] spiro

[(Methoxycarbonyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro – (1S, 3′R, 4′S, 5′S, 6′R) -6 which had been prepared in Step 5 – 3 ‘, 4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – spiro [isobenzofuran -1 (3H), 2'-[2H] pyran] Ethylene glycol dimethyl ether in solution, 2 – (2.46kg, 17.8mol), 4 butanol (25kg), anhydrous potassium carbonate – - methyl-2 were sequentially added (3.73kg, 24.9mol) ethyl phenyl boronic acid, in the reaction vessel was replaced with argon atmosphere, was bubbled with argon mixture. To the mixture – after the addition (0.72kg, 0.88mol) and palladium (II) chloride dichloromethane adduct [1,1 '-bis (diphenylphosphino) ferrocene], it was replaced with argon again inside of the vessel, one at 80 ℃ I was stirring time. After cooling, I added sequentially (0.859kg, 5.3mol) of ethylene glycol dimethyl ether (9.85kg), ethyl acetate (19kg), N-acetyl-L-cysteine in the mixture. After stirring for 2.5 h the mixture was filtered and added Celite (5.22kg), and washed with ethyl acetate (78kg) and the filter residue. The combined washings and filtrate, and the solvent is evaporated off under reduced pressure, and in addition (0.58kg, 3.6mol) and ethanol (74kg), N-acetyl-L-cysteine residue was obtained, which is heated to 70 ℃ or I was dissolved residue is then. After addition of water (9.4kg) in the solution, cooled to 60 ℃, and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more The mixture was stirred for 1 hour or more at 5 ℃ less. Centrifuge the resulting solid was washed twice with a mixture of water (35kg) and ethanol (55kg). Was dissolved at 70 ℃ ethanol (77kg) again, wet powder was obtained (10.21kg), cooled to 60 ℃ added water (9.7kg), and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more, and the mixture was stirred for 1 hour or more at 5 ℃ less. (9.45kg, dry powder rate 8.47kg, 13.7mol which was centrifuged obtained crystals were washed with a mixture of water (32kg) and ethanol (51kg), was obtained as a moist powder the title compound, 77% overall yield from the previous step).
1 H-NMR (CDCl 3) δ: 1.20 (3H, t, J = 7.5Hz), 2.60 (2H, q, J = 7.5Hz), 3.50 (3H, s), 3 .76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J = 2.8,11 .9 Hz), 4.33 (1H, dd, J = 4.5,11.9 Hz) ,4.36-4 .40 (1H, m) ,5.11-5 .20 (3H, m), 5 .41 (1H, d, J = 10.0Hz), 5.51 (1H, t, J = 10.0Hz) ,7.07-7 .11 (4H, m), 7.14 (1H, d, J = 7.8Hz), 7.19 (1H, dd, J = 1.5,7.8 Hz), 7.31 (1H, d, J = 1.5Hz).
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
Step 7: (1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-6 , 4 ‘, 5′-Preparation of triol’ – -3 [(3H), 2'-[2H] pyran isobenzofuran] spiro – (hydroxymethyl) ‘
(1S, 3′R, 4′S, 5′S, 6′R) -6 – [(4 - ethyl-phenyl) methyl] -3 ‘, 4′, 5 ‘, 6′-tetrahydro-3′, 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – wet powder spiro [(3H), 2'-[2H] pyran isobenzofuran -1] (8.92kg, In addition at 20 ℃ (4mol / L, 30.02kg, the 104.2mol) aqueous solution of sodium hydroxide, 1 hour the reaction mixture to a solution of (28kg) ethylene glycol dimethyl ether dry end conversion 8.00kg, of 12.9mol) the mixture was stirred. And the organic layer was separated by addition of water (8.0kg) in the mixture. The ethyl acetate aqueous sodium chloride solution (25 wt%, 40kg) and a (36kg) in the organic layer and the aqueous layer was removed after washing. The washed again aqueous sodium chloride solution (25 wt%, 40kg) in the organic layer was evaporated under reduced pressure. Were added and acetone (32.0kg) water (0.8kg) residue was obtained. After the solvent was evaporated under reduced pressure, dissolved in acetone (11.7kg) in water (15.8kg) and the residue obtained was cooled to below 5 ℃. Was added below 10 ℃ water (64kg) to the mixture, and the mixture was stirred for 1 hour at below 10 ℃. Centrifuge the resulting crystals were washed with a mixture of water (8.0kg) and (1.3kg) acetone. For 8 hours through-flow drying 13 ~ 16 ℃ temperature ventilation, under the conditions of 24-33% relative humidity the wet powder, the monohydrate crystal (3.94kg, 9.7mol, 75% yield) of the title compound I was obtained as: (4.502 wt% water content).
Method of measuring the amount of water:
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
MS (ESI +): 387 [M +1] +.
………………………..
Example 1 Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose Step 1: Synthesis of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one

To a solution of D-(+)-glucono-1,5-lactone (7.88 kg) and N-methylmorpholine (35.8 kg) in tetrahydrofuran (70 kg) was added trimethylsilyl chloride (29.1 kg) at 40° C. or below, and then the mixture was stirred at a temperature from 30° C. to 40° C. for 2 hours. After the mixture was cooled to 0° C., toluene (34 kg) and water (39 kg) were added thereto. The organic layer was separated and washed with an aqueous solution of 5% sodium dihydrogen phosphate (39.56 kg×2) and water (39 kg×1). The solvent was evaporated under reduced pressure to give the titled compound as an oil. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74-3.83 (3H, m), 3.90 (1H, t, J=8.0 Hz), 3.99 (1H, d, J=8.0 Hz), 4.17 (1H, dt, J=2.5, 8.0 Hz).
Step 2: Synthesis of 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene

Under a nitrogen atmosphere, to a solution of 2,4-dibromobenzyl alcohol (40 g, 0.15 mol) in tetrahydrofuran (300 ml) was added 2-methoxypropene (144 ml, 1.5 mol) at room temperature, and then the mixture was cooled to 0° C. At the same temperature, pyridinium p-toluenesulfonic acid (75 mg, 0.30 mmol) was added and the mixture was stirred for 1 hour. The reaction mixture was poured into a saturated aqueous solution of sodium hydrogen carbonate cooled to 0° C., and extracted with toluene. The organic layer was washed with a saturated aqueous solution of sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to give the titled compound as an oil in quantitative yield. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 1.44 (6H, s), 3.22 (3H, 4.48 (2H, s), 7.42 (1H, d, J=8.0 Hz), 7.44 (1H, dd, J=1.5, 8.0 Hz), 7.68 (1H, d, J=1.5 Hz).
Step 3: Synthesis of 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran

Under a nitrogen atmosphere, 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene (70 g, 207 mmol), which was obtained in the previous step, was dissolved in toluene (700 mL) and t-butylmethyl ether (70 ml), and n-butyllithium in hexane (1.65 M, 138 ml, 227 mmol) was added dropwise at 0° C. over 30 minutes. After the mixture was stirred for 1.5 hours at 0° C., the mixture was added dropwise to a solution of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one (Example 1, 108 g, 217 mol) in tetrahydrofuran (507 ml) at −78° C., and the reaction mixture was stirred for 2 hours at the same temperature. Triethylamine (5.8 ml, 41 mmol) and trimethylsilyl chloride (29.6 ml, 232 mmol) were added thereto, and the mixture was warmed to 0° C. and stirred for 1 hour to give a solution containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-bromo-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran.
The resulting solution was cooled to −78° C., and n-butyllithium in hexane (1.65 M, 263 ml, 434 mmol) was added dropwise thereto at the same temperature. After the mixture was stirred at −78° C. for 30 minutes, 4-ethylbenzaldehyde (62 ml, 455 mmol) was added dropwise at −78° C., and the mixture was stirred at the same temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the organic layer was separated, and washed with water. The solvent was evaporated under reduced pressure to give a product containing the titled compound as an oil (238 g). The product was used in the next step without further purification.
A portion of the oil was purified by HPLC (column: Inertsil ODS-3, 20 mm I.D.×250 mm; acetonitrile, 30 mL/min) to give four diastereomers of the titled compound (two mixtures each containing two diastereomers).
Mixture of Diastereomers 1 and 2:
1H-NMR (500 MHz, CDCl3) δ: −0.47 (4.8H, s), −0.40 (4.2H, s), −0.003-0.004 (5H, m), 0.07-0.08 (1314, m), 0.15-0.17 (18H, m), 1.200 and 1.202 (3H, each t, J=8.0 Hz), 1.393 and 1.399 (3H, each s), 1.44 (3H, s), 2.61 (2H, q, J=8.0 Hz), 3.221 and 3.223 (3H, each s), 3.43 (1H, t, J=8.5 Hz), 3.54 (1H, dd, J=8.5, 3.0 Hz), 3.61-3.66 (1H, m), 3.80-3.85 (3H, m), 4.56 and 4.58 (1H, each d, J=12.4 Hz), 4.92 and 4.93 (1H, each d, J=12.4 Hz), 5.80 and 5.82 (1H, each d, J=3.0 Hz), 7.14 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.50-7.57 (2H, m).
MS (ESI+): 875 [M+Na]+.
Mixture of Diastereomers 3 and 4:
1H-NMR (500 MHz, toluene-d8, 80° C.) δ: −0.25 (4H, s), −0.22 (5H, s), 0.13 (5H, s), 0.16 (4H, s), 0.211 and 0.214 (9H, each s), 0.25 (9H, s), 0.29 (9H, s), 1.21 (3H, t, J=7.5 Hz), 1.43 (3H, s), 1.45 (3H, s), 2.49 (2H, q, J=7.5 Hz), 3.192 and 3.194 (3H, each s), 3.91-4.04 (4H, m), 4.33-4.39 (2H, m), 4.93 (1H, d, J=14.5 Hz), 5.10-5.17 (1H, m), 5.64 and 5.66 (1H, each s), 7.03 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.59-7.64 (1H, m), 7.87-7.89 (1H, m).
MS (ESI+): 875 [M+Na]+.
Step 4: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

Under a nitrogen atmosphere, the oil containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran (238 g), which was obtained in the previous step, was dissolved in acetonitrile (693 ml). Water (37 ml) and 1N HCl aq (2.0 ml) were added and the mixture was stirred at room temperature for 5.5 hours. Water (693 ml) and n-heptane (693 ml) were added to the reaction mixture and the aqueous layer was separated. The aqueous layer was washed with n-heptane (693 ml×2), and water was evaporated under reduced pressure to give a product containing water and the titled compound (a diastereomer mixture) as an oil (187 g). The product was used in the next step without further purification.
1H-NMR (500 MHz, CD3OD) δ: 1.200 (3H, t, J=7.7 Hz), 1.201 (3H, t, J=7.7 Hz), 2.61 (2H, q, J=7.7 Hz), 3.44-3.48 (1H, m), 3.63-3.68 (111, m), 3.76-3.84 (4H, m), 5.09 (1H, d, J=12.8 Hz), 5.15 (1H, d, J=12.8 Hz), 5.79 (1H, s), 7.15 (2H, d, J=7.7 Hz), 7.24 and 7.25 (1H, each d, J=8.4 Hz), 7.28 (2H, d, J=7.7 Hz), 7.36 (1H, dd, J=8.4, 1.5 Hz), 7.40-7.42 (114, m).
MS (ESI+): 425 [M+Na]+.
Step 5: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (crude product)

To a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (187 g), which was obtained in the previous step, in 1,2-dimethoxyethane (693 ml) was added 5% Pd/C (26 g, 6.2 mmol, water content ratio: 53%), and the mixture was stirred in the atmosphere of hydrogen gas at room temperature for 4 hours. After filtration, the filtrate was evaporated under reduced pressure to give an oil containing the titled compound (59 g). The purity of the resulting product was 85.7%, which was calculated based on the area ratio measured by HPLC. The product was used in the next step without further purification.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (3H, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Cadenza CD-C18 50 mm P/NCD032
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Gradient operation: Eluent B: 5% to 100% (6 min), 100% (2 min)
Flow rate: 1.0 mL/min
Temperature: 35.0° C.
Detection wavelength: 210 nm
Step 6: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose
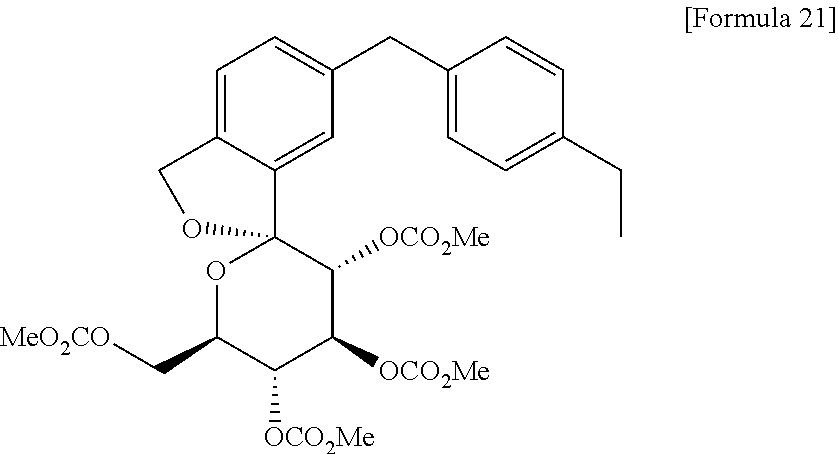
Under a nitrogen atmosphere, to a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (59 g) and 4-(dimethylamino)pyridine (175 g, 1436 mmol) in acetonitrile (1040 ml) was added dropwose methyl chloroformate (95 ml, 1231 mmol) at 0° C. The mixture was allowed to warm to room temperature while stirred for 3 hours. After addition of water, the mixture was extracted with isopropyl acetate. The organic layer was washed with an aqueous solution of 3% potassium hydrogensulfate and 20% sodium chloride (three times) and an aqueous solution of 20% sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. To the resulting residue was added ethanol (943 mL) and the mixture was heated to 75° C. to dissolve the residue. The mixture was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (472 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound (94 g). To the product (91 g) was added ethanol (1092 ml), and the product was dissolved by heating to 75° C. The solution was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (360 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound [83 g, total yield from 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene used in Step 3: 68%].
1H-NMR (CDCl3) δ: 1.20 (3H, t, J=7.5 Hz), 2.60 (2H, q, J=7.5 Hz), 3.50 (3H, s), 3.76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J=2.5, 11.8 Hz), 4.33 (1H, dd, J=4.5, 12.0 Hz), 4.36-4.40 (1H, m), 5.11-5.20 (3H, m), 5.41 (1H, d, J=10.0 Hz), 5.51 (1H, t, J=10.0 Hz), 7.07-7.11 (4H, m), 7.14 (1H, d, J=7.5 Hz), 7.19 (1H, dd, J=1.5, 7.8 Hz), 7.31 (1H, d, J=1.5 Hz).
MS (ESI+): 619 [M+1]+, 636 [M+18]+.
Another preparation was carried out in the same manner as Step 6, except that a seed crystal was not used, to give the titled compound as a crystal.
Step 7: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

To a solution of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose (8.92 kg as wet powder, corresponding to 8.00 kg of dry powder) in 1,2-dimethoxyethane (28 kg) was added a solution of sodium hydroxide (4 mol/L, 30.02 kg) at 20° C., and the mixture was stirred for 1 hour. Water (8.0 kg) was added to the mixture and the layers were separated. To the organic layer were added an aqueous solution of 25% sodium chloride (40 kg) and ethyl acetate (36 kg). The organic layer was separated, washed with an aqueous solution of 25% sodium chloride (40 kg), and the solvent was evaporated under reduced pressure. The purity of the resulting residue was 98.7%, which was calculated based on the area ratio measured by HPLC. To the resulting residue were added acetone (32.0 kg) and water (0.8 kg), and the solvent was evaporated under reduced pressure. To the resulting residue were added acetone (11.7 kg) and water (15.8 kg), and the solution was cooled to 5° C. or below. Water (64 kg) was added to the solution at 10° C. or below, and the mixture was stirred at the same temperature for 1 hour. The resulting crystal was collected by centrifugation, and washed with a mixture of acetone (1.3 kg) and water (8.0 kg). The resulting wet powder was dried by ventilation drying under a condition at air temperature of 13 to 16° C. and relative humidity of 24% to 33% for 8 hours, to give a monohydrate crystal (water content: 4.502%) of the titled compound (3.94 kg). The purity of the resulting compound was 99.1%, which was calculated based on the area ratio measured by HPLC.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (311, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Capcell pack ODS UG-120 (4.6 mm I.D.×150 mm, 3 μm, manufactured by Shiseido Co., Ltd.)
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Mobile phase sending: Concentration gradient was controlled by mixing Eluent A and Eluent B as indicated in the following table.
| TABLE 1 | ||||
| Time from | ||||
| injection (min) | Eluent A (%) | Eluent B (%) | ||
| 0 to 15 | 90→10 | 10→90 | ||
| 15 to 17.5 | 10 | 90 | ||
| 17.5 to 25 | 90 | 10 | ||
Flow rate: 1.0 mL/min
Temperature: 25.0° C.
Detection wavelength: 220 nm
Method for Measurement of Water Content:
Analysis method: coulometric titration method
KF analysis apparatus: Type KF-100 (trace moisture measuring apparatus manufactured by Mitsubishi Chemical Corporation)
Anode solution: Aquamicron AX (manufactured by Mitsubishi Chemical Corporation)
Cathode solution: Aquamicron CXU (manufactured by Mitsubishi Chemical Corporation)
…………………..
The compound of the present invention can be synthesized as shown in Scheme 1:

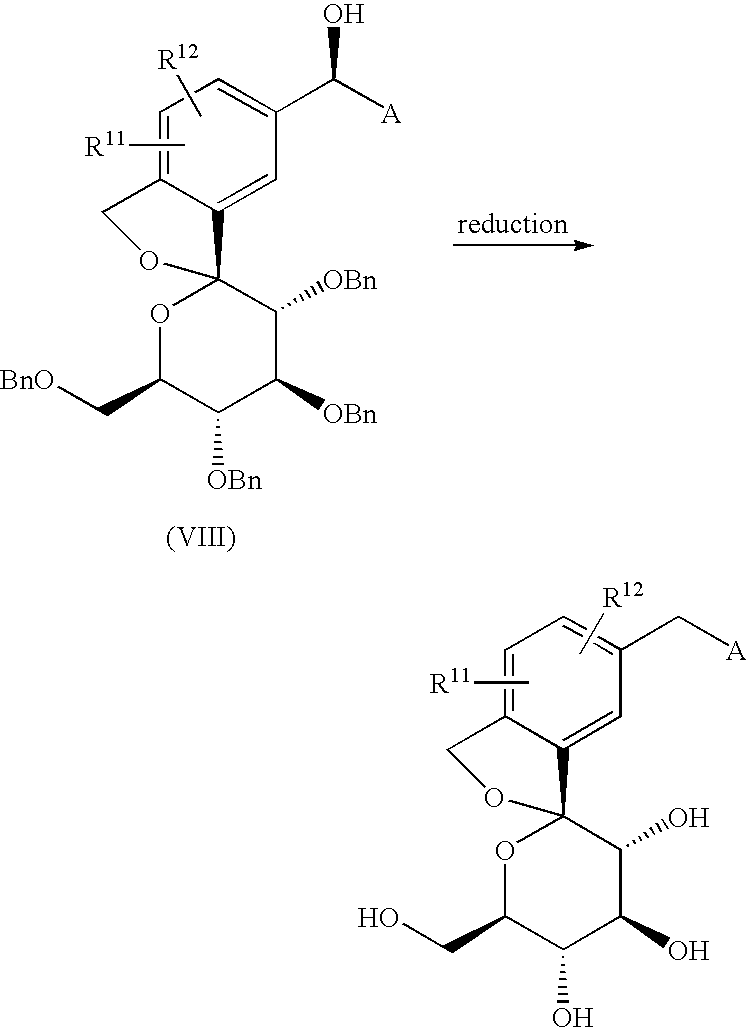
wherein R11 and R12 have the same meaning as defined above for substituents on Ar1, A is as defined above, and P represents a protecting group for a hydroxyl group.
No comments:
Post a Comment